

Background: Dimerization regulates activation of PDGF receptor in signal transduction.
Results: The transmembrane segment of PDGFR forms a left-handed helical dimer, which becomes more tilted and less stable in model membranes with decreasing lipid acyl chain lengths.
Conclusion: The membrane thickness controls the ability of the transmembrane segments to dimerize.
Significance: Receptor dimerization and activation in vivo may require relocation to thick lipid rafts.
Keywords: Circular Dichroism (CD), Molecular Dynamics, Nuclear Magnetic Resonance, Receptor Tyrosine Kinase, Solid-state NMR, Hydrophobic Mismatch, Oriented Circular Dichroism (OCD), Oriented Membrane Samples
Abstract
The platelet-derived growth factor receptor β is a member of the cell surface receptor tyrosine kinase family and dimerizes upon activation. We determined the structure of the transmembrane segment in dodecylphosphocholine micelles by liquid-state NMR and found that it forms a stable left-handed helical dimer. Solid-state NMR and oriented circular dichroism were used to measure the tilt angle of the helical segments in macroscopically aligned model membranes with different acyl chain lengths. Both methods showed that decreasing bilayer thickness (DEPC-POPC-DMPC) led to an increase in the helix tilt angle from 10° to 30° with respect to the bilayer normal. At the same time, reconstitution of the comparatively long hydrophobic segment became less effective, eventually resulting in complete protein aggregation in the short-chain lipid DLPC. Unrestrained molecular dynamics simulations of the dimer were carried out in explicit lipid bilayers (DEPC, POPC, DMPC, sphingomyelin), confirming the observed dependence of the helix tilt angle on bilayer thickness. Notably, molecular dynamics revealed that the left-handed dimer gets tilted en bloc, whereas conformational transitions to alternative (e.g. right-handed dimeric) states were not supported. The experimental data along with the simulation results demonstrate a pronounced interplay between the platelet-directed growth factor receptor β transmembrane segment and the bilayer thickness. The effect of hydrophobic mismatch might play a key role in the redistribution and activation of the receptor within different lipid microdomains of the plasma membrane in vivo.
Introduction
Receptor tyrosine kinases (RTKs)3 represent a large class of cell surface receptors that are activated by peptidic ligands: growth factors, hormones, or cytokines. They are crucial players in development, tissue repair, and maintenance of the normal cellular homeostasis. For example, the platelet-derived growth factors (PDGFs) and their receptors are involved in organ development, formation of the central nervous system, and angiogenesis (1). Pathologic tissue remodeling, various cancers and leukemias, atherosclerosis, induced restenosis, asthma, and pulmonary fibrosis have been shown to be associated with the aberrant activation of RTKs (2). Activation of PDGFRs, in particular, is implicated in cancerogenesis in lungs, renal cells, prostate, glia, etc. (3).
RTKs have a modular structure, consisting of a conserved intracellular kinase domain, a single transmembrane (TM) helix, and an extracellular ligand-binding domain that is characteristic of each subgroup. The PDGFRβ belongs to the class III subfamily of RTKs, which currently comprises five members: PDGFRα, PDGFRβ, stem cell growth factor receptor Kit, macrophage colony-stimulating factor (M-CSF) receptor, and FMS-like tyrosine kinase 3 (2). RTK activation is induced when the ligand binds simultaneously to the ectodomains of two receptors. Ligand binding results in spatial proximity and subsequent cross-activation of the intracellular kinase domains. Crystal structures have been recently published for the Ig-like domains D1–D3 of the PDGFRβ ectodomain in complex with PDGF-B, as well as for the entire ectodomain of the homologous KIT receptor in the free and ligand-bound forms. These structures revealed that the ligand makes extensive contact with D2 and D3 of both receptor monomers, thereby cross-linking them as a dimer (4, 5). The interaction is further stabilized by close homotypic contacts in the fourth and fifth Ig domains (6, 7). A comparison of the free and ligand-bound form of the KIT receptor ectodomain revealed that a conformational rearrangement takes place between D1–D3 and D4–D5 upon ligand binding, which reduces the distance between the C termini of the extracellular part from theoretically 70 Å in the free form to 15 Å in the bound form. It also moves the transmembrane segments and the adjacent cytosolic kinase domains close enough for cross-activation (5).
The traditional view that RTK dimerization is triggered exclusively by ligand interaction has been challenged by a number of reports in the last decade. Members of the ephrin (Eph) and epidermal growth factor (EGF) receptor subfamilies can dimerize in the absence of ligand, suggesting that activation of RTKs is more complex (8). Several studies have shed light on the contribution of the TM helices in the dimerization process. Inserting disulfide bridges at various positions of the TM domain (8), replacing the native TM against the TM of the constitutively active oncogenic form of the NEU receptor (9, 10), or using a simplified TM motif consisting of a stretch of valines with one or two glutamates (11), have all demonstrated that activation of the RTKs depends on a specific orientation of the TM domains with respect to each other. This indicated that ligand binding and kinase domain are actively coupled via the TM helix and that the RTK-TM dimer interface contains critical structural information to position the catalytic domains for correct phosphorylation.
Recent structural studies on the isolated TM helices of ErbB2, EphA1, EphA2, ErbB3, and the heterodimer ErbB1/ErbB2 showed that the TM helices, at least in these systems, dimerize in two different ways: either right-handed parallel or left-handed coiled-coil α-helical dimers (12–17). Ambiguity remains about what governs the arrangement in either of the two conformations or whether both conformations are relevant for activation and may in fact represent different activation states for a given RTK dimer. Based on the observation that RTK activation often occurs in special plasma membrane compartments, called microdomains (18), Arseniev and co-workers suggested that the lipid environment may control the dimerization mode (15).
