

Background: Argininosuccinate synthase (AS) is critical for endothelial nitric oxide production, yet little is known about its regulation.
Results: AS Ser-328 phosphorylation increased with calcium stimulation and decreased with PKCα interference.
Conclusion: PKCα phosphorylates AS at Ser-328 under calcium-dependent stimulatory conditions to support nitric oxide production.
Significance: Knowledge of how AS is regulated is essential in understanding nitric oxide homeostasis.
Keywords: Endothelial Cell, Nitric Oxide, Nitric-oxide Synthase, Phosphorylation, Protein kinase C (PKC), Argininosuccinate Synthase
Abstract
Endothelial nitric-oxide synthase (eNOS) utilizes l-arginine as its principal substrate, converting it to l-citrulline and nitric oxide (NO). l-Citrulline is recycled to l-arginine by two enzymes, argininosuccinate synthase (AS) and argininosuccinate lyase, providing the substrate arginine for eNOS and NO production in endothelial cells. Together, these three enzymes, eNOS, AS, and argininosuccinate lyase, make up the citrulline-NO cycle. Although AS catalyzes the rate-limiting step in NO production, little is known about the regulation of AS in endothelial cells beyond the level of transcription. In this study, we showed that AS Ser-328 phosphorylation was coordinately regulated with eNOS Ser-1179 phosphorylation when bovine aortic endothelial cells were stimulated by either a calcium ionophore or thapsigargin to produce NO. Furthermore, using in vitro kinase assay, kinase inhibition studies, as well as protein kinase Cα (PKCα) knockdown experiments, we demonstrate that the calcium-dependent phosphorylation of AS Ser-328 is mediated by PKCα. Collectively, these findings suggest that phosphorylation of AS at Ser-328 is regulated in accordance with the calcium-dependent regulation of eNOS under conditions that promote NO production and are in keeping with the rate-limiting role of AS in the citrulline-NO cycle of vascular endothelial cells.
Introduction
Nitric oxide (NO) production is strictly regulated in vascular endothelial cells. Impairment of this control is associated with risk factors that compromise endothelial function. Arginine directed to eNOS2 for purposes of NO production is maintained by the recycling of citrulline to arginine, creating a distinct pool of arginine in endothelial cells (1–4). This intracellular arginine pool that is sequestered from the bulk cytosolic arginine is generated through the action of the arginine recycling enzymes argininosuccinate synthase (AS) and argininosuccinate lyase, where catalysis by AS is rate-limiting for NO production in endothelial cells (1).
Because the endothelium represents a unique environment under the continuous influence of signaling cascades that integrate responses to physiologic cues, it was logical to propose that acute or immediate regulation of AS would be necessary to accommodate regulatory responses already known for eNOS. For example, VEGF and insulin signaling have been shown to coordinately regulate two phosphorylation sites of eNOS, increasing phosphorylation at Ser-1179 and decreasing phosphorylation at Thr-497 to effect an overall increase in eNOS activity (6). This change in eNOS phosphorylation that results from either VEGF or insulin signaling is mediated largely by calcium-independent kinases, such as Akt (7).
Despite the fact that relatively recent studies have shown that AS is a phospho-protein (8, 9), no physiologically relevant phosphorylation sites have been reported. In 2008, an automated phospho-proteome analysis revealed that AS is phosphorylated at serine 352 in HeLa cells, but the physiologic significance was not established (9). Later that same year, Corbin et al. (8) also demonstrated that AS was a phospho-protein and that VEGF stimulation of NO production affected the phosphorylation of AS via a PKA pathway, but no phosphorylation site was identified. In this study, we now show that phosphorylation of AS at Ser-328 occurs in response to the calcium-dependent activation of eNOS and is mediated by the α isotype of PKC (PKCα).
EXPERIMENTAL PROCEDURES
Cell Culture
BAECs were isolated from whole bovine aorta as described in Ref. 10 with modifications. Briefly, aortas were harvested and washed with PBS without magnesium or calcium to remove blood, and lumenal surfaces were exposed by longitudinal dissection followed by incubation with 0.1% collagenase for 10 min. Endothelial cell isolation was achieved by rolling sterile swabs across the surface and then releasing cells by twirling swabs in 5 ml of enriched Dulbecco's modified Eagle's medium (DMEM, 1 g/liter glucose, Invitrogen) containing 10% FBS (HyClone) with 100 IU/ml penicillin and streptomycin and 250 ng/ml amphotericin B (Cellgro) in 15-ml conical tubes. Cells were pelleted, resuspended in 6 ml of DMEM + 10% FBS, and plated in 25-cm2 treated cell culture flasks. Authentication of isolation of endothelial cells in culture was confirmed by flow cytometry.