Here, we find that also PDGFRβ-TM has a propensity to dimerize by itself, which adds a further example to the above mentioned group of RTKs. We have combined a high resolution NMR structure analysis in dodecylphosphocholine (DPC) micelles with a study on the orientation of the TM dimer in lipid bilayers with different thickness by oriented circular dichroism and solid-state NMR spectroscopy. Because DPC micelles are considered a nonideal membrane mimicking environment, we also used molecular dynamics (MD) simulations to assess the conformational behavior of the dimer in the different lipid bilayer systems. The theoretical calculations agree with the experimental tilt angle estimations, showing that the transition from long-chain to short-chain lipids is associated with an increase in the tilt angle of the entire dimer, which is at the same time accompanied by a less efficient reconstitution and reduced stability. These findings indicate that bilayer mismatch plays a key role in the ability of RTK-TM segments to dimerize.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of PDGFRβ-TM
Amino acids 526–563 of human PDGFRβ were cloned into a pMMHb vector (gift of Peter Kim, Howard Hughes Medical Institute, Chevy Chase, MD), adding an N-terminal Trp-ΔLE sequence, a His9 tag, and adjacent Asn and Gly residues for cleavage with hydroxylamine. The protein was expressed in Escherichia coli BL21(DE3) pLysS cells (Novagen). Expression and purification were carried out as described in Ref. 19. Reconstitution of PDGFRβ-TM into detergent micelles and lipid vesicles also followed the previously established protocol.
Chemical Cross-linking with Bis(sulfosuccinimidyl)suberate (BS3)
The cross-linking reagent BS3 (Thermo Scientific) was added in a twentyfold excess to PDGFRβ-TM. The reaction was allowed to proceed for 2 h at room temperature before the products were analyzed by HPLC. Cross-linked and non-cross-linked peptide samples were separated by HPLC using an analytical (250 × 4.6 mm) reversed phase C18 polymer column (Grace). The identities of the collected fractions were determined by MADLI-TOF MS.
Liquid-state NMR Spectroscopy
Triple-resonance experiments for assignment and 13C NOESY experiments were recorded on 1 mm uniformly 15N-labeled or 15N13C-labeled PDGFRβ-TM peptide in 200 mm deuterated DPC micelles (Bruker Avance 600 spectrometer equipped with a TBI triple-resonance probe head). Intermonomer NOEs were acquired from a three-dimensional 13C-filtered 13C-edited NOESY measured on a 1:1 mixture of uniformly 15N13C-labeled and unlabeled peptide dissolved in D2O (Bruker Avance 900 spectrometer). ARIA1.2-CNS was used for NOE calibration and structure calculation (20). Details are found in the supplemental Materials and Methods.
Solid-state NMR Spectroscopy
15N NMR spectra of oriented samples (for sample and experiment details, see supplemental Materials and Methods) were measured on a Bruker Avance III 500 MHz spectrometer at 20 °C (DEPC, POPC, and DLPC) or 30 °C (DMPC). The quality of the membrane alignment was checked by 31P NMR. For details, see the supplemental Materials and Methods.
Oriented CD (OCD)
OCD was carried out as described previously (19), using the same kind of oriented samples as for solid-state NMR.
MD Simulation in Lipid Bilayers
The initial conformation for the left-handed dimer (“expLH”) was taken from the NMR set. A three-dimensional model of a right-handed dimer (“modelRH”) was predicted de novo using the original PREDDIMERH approach (21). Both structures were subjected to series of MD simulations in full atom DMPC, POPC, DEPC, and sphingomyelin (SM) bilayers surrounded by explicit water molecules. For each conformation in each bilayer five independent equilibrations followed by five unconstrained 50-ns long MD runs were performed, which gave total statistics over 1.5 μs. The relative stability of the dimer conformations was estimated according to the average time autocorrelation functions calculated for residual contact maps of the dimer in the TM region. For MD protocols and analysis, see supplemental Materials and Methods, supplemental Tables S3 and S4, and supplemental Fig. S5.
RESULTS
PDGFRβ-TM Predominantly Forms Dimers in Detergent Micelles and Lipid Vesicles
A propensity for oligomerization was evident from SDS-PAGE analysis where PDGFRβ-TM migrated as monomers and oligomers. To assess the oligomerization state of recombinantly expressed PDGFRβ-TM, we chemically cross-linked PDGFRβ-TM with BS3. The reaction products were analyzed by RP-HPLC and characterized by MALDI-TOF MS (supplemental Fig. S1). PDGFRβ-TM has four reactive groups that can bind BS3: the ϵ-amine groups of lysines 531, 558, and 559, and the N terminus. All four are located in the unstructured N- or C-terminal regions, which are accessible to the water-soluble cross-linker, even in the presence of a protecting micellar or lipid bilayer environment. Consequently, up to four potential binding events are possible for each molecule in a micellar environment. In contrast, no more than two are possible in unilamellar lipid vesicles, where either the N- or C-terminal region is protected in the interior.
PDGFRβ-TM was reconstituted in DPC micelles and in lipid vesicles (DMPC (di-C14:0), POPC (C16:0/C18:1), or DEPC (di-C20:1 (cis))). Incubation with the bifunctional cross-linker yielded only monomers and dimers, as summarized in Table 1. Also, some hydrolysis of the sulfonyl groups occurred as a competing reaction, leading to a corresponding increase in mass by one unit of BS3. On average, one or two cross-linker molecules per peptide can be detected, which amounts to <50% of the theoretically possible binding events. This observation may suggest that the reaction was not very efficient, as also observed by others (22). However, it may also stem from the fact that not all potential ϵ-amine groups of lysines lie within the cross-linking radius in the dimer. In the experimental dimer structure (see below), Lys-531 points away from the dimer interface. In a potential trimer or tetramer, however, one might expect both side chains to experience more conformational freedom. We therefore interpret the absence of higher oligomer masses as a sign that the predominant form of PDGFRβ-TM is dimeric. As a control, the reaction was also studied in 80% TFE/buffer, where a trimer could also be detected.
TABLE 1.