Prior to treatment, confluent BAECs (between passages 3 and 9) were serum-starved for 16 h in DMEM (1 g/liter glucose, minus phenol red, Invitrogen) supplemented with 0.1% BSA and 1 mm glutamine. Treatments included incubation with 100 nm insulin (Sigma), 100 ng/ml VEGF (R&D Systems), 0.5 μm calcium ionophore A23187 (Sigma), 10 μm bradykinin (Sigma), 42 μm rottlerin, or 2.5 μm bisindolylmaleimide I (Sigma) for PKC inhibition or 50 μm BAPTA-AM (Calbiochem). Incubation times are indicated in the figure legends. Controls were treated with serum starvation media only.
Generation of AS Variants and Transient Transfections
AS cDNA was cloned into pcDNA 3.1/V5-His, B expression vector (Invitrogen). The AS construct was then subjected to site-directed mutagenesis via the QuikChange kit (Stratagene) per the manufacturer's instructions. Briefly, Ser-328 was mutated to alanine to mimic a nonphosphorylated state and to aspartic acid to mimic a constitutively phosphorylated state. Primers used for S328A mutation were sense: ACGGGTTTCTGGCACGCGCCCGAGTGTGAATTT and antisense: AAATTCACACTCGGGCGCGTGCCAGAAACCCGT. Primers used for S328D mutation were sense: ACGGGTTTCTGGCACGACCCCGAGTGTGAATTT and antisense: AAATTCACACTCGGGGTCGTGCCAGAAACCCGT. Experimental plasmids (1 μg DNA/well of a 6-well dish) were transiently transfected into BAECs using Lipofectamine 2000 (Invitrogen) in serum-free Opti-MEM I (Invitrogen). After 4 h, medium was replaced with DMEM containing 10% serum, and cells were cultured for 6 or 24 h. Cells were stimulated with calcium ionophore A23187 and sodium orthovanadate for 4 h, and then culture medium was assayed for NO production via the 2,3-diaminonaphthalene (DAN) assay.
Mass Spectrometry Analysis
BAECs transiently transfected with AS were incubated for 24 h, serum-starved for 16 h, and treated with 10 μm bradykinin or 50 nm okadaic acid for 30 min. Overexpressed AS was purified by its fused His6 tag using nickel-nitrilotriacetic acid-agarose magnetic beads (Qiagen). Proteins were separated by SDS-PAGE to identify the AS band (51 kDa). A duplicate gel was run to confirm expression of the AS plasmid by Western blot. Gel bands of interest were excised and destained. The protein disulfides were reduced with triscarboxyethylphosphine, and the cysteines were alkylated with iodoacetamide. In-gel trypsin digestion was used for proteolysis. Nanoflow reverse phase liquid chromatography was used to separate the peptides by hydrophobicity (LC Packings, Dionex, Sunnyvale, CA). Online detection was accomplished with an electrospray linear ion trap mass spectrometer (LTQ, Thermo Scientific). Tandem mass spectra were assigned to peptide sequences using Mascot and SEQUEST database search algorithms. Sequence assignments were validated by manual inspection of the data.
Argininosuccinate Synthase Phospho-serine Ser-328 Specific Antibody
A phospho-specific antibody against Ser(P)-328 AS was generated by 21st Century Biochemicals using the phospho-peptide AS sequence 322–335: RR-Ahx-YGFWH[pS]PECEFVR-amide (where pS indicates phospho-serine). The phospho-AS antibody specificity was determined by performing phospho-peptide competition and immunoprecipitation, followed by Western blotting.
Western Blot Analysis
BAECs were lysed in radio immunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Pierce) followed by scraping for collection. Whole cell lysates were clarified, and total protein was quantitated using the BCA protein assay (Pierce). Equal amounts (20 μg) of protein were resolved on 4–15% Tris-HCl TGX precast gels (Bio-Rad) and blotted onto Immobilon-P polyvinylidene difluoride membranes. Western blotting was performed as described previously (11). Membranes were blocked in 100% StartingBlock (Pierce) and incubated with primary antibody, 1:2500 anti-AS (BD Transduction Laboratories), in 20% StartingBlock. Secondary antibody used was peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch Laboratories) at 1:50,000 dilution. Blots were visualized by chemiluminescence using West Pico reagent (Pierce) and exposed to film. Band intensities were quantitated using the Quantity-One software (Bio-Rad).
Nitric Oxide Determinations
Nitrite was measured in the medium as an indicator of cellular NO using the fluorescent DAN (Sigma) assay as described previously (12). Reactions were carried out in 96-well black plates, and fluorescence was read on a BMG FLUOstar Galaxy spectrofluorometer plate reader exciting at 360 nm and detecting emission at 405 nm. Data are described as pmol of nitrite per microgram protein.
PKCα Knockdown Assay
BAECs, 90% confluent, were transfected for 48 h with dicer substrate siRNA (DsiRNA) directed against the fifth exon of PKCα (Integrated DNA Technologies) using Lipofectamine 2000 (Invitrogen) performed as described previously for AS constructs. The duplex sequence used to silence the gene expression was the following: 5′-CGAGGAGCAAGCACAAGUUCAAGAT-3′ and 5′-AUCUUGAACUUGUGCUUGCUCCUCGGG-3′. Transfected BAECs were then treated with calcium ionophore (A23187, Sigma) and lysed followed by Western blotting. Relative amounts of indicated proteins were quantitated as described above.