Cross-linking by BS3 of PDGFRβ-TM reconstituted in various membrane(-like) environments
| Environment | +BS3 | −BS3 (control) |
|---|---|---|
| DMPC | Monomer + La | Monomer, dimer |
| POPC | Monomer, dimer | Monomer |
| Monomer + L | Dimer | |
| Dimerb | ||
| Dimerb + L | ||
| DEPC | Monomer, dimer | Monomer |
| Monomer + L | Dimer | |
| Dimerb | Trimer | |
| Dimerb + L | Tetramer | |
| Pentamerc | ||
| DPC | Monomer | Monomer |
| Monomer + L | Dimer | |
| Dimerb | Trimer | |
| Dimerb + L | Tetramer | |
| Dimerd | Pentamer | |
| Dimerd + L | Hexamerc | |
| 80% TFE | Monomer + L | Monomer |
| Dimerd | Dimer | |
| Dimere | Trimer | |
| Dimerb + L | Tetramer | |
| Trimere + L | Pentamerc |
a L, hydrolysis of the terminal sulfonyl substitutents occurred as a competing reaction, so one or more cross-linkers are attached to these peptides.
b One cross-linking event with the dimer.
c Mass not exactly determined due to low signal/noise ratio; hence questionable.
d Two cross-linking events within the dimer.
e Three cross-linking events within the dimer or trimer.
Interestingly, analysis of the control fractions without cross-linking reagent showed that noncovalently assembled dimers, trimers, and tetramers (even pentamers and hexamers) were detected by MALDI-TOF (Table 1). However, it is not possible to determine a priori whether these oligomers already existed in the lipid/detergent environment, whether they formed in the HPLC solvent or built up under MALDI conditions.
Three-dimensional Structure Characterization by Liquid-state NMR Spectroscopy
The NMR structure of the recombinantly expressed PDGFRβ-TM dimer (amino acids 526–563) was determined in DPC micelles (supplemental Fig. S2A). Full chemical shift assignment was obtained from 15N13C triple-resonance experiments and 13C NOESY spectra at two different temperatures. The structure calculation of the PDGFRβ-TM dimer was performed in two steps. First, the structure of the monomer was solved from 13C- and 15N-edited NOE-derived distance restraints and ϕψ-dihedral angles from TALOS analysis. The peptide forms a continuous slightly curved α-helix, extending from Phe-530 to Trp-556 (Fig. 1A). The N- and C-terminal regions are disordered due to a lack of 13C-detected NOEs. 15N relaxation spectroscopy corroborated that the C terminus from Lys-527 onwards is highly flexible (supplemental Fig. S2C). In the second step we had to break the symmetry of the homodimer by differential isotope labeling, to be able to unambiguously assign intermonomer NOEs that are required for calculating the dimer structure. 46 intermonomer NOEs were assigned in a 13C-filtered 13C-edited NOESY experiment on a sample consisting of equal amounts of uniformly 15N13C-labeled and unlabeled PDGFRβ-TM peptides. Despite the high resolution achieved at the field strength of 21.1 Tesla (900 MHz), the chemical shift degeneracy in the proton and carbon dimension for equal types of amino acids rendered the assignment of the intermonomer NOEs nontrivial. On the basis of the 13C resonances it was apparent that the dimer interface is lined by alanines, valines, isoleucines, and leucines and by the methyl group of a methionine (supplemental Fig. S2B). Because Met-554, the only methionine in the sequence, is located on the same face of the helix as Ala-537 and Ala-540, this provided an anchoring point to assign the inter-NOEs and calculate the dimer structure (Fig. 1, B and C, and supplemental Table S1). The helix interface consists of the “a” and “d” positions of a canonical heptad repeat: Ala-537, Ala-540, Leu-544, Ile-547, Ile-551, and Met-554. Val-533 on a further “d” position and Val-543 on position “g” at the edge of the interface also make hydrophobic contact across the interface (supplemental Fig. S2C). The two monomers are in intimate contact along the C-terminal half of the TM helix, whereas the two alanines in the N-terminal region experience a less tight packing at the interface. In this left-handed coiled-coil structure the dimer crossing angle χ between the two helices is about 20°.
FIGURE 1.

A and B, superposition of the 10 best NMR structures of the PDGFRβ-TM dimer (amino acids 526–563). A, backbone representation, with disordered N and C termini, illustrating the dimer crossing angle of ∼20°. The side chains of Phe-530 and Trp-556 are shown in blue for visual orientation. B, details of the interface after a rotation of 30° around the z axis. The contributing residues are depicted in red and green, and the disordered termini are removed for clarity. C, ribbon representation of the helical regions after rotation of the dimer by 90° around the x axis.
We examined which parts are stably inserted in the micelle by proton-deuterium exchange experiments, which showed the backbone from Ile-538 to Met-554 to be protected from solvent exchange (supplemental Fig. S2D). Based on the observation of NOE contacts from amide protons to bulk water, the solvent-protected part of the helix is extended by one turn on each side (N-terminally until Ile-535, and C-terminally until Trp-556, excluding only the Hϵ1 ring proton of Trp-556). This 22-amino acid stretch fits to the predicted length of the TM helix of 23 residues and places the aromatic ring of Trp-556 at the micelle-water interface.
For several RTK-TM dimers it has been noted that the sequence contains two alternative dimerization motifs, namely a left-handed leucine zipper like motif and a right-handed GX3G motif (12). In the latter glycines are often replaced by other small residues, such as alanine, serine, and threonine. Thus, the stretch of Ser-536 (Ala, Ile, Leu) Ala-540 in PDGFRβ-TM might be regarded as an equivalent motif. However, serine and alanine do not pack as well as glycine, to favor the knobs-into-holes assembly observed for the GX3G motif, which may thus destabilize the corresponding right-handed dimer conformation. In the present left-handed conformation of PDGFRβ-TM, Ser-536 on position “f” of the heptad repeat is at the edge of the interface, whereas Thr-545 on position “e” of the helical wheel putatively forms an internal hydrogen bond with Ser-548. That this position faces the micellar environment supports the fact that a mutation threonine to leucine had no effect on the receptor activation (23).
Solid-state 15N NMR Analysis in Lipid Bilayers
To determine the alignment of the PDGFRβ-TM helix in a proper lipid environment, one-dimensional solid-state 15N NMR spectra were acquired in macroscopically oriented membrane samples, as they can reveal the molecular orientation and mobility (24, 25). By using lipids with different acyl chain lengths for reconstitution, we studied the influence of the membrane thickness on the helix orientation and on the stability of the peptide in the bilayer. To this aim, PDGFRβ-TM was reconstituted at a peptide:lipid ratio of 1:200 in the short-chain lipid DLPC (di-C12:0), in the slightly longer DMPC, in the intermediate POPC, and in the long-chain DEPC. The corresponding bilayers have hydrophobic widths of about 20.9, 25.4, 27.1, and 31.5 Å, respectively (26, 27), and all were liquid-crystalline at the experimental temperatures. The 15N NMR spectra (Fig. 2, A–D) exhibit contributions of well oriented transmembrane peptides (∼200 ppm), besides some nonoriented peptides that give rise to a broad powder spectrum with a characteristic maximum at ∼80 ppm. The lack of orientation can be attributed to both transmembrane helices in a poorly oriented bilayer environment and to a fraction of peptides that were not properly reconstituted in their helical state and have thus aggregated.
FIGURE 2.

Solid state 15N NMR (A–D) and 31P NMR spectra (I–L) of PDGFRβ-TM in macroscopically oriented bilayers in the liquid crystalline state. The peptide was reconstituted in lipids of different thickness, namely in DEPC (A and I), POPC (B and J), DMPC (C and K), and DLPC (D and L) at a peptide:lipid ratio of 1:200. The 15N NMR spectra were fitted by a sum of three contributions: (i) well aligned “oriented” peptides with a transmembrane orientation (resonances between ∼220 and 170 ppm depending on the local tilt angle τ), (ii) reconstituted transmembrane peptides that are present in “nonoriented” parts of the bilayer sample, plus (iii) “aggregated” peptides that were not properly reconstituted (for the latter contributions (ii) and (iii): resonances between 220 and 60 ppm. The sum of all three contributions is shown as a thick solid line, and the contribution of the well oriented peptide (i) as a dashed line. The fraction of properly reconstituted yet nonoriented peptides (ii) was determined from the 31P NMR spectra (I–L) of the phospholipids. These lineshapes were fitted by a sum of two contributions of oriented membranes (dashed line) plus nonoriented membranes. The 15N NMR spectra (A–D) were simulated for varying tilt angles τ and with a varying fraction of aggregated peptides, to be compared with the experimental lineshapes in the form of root mean square deviation maps (E–H). These maps are thus displayed as a function of the tilt angle τ and the fraction of aggregated peptide. Neighboring root mean square difference steps differ by a factor of 1.2, showing the best fit area in black. The 15N NMR spectrum of DLPC (D) could not be reliably deconvoluted into the different contributions due to excessive aggregation. In the other lipid environments, a decrease in helix tilt angle and in the aggregated fraction was observed for increasing bilayer thickness (E–G).
To determine the helix tilt angle τ of the well oriented transmembrane peptides and to extract the fraction of aggregated peptides, the spectral lineshapes were deconvoluted into three contributions: (i) “oriented” peptides, (ii) properly reconstituted but “nonoriented” peptides, and (iii) nonreconstituted “aggregated” peptides. To ensure an unbiased analysis, the ratio of fraction (i) to (ii) was calculated independently for the phospholipids from the corresponding 31P NMR spectra (Fig. 2, I–L, supplemental Materials and Methods, and Table S2). Our results show that the helix tilt angle of PDGFRβ-TM increases from τ ≈ 10° in DEPC to ∼20° in POPC, and to ∼30° in DMPC bilayers (Fig. 2, E–H, and Table 2). At the same time, the aggregated fraction of peptides increases in the order of DEPC < POPC < DMPC, from about 5% to 25% to 50%, respectively. In DLPC bilayers, finally, the 15N NMR spectra showed essentially only a powder lineshape, meaning that the comparatively long hydrophobic segment was not at all amenable to reconstitution in the thin lipid bilayer and had simply aggregated.
TABLE 2.
Best fit values of the order parameter (S), helix tilt angle (τ), and the fraction of aggregated nonreconstituted peptide obtained by solid-state NMR analysis of PDGFRβ-TM in different bilayers
| Peptide | Thickness | τ | Aggregated | S | r.m.s.d.a |
|---|---|---|---|---|---|
| DLPC | 20.9 Å | NDb | >0.70 | 0.98 | 0.00031 |
| DMPC | 25.4 Å | 31° | 0.54 | 0.94 | 0.00030 |
| POPC | 27.1 Å | 17° | 0.26 | 1.00 | 0.00024 |
| DEPC | 31.5Å | 10° | 0.04 | 0.98 | 0.00020 |
a Root mean square deviation.
b Due to the low fraction of oriented peptide, a meaningful result for the tilt angle was not obtained.
Oriented CD
The most informative feature to discriminate between a parallel or perpendicular orientation of the helix with respect to the bilayer normal is a positive (tilt angle 0°) or negative (tilt angle 90°) band at 208 nm (28, 29). The OCD lineshape of PDGFRβ-TM showed a pronounced variation in bilayers with different thickness (Fig. 3). The intensity at 208 nm changed steadily from slightly negative in DMPC, via zero in POPC, to a positive band in DEPC. These lineshapes indicate that the peptide is inserted as a transmembrane helix in all three bilayers, but the actual tilt angle varies with the lipid employed. Vogel (28) and Clayton and Sawyer (29) proposed a formula to estimate the helix tilt angle from the intensity of the band at 208 nm. On this basis, we calculated a helix tilt angle of τ ≈ 30° in DMPC, ∼20° in POPC, and ∼10° in DEPC. These values correlate very well with those obtained from the independent solid-state NMR analysis.
FIGURE 3.