In Vitro Kinase Assay
Assays were performed as described by Corbin et al. (8). Briefly, purified recombinant AS was generated via subcloning bovine AS (NP_776317) into the pET-28(c)+ vector (Novagen) and expressed in Escherichia coli. The protein was subsequently purified via the His·Bind resin per the manufacturer's instructions (Novagen). Successful purification was verified by SDS-PAGE. The positive control used to determine kinase activity for each kinase contained applicable peptide substrate, whereas negative control contained all components except peptide. The addition of [33P]ATP to each reaction mixture initiated the assays incubated at 30 °C for 30 min. Assay termination was achieved by reaction spotting on phospho-cellulose P81 plates followed by three washes, and then radioactivity was read via scintillation counting. Imaged in vitro kinase reactions were terminated by the addition of sample buffer and then fractionated on 10% polyacrylamide gels.
RESULTS
Identification of the Argininosuccinate Synthase Ser-328 Phosphorylation Site
To establish biologically significant sites of phosphorylation in the AS sequence, we carried out a proteomic analyses by applying mass spectrometry (LC-MS/MS) on overexpressed AS purified from cultured BAECs treated with bradykinin or okadaic acid (protein phosphatase 2A (PP2A) and PP1 phosphatase inhibitor). Mass spectrometry analysis revealed three highly conserved serine/threonine phosphorylation sites that were distinguished from background ion signal. These included Thr-131/Ser-134, Ser-180/Ser-189, and Ser-328 (Fig. 1). The proximity of Thr-131 to Ser-134 and Ser-180 to Ser-189 made these individual signals indistinguishable and therefore described as Thr-131/Ser-134 and Ser-180/Ser-189.
FIGURE 1.
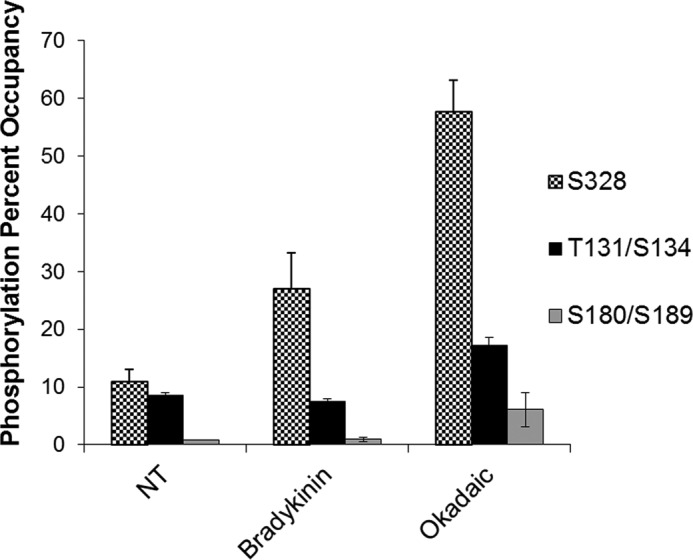
Ser-328 (S328) is an exposed site accessible by kinases and shows change in phosphorylation relative to treatment. BAECs were transiently transfected with AS expression vector and then treated with 10 μm bradykinin or 50 nm okadaic acid. AS was His tag-purified and then subjected to SDS-PAGE. The band corresponding to AS was excised and trypsin-digested before tandem mass spectrometry analysis. Results represent one experiment with samples analyzed in triplicate with S.E. represented by error bars. NT, nontreated.
Of the sites identified, Ser-328 showed the greatest change in signal between cells treated with phosphatase inhibitor (okadaic acid) and nontreated cells. In addition, Ser-328 showed substantial change in signal with bradykinin treatment relative to nontreated control cells (Fig. 1). Furthermore, in silico modeling showed that the most accessible sites for kinase interaction were Thr-131/Ser-134 and Ser-328, whereas Ser-180/Ser-189 appeared buried near the active site of the enzyme (data not shown). Based on these results, Ser-328 was selected as being the strongest candidate for phosphorylation in vivo. Consequently, a phospho-specific antibody directed against Ser(P)-328 was developed for further investigation (see “Experimental Procedures”).
Calcium-independent Stimulation of BAECs Decreases Phosphorylation at Ser-328
To determine whether phosphorylation at Ser-328 had physiological significance, changes in Ser-328 phosphorylation were examined relative to treatment by physiological cues that alter NO production. In particular, both VEGF and insulin stimulation of NO production were chosen for two reasons. First, there is evidence that AS phosphorylation changed in response to VEGF treatment, which was mediated via PKA signaling (8). Second, both VEGF and insulin are known to stimulate endothelial NO production by promoting phosphorylation of Ser-1179 of eNOS (13, 14).
As shown in Fig. 2, and consistent with the literature (13), we found that insulin and VEGF promoted an increase in phosphorylation by roughly 50% of eNOS at Ser-1179 (Western blot, Fig. 2, A and C; densitometry, Fig. 2, B and D), with a corresponding increase in NO production (Fig. 2E). There was also concomitant decrease in phosphorylation of eNOS at Thr-497 by ∼40% (Western blot, Fig. 2, A and C; densitometry, Fig. 2, B and D) (15–17). Surprisingly, phosphorylation at Ser-328 was markedly reduced by treatment with either VEGF (85%) or insulin (40%) (Western blot, Fig. 2, A and C; densitometry, Fig. 2, B and D).
FIGURE 2.

Calcium-independent stimulation of NO production decreased phosphorylation at argininosuccinate synthase Ser-328 (S328). A and C, BAECs were treated with insulin (Ins) at 100 nm (A) or VEGF at 100 ng/ml (C) for 20 min. Phosphorylation (P) was assayed by Western blotting using phospho-specific antibodies and reprobed with eNOS and AS specific antibodies. Blots are representative of three separate experiments. NT, nontreated. B and D, densitometric values represent the percentage of change from nontreated controls normalized to total eNOS or AS, respectively. Densitometric changes are significant with S.E. represented by error bars (n = 3, p < 0.05). E, culture medium from BAECs treated with insulin or VEGF was assayed for nitrite with the DAN assay with S.E. represented by error bars (*, n = 3, p < 0.05).
Calcium-dependent Stimulation of BAECs Increases Phosphorylation at Ser-328
Insulin and VEGF mediate eNOS phosphorylation at Ser-1179 via the PI3K/Akt pathway, which is largely calcium-independent (18–20). Because calcium-independent signaling mediated by either insulin or VEGF promoted the apparent dephosphorylation of AS Ser-328, we then questioned whether phosphorylation at Ser-328 was involved with calcium-dependent signaling. To initially explore this question, site-directed mutagenesis analysis was carried out. Three constructs were used, one containing wild type (WT) AS, one containing an Ala substitution for Ser-328 (phospho-null), and one containing an Asp substitution for Ser-328 (phospho-mimetic). As shown in Fig. 3A, the overexpression of phospho-mimetic S328D AS enhanced NO production similar to WT AS when stimulated with calcium ionophore. Conversely, overexpression of phospho-null S328A AS failed to enhance NO production in response to the same treatment. Consistent expression of the constructs is shown by Western blotting (Fig. 3B). The observation that only S328D overexpression increased NO production to the level of WT suggested that phosphorylation at Ser-328 supports arginine delivery to eNOS under calcium-dependent stimulatory conditions.
FIGURE 3.

Overexpressed Ser-328 phospho-null mutation to alanine produced significantly less stimulated NO than WT or phospho-mimetic (S328D). A, BAECs were transiently transfected with WT, phospho-null serine to alanine 328 (S328A), or phospho-mimetic serine to aspartate (S328D) AS and then stimulated with 0.5 μm calcium ionophore A23187 and 50 μm sodium orthovanadate. Results are representative of four different experiments with S.E. noted by error bars. (*, p < 0.05 relative to WT; †, p < 0.05 relative to S328D). B, representative Western blot confirming WT, S328A, and S328D overexpression.
Because these findings were consistent with calcium dependence, experiments were then carried out to determine whether calcium-dependent changes in Ser-328 could be demonstrated for endogenous AS. For these experiments, BAECs were treated with either the calcium ionophore, A23187, or the endoplasmic reticulum ATPase inhibitor, thapsigargin. Both agents are known to promote an intracellular increase in calcium (21) and stimulate NO production. As shown in Fig. 4, A and B, treatment with either agent promoted phosphorylation of eNOS at Ser-1179 by nearly 50% with a subsequent decrease in phosphorylation of eNOS at Thr-497 by 40%, consistent with reported values (21, 22). However, in this case, treatment with calcium ionophore or thapsigargin demonstrated an increase in phosphorylation at AS Ser-328 (∼60%) (Fig. 4, A and B).
FIGURE 4.

Calcium-dependent stimulation of NO production increased phosphorylation at argininosuccinate synthase Ser-328. A, Western blot analysis of changes in phosphorylation (P) relative to 20 min of treatment with calcium ionophore (A23187) and thapsigargin (Thap) in the presence or absence of intracellular calcium chelator BAPTA-AM. Blots are representative of five separate experiments. B, densitometric values represent the percentage of change from nontreated (NT) controls normalized to total eNOS or AS, respectively. Densitometric changes are significant with S.E. represented by error bars. C, culture medium from BAECs treated with calcium ionophore (A23187) and thapsigargin in the presence or absence of BAPTA-AM was assayed for nitrite with the DAN assay. (n = 5, significantly increased from nontreated: *, p < 0.05, significantly decreased from A23187 alone ‡, p < 0.05, significantly decreased from thapsigargin alone; †, p < 0.05).