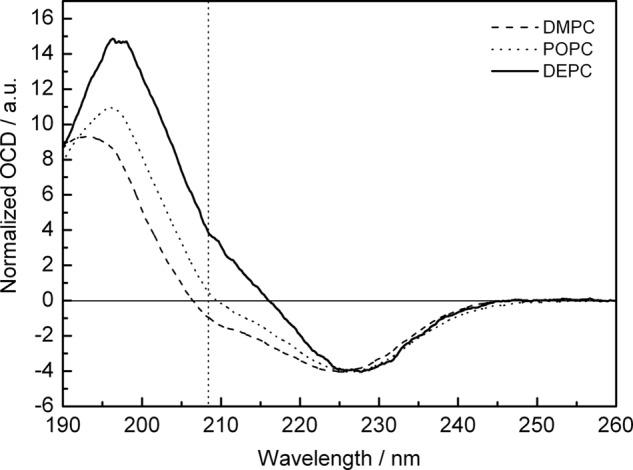
OCD spectra of PDGFRβ-TM reconstituted in macroscopically aligned liquid-crystalline bilayers at a peptide:lipid ratio of 1:50. The dotted line marks the intensity at 208 nm that is used to determine the alignment of the peptides.
Control measurements by conventional CD in unilamellar vesicle suspensions confirmed that PDGFRβ-TM is α-helical in all three lipids (supplemental Fig. S3A). Secondary structure estimation with the CONTIN-LL (30–32) algorithm showed that the overall helicity of the protein under these conditions is ∼65%. The corresponding sample in DLPC, however, was extensively aggregated at a peptide:lipid ratio of 1:50, which caused light scattering and prevented a reliable reproduction of the CD and OCD spectra (supplemental Fig. S3). To assess the general influence of peptide concentration, a series with different peptide:lipid ratios of 1:200, 1:100, and 1:50 was measured in DMPC, revealing no changes in the CD or OCD spectra.
Molecular Modeling
The liquid-state NMR structure of the left-handed coiled-coil PDGFRβ-TM dimer was obtained in DPC micelles, which may be considered as a nonideal membrane mimicking environment. We therefore performed a series of all-atom MD simulations of the experimentally determined left-handed dimer, denoted as expLH dimer. For comparison, we also generated an alternative right-handed conformation, denoted as modelRH dimer, with a crossing angle of χ = 43° (and an interhelical Ser-536–Ser-536 H-contact), using the PREDDIMERH approach (21). Both models were incorporated in DMPC, POPC, and in DEPC bilayers. For each conformation in each bilayer we ran five independent 50-ns long MD simulations (supplemental Fig. S4). Altogether, for these three membranes we collected statistics over 1.5 μs. The resulting average helix tilt angles are summarized as histograms in Fig. 4A and listed in Table 3. For the expLH dimer they follow the same trend as the experimental results, decreasing steadily with increasing bilayer thickness. The absolute values were slightly larger in MD than in solid-state NMR and OCD, a situation also observed in other simulations (33). However, the dependence of the helix tilt angle on bilayer thickness was much less pronounced for the modelRH dimer, where almost no difference between POPC and DEPC was found, in contrast to our experiments. Remarkably, the monomeric peptide showed almost no response in its tilt angle to changing the membrane environment (Table 3). Also, we note that no conformational conversion between the expLH and modelRH conformations was observed during the time of the simulation.
FIGURE 4.

Comparison of structural and dynamic properties of expLH (left panels) and modelRH (right panels) conformations of the PDGFRβ-TM dimer obtained via MD simulations in DMPC (black), POPC (gray), and DEPC bilayers (dashed). Each curve corresponds to statistics over 5. A and B, summary histograms of MD values obtained over five independent MD runs of 50 ns each in each bilayer. A, average tilt angle of helices and B, helix-helix crossing angle. C, time autocorrelation functions (ACF) for contact maps of the TM regions of the dimer.
TABLE 3.
Structural parameters of PDGFRβ-TM obtained by MD simulations in different bilayers
Dpp: distance between phosphorus planes formed by lipids in both bilayer leaflets; helix-helix crossing angle χ: angle between the helix-axes of the TM segments; tilt angle τ: average angle between the long-axis of each TM helix and the bilayer normal. All data were averaged over 5 independent 50 ns MD runs in each case.
| MD simulation | DMPC | POPC | DEPC |
|---|---|---|---|
| ExpLH (left-handed) | |||
| Dpp, Å | 32.1 ± 1.4 | 38.4 ± 1.4 | 42.5 ± 2.3 |
| Helix-helix crossing angle χ, degrees | 31.4 ± 5.3 | 28.0 ± 4.7 | 26.9 ± 3.4 |
| Tilt angle τ, degrees | 38.2 ± 6.1 | 27.2 ± 5.6 | 21.6 ± 5.9 |
| ModelRH (right-handed) | |||
| Dpp, Å | 32.7 ± 1.4 | 38.4 ± 1.6 | 41.4 ± 2.4 |
| Helix-helix crossing angle χ, degrees | −40.0 ± 5.3 | −39.7 ± 6.2 | −37.6 ± 8.3 |
| Tilt angle τ, degrees | 31.7 ± 8.4 | 22.5 ± 3.6 | 20.9 ± 4.8 |
| Monomer | |||
| Tilt angle τ, degrees | 23.9 ± 7.3 | 25.6 ± 10.7 | 20.1 ± 6.9 |
Although the expLH conformation was slightly more stable in DEPC than in DMPC, yet more flexible in POPC, the stability of the modelRH dimer decreased with increasing bilayer thickness (Fig. 4C). Interestingly, the ensemble heterogeneity of the dimer in the expLH conformation also seemed to be affected by changing the lipid environment, as the distribution of the helix-helix crossing angle became narrower with increasing of bilayer thickness (Fig. 4B). In DEPC lipid bilayers the situations starts to resemble the relaxed state that was experimentally observed in soft DPC micelles.
The helix-helix crossing angle χ in the dimer can be related to the tilt angle τ, which describes each single helix with respect to the membrane normal. When the observed tilt is larger than half the crossing angle, this means that the entire dimer tilts en bloc and thereby contributes to the value of the helix tilt. We observed this situation in all MD simulations of the expLH dimer, suggesting that the PDGFRβ-TM dimer is tilted in the membrane as a whole (Fig. 5). Because the crossing angle in the modelRH dimer was per se considerably larger, a tilt of the entire modelRH dimer would only need to be assumed in the DMPC bilayer.