To further substantiate the calcium dependence of AS Ser-328 phosphorylation, BAECs were treated with the intracellular calcium chelator BAPTA-AM. As expected, calcium ionophore and thapsigargin rescued phosphorylation at eNOS Ser(P)-1179 that diminished with BAPTA-AM treatment alone. Importantly, as shown in Fig. 4A, treatment with 50 μm BAPTA-AM completely abolished phosphorylation at Ser-328, as well as the phosphorylation at eNOS Ser-1179. Thus, phosphorylation of AS Ser-328 appeared to be coordinately regulated with the phosphorylation of eNOS Ser-1179 through calcium-dependent signaling.
To demonstrate physiological relevance, NO measurements were taken after cells were treated with BAPTA-AM for 30 min followed by stimulation with calcium ionophore A23187 for 20 min. A23187 and thapsigargin significantly increased NO production relative to nontreated or BAPTA-AM-treated BAECs (Fig. 4C). In addition, BAPTA-AM treatment significantly decreased A23187- or thapsigargin-stimulated NO production by 2.5- and 2-fold, respectively.
PKC Is Involved in Mediating Ser-328 Phosphorylation in BAECs
To further elucidate the signaling pathway involved in the calcium-dependent phosphorylation of AS at Ser-328, a series of experiments was carried out using kinase inhibitors. Because the classical PKC isotypes are calcium-dependent kinases and are not involved in insulin or VEGF signaling (23, 24), we hypothesized that a member of the PKC family may be involved in the calcium-dependent phosphorylation of AS Ser-328. To test this hypothesis, non-isotype-specific PKC inhibition studies were carried out. As a first approach, the effects of PKC inhibition were directly measured by examining AS Ser-328 phosphorylation. As shown in Fig. 5A and B, treatment of BAECs with either the PKC inhibitor rottlerin or the PKC inhibitor bisindolylmaleimide I, followed by stimulation with calcium ionophore, decreased AS Ser-328 phosphorylation by ∼60% relative to nontreated controls. These results were taken to be consistent and supportive of the role of a classical PKC isotype promoting phosphorylation of AS at Ser-328.
FIGURE 5.

Non-isotype-specific inhibition of PKC with 42 μm rottlerin or 2.5 μm bisindolylmaleimide I decreased phosphorylation (P) at AS Ser-328. BAECs were pretreated with 42 μm rottlerin (Rott) or 2.5 μm bisindolylmaleimide I (Bis) for 30 min prior to stimulation with calcium ionophore (A23187) for 20 min. A, representative Western blot of three experiments. B, densitometric values represent the percentage of change from nontreated (NT) controls normalized to total eNOS or AS, respectively. Densitometric changes are significant with S.E. represented by error bars (n = 3, p < 0.05). C, culture medium from treated BAECs was assayed for nitrite with the DAN assay. A23187 significantly increased NO production with or without bisindolylmaleimide I (significance relative to nontreated: *, n = 3, p < 0.05; significance relative to A23187: ‡, n = 3, p < 0.05).
Interestingly, phosphorylation at Thr-497 or Ser-1179 of eNOS also decreased with treatment of rottlerin by ∼60%, irrespective of calcium stimulation. In contrast, when BAECS were treated with bisindolylmaleimide I alone, phosphorylation at Ser-1179 and Thr-497 of eNOS decreased by only 20%. However, upon calcium stimulation, phosphorylation at eNOS Ser-1179 increased to levels similar to calcium ionophore treatment alone. Nevertheless, phosphorylation at Ser-328 was unaffected by calcium in the presence of bisindolylmaleimide I (Fig. 5A).
To demonstrate the physiological relevance of the system in response to inhibitors, we measured NO production for each treatment (Fig. 5C). PKC inhibition with bisindolylmaleimide I has little effect on NO production by BAECs; however, rottlerin significantly reduced calcium ionophore-stimulated NO production by over 2-fold. These results suggest that rottlerin may affect the activity of several kinases, and bisindolylmaleimide I clearly decreased phosphorylation at eNOS Thr-497 and thus promoted NO production similar to calcium ionophore alone.
To further examine the potential involvement of other calcium-dependent kinases that could phosphorylate Ser-328, additional kinase inhibition studies were carried out. ERK1/2 was inhibited with PD98059, and Ca2+/calmodulin-dependent protein kinase II (CaMKII) was inhibited with KN93. In either case, no inhibitor effect was observed on AS Ser-328 phosphorylation when BAECs were co-treated with calcium ionophore or thapsigargin (data not shown). These results suggest that PKC, but not ERK1/2 or Ca2+/calmodulin-dependent protein kinase II, is responsible for the calcium-stimulated phosphorylation of AS Ser-328.
Because PKCα is a calcium-dependent kinase that has been shown to be active during calcium-dependent stimulation of eNOS (25), we sought to determine whether this is the isotype responsible for phosphorylating Ser-328 of AS. To test this, we examined the effects of direct knockdown of PKCα with DsiRNA. DsiRNA is different from traditional siRNA used for gene silencing in that the RNA duplex used to silence not only mimics dicer products, but is also optimized for dicer processing by the RNA-induced silencing complex (RISC) to result in a more potent knockdown (26). As shown in Fig. 6, significant knockdown of PKCα was achieved. DsiRNA directed against PKCα allowed for 50 and 75% knockdown at 1 and 5 nm DsiRNA, respectively. Knockdown of PKCα followed by treatment with calcium ionophore showed a decrease in phosphorylation of Ser-328 by 50 and 60% with 1 or 5 nm, respectively, and prevented increased phosphorylation in response to calcium ionophore treatment. Phosphorylation of eNOS at Ser-1179 was largely unaffected by PKCα knockdown under these conditions, whereas eNOS Thr-497 dropped ∼60% when PKCα was knocked down by 80%. These results suggest that PKCα is a kinase that phosphorylates Ser-328 of AS.
FIGURE 6.

Knockdown of PKCα decreased phosphorylation (P) at Ser-328 but not eNOS Ser-1179. BAECs were treated with 1 or 5 nm DsiRNA directed against the fifth exon of PKCα and allowed to express for 24 h, followed by stimulation with calcium ionophore A23187 for 20 min, and compared with nontreated BAECs. A, representative Western blot of three experiments. B, densitometric values represent the percentage of change from nontreated (NT) controls normalized to total eNOS or AS, respectively. Densitometric changes are significant (except for A2317 effect on PKCα expression or 1 nm DsiRNA treatment on pS1179 eNOS) with S.E. represented by error bars (n = 3, p < 0.05) (significance relative to nontreated or DsiRNA alone: *, n = 3, p < 0.05).
PKCα Directly Phosphorylates AS
Because an observed decrease in phosphorylation due to kinase knockdown may be the result of inhibition of an upstream kinase in a signaling cascade, a targeted in vitro kinase assay was performed to determine whether PKCα directly phosphorylates AS. Akt1 was used as a negative control based on the lack of a consensus Akt phosphorylation motif (RXRXX(S/T)) (27) in the AS protein sequence.
As shown in Fig. 7A, PKCα clearly demonstrated the capacity to phosphorylate AS in vitro. Although there was an increase in counts above the blank reaction with Akt1, this was below the threshold considered significant. SDS-PAGE fractionation of 32P-labeled kinase reaction products and visualization by autoradiography film exposure confirmed that PKCα, but not Akt1, phosphorylates AS (Fig. 7B).
FIGURE 7.

PKCα, but not Akt, phosphorylate AS in vitro. A, in vitro kinase assays. Gray bars represent the incubation of purified AS minus kinase (Blank). Black bars represent the incubation of purified AS with the indicated kinase (Kinase). Kinase activity is represented as cpm with error bars representing S.E. (n = 3, *, p < 0.05). B, representative autoradiograph of in vitro kinase reactions separated by SDS-PAGE as described in A.
DISCUSSION
Vascular endothelial NO production is a dynamic and targeted process, where multilevel regulation of eNOS is well established (28). Because AS is required for NO production, it was logical to speculate that the post-translational regulation of AS would play a significant role in maintaining NO homeostasis. Indeed, recent work using mass spectrometry analysis showed that AS is phosphorylated at site Ser-352 (human sequence) (9); however, the biological significance of this phosphorylation was not defined. In addition, Corbin et al. (8) showed that phosphorylation of AS was altered in response to VEGF treatment, yet the phosphorylation site was unknown. In this study, we demonstrated that AS Ser-328 phosphorylation supports the calcium-dependent stimulation of NO production in vascular endothelial cells and is mediated by PKCα.
The involvement of AS in catalyzing the rate-limiting step of the arginine recycling side of the citrulline-NO cycle is critical in sustaining NO production in endothelial cells (29). Consistent with the established transcriptional co-regulation between AS and eNOS (30–32), work described in this study, using Western blot analysis, showed that AS is co-regulated post-translationally with eNOS under conditions where the stimulation of NO production is dependent on calcium-mediated signaling. Further studies showed that calcium stimulation of BAECs overexpressing the phospho-mimetic (S328D) AS increased NO production to that of the overexpressed WT AS, whereas the overexpressed phospho-null (S328A) AS was unable to support this increase in NO production relative to the WT control (Fig. 3). Moreover, the finding that under basal conditions both the null and the phospho-mimetic AS mutant constructs were able to enhance NO production to that of the wild type construct also verified that both overexpressed AS mutants were metabolically active (data not shown). Finally, through kinase inhibitor studies and knockdown analyses, we were able to establish that the phosphorylation of AS Ser-328 under calcium-mediated stimulation of NO production was facilitated by PKCα.
Considering the essential role that AS plays in NO production and the spatial and temporal manner in which eNOS is regulated by calcium levels, it is not surprising that AS is susceptible to a regulatory mechanism governed by changes in calcium. Indeed, the fact that the calcium-independent activation of eNOS by VEGF or insulin resulted in a decrease in phosphorylation at AS Ser-328 further differentiated AS Ser-328 phosphorylation from the calcium-dependent pathway for eNOS activation.