FIGURE 5.

Representative MD snapshots of the PDGFRβ-TM dimer, illustrating how the expLH adapts its tilt angle en bloc to the membrane thickness in DMPC, POPC, DEPC, and SM bilayers. The peptide is shown in schematic form, and lipids as transparent spheres of different colors (gray, red, and orange for carbon, oxygen, and phosphorus atoms, respectively). Water molecules are shown by blue lines.
In every bilayer simulation the expLH dimer turned out to be more tilted than the modelRH. A possible explanation could be that the left-handed conformation is more compact in all cases and this dimer is able to tilt as whole entity, whereas each helix in the right-handed conformation can move relatively independently with respect to the bilayer normal.
DISCUSSION
The plasma membrane can by no means be regarded as homogeneous. It is not only heterogeneous on the compositional level (lipids, integral proteins, glycoproteins, etc.) but also within the lipid bilayer itself. Multiple dynamic, laterally segregated microdomains are present (also denoted as lipid rafts, or SM-cholesterol-rich domains, etc.) (34). Biologically, these microdomains appear to serve as a key organizing principle to confine cellular processes to certain membrane areas. RTK activation, in particular, has been reported to be associated with lipid microdomains (for a review, see Ref. 18). Lipid rafts are characterized by a specific lipid composition (e.g. SM, certain glycolipids and cholesterol) (35), leading in general to thicker, more ordered membranes. They contain a distinct subset of proteins compared with the rest of the plasma membrane.
There is no consensus view on the criteria that select proteins to these compartments. Apart from specific protein-protein or protein-lipid interactions (36–38) which are likely to retain proteins in raft-like domains, the general physicochemical properties of the microdomains appear to play an essential role. Especially bilayer fluidity and membrane thickness may provide the energetic constraints to select for certain proteins with optimally adapted transmembrane segments. One such property proposed in this context is the match between the hydrophobic thickness of the lipid bilayer and the length of the TM domains similar to the thickness-dependent TM sorting between ER and Golgi membranes on the one hand and the plasma membrane on the other (39). For the lateral sorting within the plasma membrane, a similar mechanism may drive TMs of a matched (generally longer) length into a thicker raft, whereas shorter helices would possess a higher preference toward the nonraft bilayer regions.
The sequences of RTK-TM domains are highly conserved across species in length and amino acid composition. In Fig. 6 we have compared the TM sequences of the most studied RTKs in human (Homo sapiens), mouse (Mus musculus), chicken (Gallus gallus), zebrafish (Danio rerio), and claw frog (Xenopus laevis). From the sequence point of view, PDGFRβ has one of the longest TM helices (26 residues), therefore it should be geometrically apt to reside in thicker membrane sections to avoid any energetically unfavorable bilayer mismatch. Notably, left-handed dimerization with a small helix crossing angle further increases (by about 2 Å) the hydrophobic length compared with a right-handed dimer as it was found, e.g. in the EGF family for the ErbB2-TM and EphA1-TM homodimers or the ErbB1-TM/ErbB2-TM heterodimer.
FIGURE 6.

Multiple sequence alignment of RTK-TM domains. The TM domain (experimentally determined) (12, 14–16) (or predicted) is depicted in bold. GX3G or GX3GX3G motifs involved in dimerization are highlighted. h.s., H. sapiens; m.m., M. musculus; g.g., G. gallus; d.r., D. rerio; x.l., X. laevis.
As a consequence, insertion into unfavorably thin membranes should lead to an adjustment of the tilt of the entire TM part. In accordance with theory and previous observations (24, 25, 40) PDGFRβ-TM indeed adjusts its tilt according to bilayer thickness from 30° in DMPC, to 20° in POPC, to 10° in DEPC. The latter value matches well with the helix crossing angle χ of ∼20° of the PDGFRβ-TM dimer structure determined by liquid-state NMR in DPC micelles (Fig. 1A). We thus consider this structure to be representative of the structure in the thick DEPC bilayers. Instead of adjusting the tilt of the dimer the higher tilt angles in the thinner POPC and DMPC lipids could also be explained by a change in conformation to a right-handed dimer with a helix crossing angle of >40°. Such a structural conversion would correspond on a molecular level to the rotation-coupled activation hypothesis (8) and was suggested by Arseniev and co-workers (15). The changes in tilt angle could finally also be attributed to a disassembly into unconstrained monomers that might adapt more easily to the environment
Unrestrained MD simulations were performed to differentiate between the different scenarios. The calculations showed that within the time scale of the simulation in all three bilayers (50 ns) both the left-handed (expLH) and right-handed dimer (modelRH) were stable, and their tilt showed a marked dependence on the membrane thickness. However, the tilt dependence followed the observed experimental tendency only for the expLH conformation (Table 3). Taken together, experiments and MD simulations on PDGFRβ argue against a conformational change from left- to right-handed conformation. This was suggested for ErbB2, EphA1, and EphA2 because their TM domains contain additional GX3G motifs that are not involved in the dimer interface (15). However, our sequence alignment shows that neither the alternative glycine zipper motif in EphA2 nor the C-terminal GX3G in ErbB2 is conserved across the species, as illustrated in Fig. 6.
In the present study it is remarkable to see that the membrane-bound dimer can adapt to hydrophobic mismatch in the simulations by increasing its tilt angle en bloc. On the experimental timescale, much longer than simulated by MD, we observed, however, that concomitant to the increase in tilt angle, the aggregated powder part dramatically increases going from DEPC to POPC and DMPC, arguing for a destabilization of the system in shorter lipids. This suggests that these lipids represent increasingly unfavorable environments for the PDGFRβ-TM dimer, a situation that was most extreme in DLPC. Also others have noticed that protein aggregation can occur in unfavorable cases of extreme mismatch as this reduces the area of the membrane that needs to be deformed (41–43).