Previously, our laboratory had shown that AS resides in caveolae with eNOS (33). Because PKCα is associated with the plasma membrane when activated (34), a local functional relationship between PKCα and AS may be supported. In addition, it has been reported that changes in intracellular calcium promote the cytosolic relocalization of eNOS (5). Therefore, it is also tempting to speculate that the phosphorylation of AS at Ser-328 may promote cytosolic AS relocalization in a manner consistent with eNOS in response to calcium-mediated signaling. This would then ensure maintenance of the functional relationship between arginine recycling and the translocated eNOS. Experiments are underway to test this hypothesis.
In summary, these results represent the first demonstration of a biologically relevant phosphorylation site for AS and are consistent with previous findings demonstrating the coordinate regulation of eNOS and AS relative to vascular endothelial NO production. Given the critical role of AS in vascular endothelial NO production and previous work from our laboratory showing that the overall phosphorylation of AS changes in response to VEGF treatment (8), these results also suggest that other sites of phosphorylation are in involved in AS regulation and that these sites merit further consideration.
Acknowledgments
We thank Drs. Wayne Guida and Daniel Santiago from the University of South Florida Department of Chemistry for expertise with in silico modeling, Dr. John Kooman from the H. Lee Moffitt Cancer Center and Research Institute, Proteomics Core Facility for the mass spectrometry analysis, and Dr. Denise Cooper for guidance with PKC inhibition.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL083153-01A2 (to Duane C. Eichler). This work was also supported by the James Esther King Biomedical Research Program Grant, Florida Department of Health (to Duane C. Eichler).
This article was selected as a Paper of the Week.
- eNOS
- endothelial nitric-oxide synthase
- AS
- argininosuccinate synthase
- BAEC
- bovine aortic endothelial cell
- BAPTA-AM
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester)
- DAN
- 2,3-diaminonaphthalene
- DsiRNA
- dicer substrate siRNA.
REFERENCES
- 1. Flam B. R., Eichler D. C., Solomonson L. P. (2007) Endothelial nitric oxide production is tightly coupled to the citrulline-NO cycle. Nitric Oxide 17, 115–121 [DOI] [PubMed] [Google Scholar]
- 2. Haines R. J., Pendleton L. C., Eichler D. C. (2011) Argininosuccinate synthase: at the center of arginine metabolism. Int. J. Biochem. Mol. Biol. 2, 8–23 [PMC free article] [PubMed] [Google Scholar]
- 3. Li H., Meininger C. J., Hawker J. R., Jr., Haynes T. E., Kepka-Lenhart D., Mistry S. K., Morris S. M., Jr., Wu G. (2001) Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 280, E75–82 [DOI] [PubMed] [Google Scholar]
- 4. Simon A., Plies L., Habermeier A., Martiné U., Reining M., Closs E. I. (2003) Role of neutral amino acid transport and protein breakdown for substrate supply of nitric-oxide synthase in human endothelial cells. Circ. Res. 93, 813–820 [DOI] [PubMed] [Google Scholar]
- 5. Prabhakar P., Thatte H. S., Goetz R. M., Cho M. R., Golan D. E., Michel T. (1998) Receptor-regulated translocation of endothelial nitric-oxide synthase. J. Biol. Chem. 273, 27383–27388 [DOI] [PubMed] [Google Scholar]
- 6. Fleming I. (2010) Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 459, 793–806 [DOI] [PubMed] [Google Scholar]
- 7. Michell B. J., Chen Z. p., Tiganis T., Stapleton D., Katsis F., Power D. A., Sim A. T., Kemp B. E. (2001) Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 276, 17625–17628 [DOI] [PubMed] [Google Scholar]
- 8. Corbin K. D., Pendleton L. C., Solomonson L. P., Eichler D. C. (2008) Phosphorylation of argininosuccinate synthase by protein kinase A. Biochem. Biophys. Res. Commun. 377, 1042–1046 [DOI] [PubMed] [Google Scholar]
- 9. Imami K., Sugiyama N., Kyono Y., Tomita M., Ishihama Y. (2008) Automated phospho-proteome analysis for cultured cancer cells by two-dimensional nanoLC-MS using a calcined titania/C18 biphasic column. Anal. Sci. 24, 161–166 [DOI] [PubMed] [Google Scholar]
- 10. Gospodarowicz D., Moran J., Braun D., Birdwell C. (1976) Clonal growth of bovine vascular endothelial cells: fibroblast growth factor as a survival agent. Proc. Natl. Acad. Sci. U.S.A. 73, 4120–4124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pendleton L. C., Goodwin B. L., Flam B. R., Solomonson L. P., Eichler D. C. (2002) Endothelial argininosuccinate synthase mRNA 5′-untranslated region diversity: infrastructure for tissue-specific expression. J. Biol. Chem. 277, 25363–25369 [DOI] [PubMed] [Google Scholar]
- 12. Misko T. P., Schilling R. J., Salvemini D., Moore W. M., Currie M. G. (1993) A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 214, 11–16 [DOI] [PubMed] [Google Scholar]
- 13. Brouet A., Sonveaux P., Dessy C., Balligand J. L., Feron O. (2001) Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J. Biol. Chem. 276, 32663–32669 [DOI] [PubMed] [Google Scholar]
- 14. Montagnani M., Chen H., Barr V. A., Quon M. J. (2001) Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser-1179. J. Biol. Chem. 276, 30392–30398 [DOI] [PubMed] [Google Scholar]
- 15. Kroll J., Waltenberger J. (1999) A novel function of VEGF receptor-2 (KDR): rapid release of nitric oxide in response to VEGF-A stimulation in endothelial cells. Biochem. Biophys. Res. Commun. 265, 636–639 [DOI] [PubMed] [Google Scholar]
- 16. Montagnani M., Ravichandran L. V., Chen H., Esposito D. L., Quon M. J. (2002) Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol. Endocrinol. 16, 1931–1942 [DOI] [PubMed] [Google Scholar]
- 17. Papapetropoulos A., García-Cardeña G., Madri J. A., Sessa W. C. (1997) Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 100, 3131–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeng G., Quon M. J. (1996) Insulin-stimulated production of nitric oxide is inhibited by wortmannin: direct measurement in vascular endothelial cells. J. Clin. Invest. 98, 894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dimmeler S., Dernbach E., Zeiher A. M. (2000) Phosphorylation of the endothelial nitric-oxide synthase at Ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 477, 258–262 [DOI] [PubMed] [Google Scholar]
- 20. Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., Zeiher A. M. (1999) Activation of nitric-oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 [DOI] [PubMed] [Google Scholar]
- 21. Xiao Z., Wang T., Qin H., Huang C., Feng Y., Xia Y. (2011) Endoplasmic reticulum Ca2+ release modulates endothelial nitric-oxide synthase via extracellular signal-regulated kinase (ERK) 1/2-mediated serine 635 phosphorylation. J. Biol. Chem. 286, 20100–20108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tran Q. K., Watanabe H. (2006) Calcium signaling in the endothelium. Handb. Exp. Pharmacol. 145–187 [DOI] [PubMed] [Google Scholar]
- 23. Rask-Madsen C., King G. L. (2007) Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat. Clin. Pract. Endocrinol. Metab. 3, 46–56 [DOI] [PubMed] [Google Scholar]
- 24. Rask-Madsen C., King G. L. (2008) Differential regulation of VEGF signaling by PKC-α and PKC-ϵ in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 919–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Partovian C., Zhuang Z., Moodie K., Lin M., Ouchi N., Sessa W. C., Walsh K., Simons M. (2005) PKCα activates eNOS and increases arterial blood flow in vivo. Circ. Res. 97, 482–487 [DOI] [PubMed] [Google Scholar]
- 26. Collingwood M. A., Rose S. D., Huang L., Hillier C., Amarzguioui M., Wiiger M. T., Soifer H. S., Rossi J. J., Behlke M. A. (2008) Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs. Oligonucleotides 18, 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yaffe M. B., Leparc G. G., Lai J., Obata T., Volinia S., Cantley L. C. (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 19, 348–353 [DOI] [PubMed] [Google Scholar]
- 28. Dudzinski D. M., Michel T. (2007) Life history of eNOS: partners and pathways. Cardiovasc Res. 75, 247–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goodwin B. L., Solomonson L. P., Eichler D. C. (2004) Argininosuccinate synthase expression is required to maintain nitric oxide production and cell viability in aortic endothelial cells. J. Biol. Chem. 279, 18353–18360 [DOI] [PubMed] [Google Scholar]
- 30. Goodwin B. L., Corbin K. D., Pendleton L. C., Levy M. M., Solomonson L. P., Eichler D. C. (2008) Troglitazone up-regulates vascular endothelial argininosuccinate synthase. Biochem. Biophys. Res. Commun. 370, 254–258 [DOI] [PubMed] [Google Scholar]
- 31. Goodwin B. L., Pendleton L. C., Levy M. M., Solomonson L. P., Eichler D. C. (2007) Tumor necrosis factor-α reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 293, H1115–H1121 [DOI] [PubMed] [Google Scholar]
- 32. Haines R. J., Corbin K. D., Pendleton L. C., Meininger C. J., Eichler D. C. (2012) Insulin transcriptionally regulates argininosuccinate synthase to maintain vascular endothelial function. Biochem. Biophys. Res. Commun. 421, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flam B. R., Hartmann P. J., Harrell-Booth M., Solomonson L. P., Eichler D. C. (2001) Caveolar localization of arginine regeneration enzymes, argininosuccinate synthase, and lyase, with endothelial nitric-oxide synthase. Nitric Oxide 5, 187–197 [DOI] [PubMed] [Google Scholar]
- 34. Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 [DOI] [PubMed] [Google Scholar]