The native PDGFRβ has been shown to be located preferentially in caevolae (44). In caveolae rafts the bilayer is considerably thicker than in typical POPC-like plasma-membranes and is enriched in cholesterol and sphingomyelin. Thus, we have performed further MD simulations in typical sphingomyelin (C16:0/C18:0) bilayers (Figs. 4C and Fig. 5 and supplemental Table S5). As one can see from Fig. 4C, the expLH conformation displays a slightly more stable behavior in SM than in the other studied membrane systems, whereas modelRH is most unstable in this bilayer. Thus, SM makes the membrane most sensitive to a particular conformation of the dimer within the whole series of lipids. Assuming that the tilt angle found in thick DEPC bilayers resembles most closely the situation in SM-rich microdomains, a raft-like environment would considerably stabilize the dimer conformation. Any thinner membranes would, on the other hand, disfavor the dimer conformation by hydrophobic mismatch and thereby prevent premature activation of the receptor. Interestingly, it has been shown that EGF receptor activation is stimulated by cholesterol depletion of plasma membranes (45–47), It is therefore tempting to speculate that the match of the shorter hydrophobic right-handed dimer with the thinner plasma membrane sections could be the underlying molecular reason for this behavior.
The structure found here has implications not only for homodimerization, but also for the interaction of PDGFRβ with other membrane TM proteins. For example, PDGFRβ is activated by the viral oncoprotein E5 via specific interactions: hydrogen-bonding between Gln-17 of E5 and Thr-545 of the receptor, and electrostatic interactions between Asp-33 of E5 and Lys-531 of the receptor (23). Both amino acids are located in the NMR structure on the outer face of the dimer (Fig. 1B). We thus predict the PDGFRβ-TM structure to be compatible with a side-by-side arrangement of the PDGFRβ-dimer and the E5-dimer counterpart. Moreover, binding of E5 might simply act as a mismatch compensator, allowing PDGFRβ to move into membrane regions of otherwise inappropriate thickness.
In summary, we have demonstrated that the thickness of the membrane environment has a significant impact on the TM-helix arrangement of PDGFRβ, in full agreement with the hydrophobic matching hypothesis. Our results suggest that the TM domain contributes considerably to the stability of the dimer of the full-length receptor. It thus appears conceivable that the inactive and activated states of a receptor are characterized by different helix-helix packing stabilities, which may in turn be the trigger for their lateral redistribution between different membrane microdomains.
Supplementary Material
Acknowledgment
We thank the DFG Center for Functional Nanostructures (TP E1.2) for the NMR infrastructure.
High-field NOESY spectra were recorded at the Frankfurt Facility for Biomolecular NMR, which is funded by the European Union Project “EU-NMR-European Network of Research Infrastructures for Providing Access and Technological Advancement in Bio-NMR” FP-2005-RII3 Contract 026145. The modeling part of the study was supported by Russian Ministry of Science and Education Grant MK-8439.2010.4 and Austrian Science Fund FWF START Grant Y 514-B11.

This article contains supplemental Materials and Methods, additional references, Tables S1–S5, and Figs. S1–S5.
The atomic coordinates and structure factors (code 2L6W) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/). The chemical shifts have been deposited with the BioMagResBank (www.bmrb.wisc.edu) under accession number 17966.
- RTK
- receptor tyrosine kinase
- BS3
- bis(sulfosuccinimidyl)suberate
- DPC
- dodecylphosphocholine
- DEPC
- 1,2-dieicosenoyl-sn-3-glycero-phosphocholine
- DLPC
- 1,2-dilauroyl-sn-3-glycero-phosphocholine
- DMPC
- 1,2-dimyristoyl-sn-3-glycero-phosphocholine
- Eph
- ephrin
- expLH
- experimentally determined left-handed
- MD
- molecular dynamics
- modelRH
- model of a right-handed dimer
- OCD
- oriented circular dichroism
- PDGFR
- PDGF receptor
- POPC
- 1-palmitoyl-2-oleoyl-sn-3-glycero-phosphocholine
- SM
- sphingomyelin
- TM
- transmembrane.
REFERENCES
- 1. Andrae J., Gallini R., Betsholtz C. (2008) Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 3. Pietras K., Sjöblom T., Rubin K., Heldin C. H., Ostman A. (2003) PDGF receptors as cancer drug targets. Cancer Cell 3, 439–443 [DOI] [PubMed] [Google Scholar]
- 4. Shim A. H., Liu H., Focia P. J., Chen X., Lin P. C., He X. (2010) Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl. Acad. Sci. U.S.A. 107, 11307–11312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yuzawa S., Opatowsky Y., Zhang Z., Mandiyan V., Lax I., Schlessinger J. (2007) Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130, 323–334 [DOI] [PubMed] [Google Scholar]
- 6. Isozaki K., Hirota S. (2006) Gain-of-function mutations of receptor tyrosine kinases in gastrointestinal stromal tumors. Curr. Genomics 7, 469–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y., Yuzawa S., Schlessinger J. (2008) Contacts between membrane proximal regions of the PDGF receptor ectodomain are required for receptor activation but not for receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 105, 7681–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moriki T., Maruyama H., Maruyama I. N. (2001) Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 311, 1011–1026 [DOI] [PubMed] [Google Scholar]
- 9. Petti L. M., Irusta P. M., DiMaio D. (1998) Oncogenic activation of the PDGFβ receptor by the transmembrane domain of p185neu*. Oncogene 16, 843–851 [DOI] [PubMed] [Google Scholar]
- 10. He L., Shobnam N., Hristova K. (2011) Specific inhibition of a pathogenic receptor tyrosine kinase by its transmembrane domain. Biochim. Biophys. Acta 1808, 253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bell C. A., Tynan J. A., Hart K. C., Meyer A. N., Robertson S. C., Donoghue D. J. (2000) Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol. Biol. Cell 11, 3589–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bocharov E. V., Mineev K. S., Volynsky P. E., Ermolyuk Y. S., Tkach E. N., Sobol A. G., Chupin V. V., Kirpichnikov M. P., Efremov R. G., Arseniev A. S. (2008) Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J. Biol. Chem. 283, 6950–6956 [DOI] [PubMed] [Google Scholar]
- 13. Artemenko E. O., Egorova N. S., Arseniev A. S., Feofanov A. V. (2008) Transmembrane domain of EphA1 receptor forms dimers in membrane-like environment. Biochim. Biophys. Acta 1778, 2361–2367 [DOI] [PubMed] [Google Scholar]
- 14. Bocharov E. V., Mayzel M. L., Volynsky P. E., Goncharuk M. V., Ermolyuk Y. S., Schulga A. A., Artemenko E. O., Efremov R. G., Arseniev A. S. (2008) Spatial structure and pH-dependent conformational diversity of dimeric transmembrane domain of the receptor tyrosine kinase EphA1. J. Biol. Chem. 283, 29385–29395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bocharov E. V., Mayzel M. L., Volynsky P. E., Mineev K. S., Tkach E. N., Ermolyuk Y. S., Schulga A. A., Efremov R. G., Arseniev A. S. (2010) Left-handed dimer of EphA2 transmembrane domain: helix packing diversity among receptor tyrosine kinases. Biophys. J. 98, 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mineev K. S., Bocharov E. V., Pustovalova Y. E., Bocharova O. V., Chupin V. V., Arseniev A. S. (2010) Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J. Mol. Biol. 400, 231–243 [DOI] [PubMed] [Google Scholar]
- 17. Mineev K. S., Khabibullina N. F., Lyukmanova E. N., Dolgikh D. A., Kirpichnikov M. P., Arseniev A. S. (2011) Spatial structure and dimer-monomer equilibrium of the ErbB3 transmembrane domain in DPC micelles. Biochim. Biophys. Acta 1808, 2081–2088 [DOI] [PubMed] [Google Scholar]
- 18. Pike L. J. (2005) Growth factor receptors, lipid rafts, and caveolae: an evolving story. Biochim. Biophys. Acta 1746, 260–273 [DOI] [PubMed] [Google Scholar]
- 19. Windisch D., Hoffmann S., Afonin S., Vollmer S., Benamira S., Langer B., Bürck J., Muhle-Goll C., Ulrich A. S. (2010) Structural role of the conserved cysteines in the dimerization of the viral transmembrane oncoprotein E5. Biophys. J. 99, 1764–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linge J. P., O'Donoghue S. I., Nilges M. (2001) Automated assignment of ambiguous nuclear Overhauser effects with ARIA. Methods Enzymol. 339, 71–90 [DOI] [PubMed] [Google Scholar]
- 21. Polyansky A. A., Volynsky P. E., Efremov R. G. (2011) Structural, dynamic, and functional aspects of helix association in membranes: a computational view. Adv. Protein Chem. Struct. Biol. 83, 129–161 [DOI] [PubMed] [Google Scholar]
- 22. Tertoolen L. G., Blanchetot C., Jiang G., Overvoorde J., Gadella T. W., Jr., Hunter T., den Hertog J. (2001) Dimerization of receptor protein-tyrosine phosphatase α in living cells. BMC Cell Biol. 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petti L. M., Reddy V., Smith S. O., DiMaio D. (1997) Identification of amino acids in the transmembrane and juxtamembrane domains of the platelet-derived growth factor receptor required for productive interaction with the bovine papillomavirus E5 protein. J. Virol. 71, 7318–7327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim T., Im W. (2010) Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys. J. 99, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strandberg E., Esteban-Martín S., Salgado J., Ulrich A. S. (2009) Orientation and dynamics of peptides in membranes calculated from 2H NMR data. Biophys. J. 96, 3223–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathai J. C., Tristram-Nagle S., Nagle J. F., Zeidel M. L. (2008) Structural determinants of water permeability through the lipid membrane. J. Gen. Physiol. 131, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu Q., Kim M., Ho K. W., Lachowicz P., Fanucci G. E., Cafiso D. S. (2008) Membrane hydrocarbon thickness modulates the dynamics of a membrane transport protein. Biophys. J. 95, 2849–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vogel H. (1987) Comparison of the conformation and orientation of alamethicin and melittin in lipid membranes. Biochemistry 26, 4562–4572 [DOI] [PubMed] [Google Scholar]
- 29. Clayton A. H., Sawyer W. H. (2000) Oriented circular dichroism of a class A amphipathic helix in aligned phospholipid multilayers. Biochim. Biophys. Acta 1467, 124–130 [DOI] [PubMed] [Google Scholar]
- 30. Provencher S. W., Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 31. van Stokkum I. H., Spoelder H. J., Bloemendal M., van Grondelle R., Groen F. C. (1990) Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 191, 110–118 [DOI] [PubMed] [Google Scholar]
- 32. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 33. Monticelli L., Tieleman D. P., Fuchs P. F. (2010) Interpretation of 2H NMR experiments on the orientation of the transmembrane helix WALP23 by computer simulations. Biophys. J. 99, 1455–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindner R., Naim H. Y. (2009) Domains in biological membranes. Exp. Cell Res. 315, 2871–2878 [DOI] [PubMed] [Google Scholar]
- 35. Simons K., Gerl M. J. (2010) Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688–699 [DOI] [PubMed] [Google Scholar]
- 36. Couet J., Sargiacomo M., Lisanti M. P. (1997) Interaction of a receptor tyrosine kinase, EGF-R, with caveolins: caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 272, 30429–30438 [DOI] [PubMed] [Google Scholar]
- 37. Lajoie P., Partridge E. A., Guay G., Goetz J. G., Pawling J., Lagana A., Joshi B., Dennis J. W., Nabi I. R. (2007) Plasma membrane domain organization regulates EGFR signaling in tumor cells. J. Cell Biol. 179, 341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawashima N., Yoon S. J., Itoh K., Nakayama K. (2009) Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J. Biol. Chem. 284, 6147–6155 [DOI] [PubMed] [Google Scholar]
- 39. Sharpe H. J., Stevens T. J., Munro S. (2010) A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park S. H., Opella S. J. (2005) Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 350, 310–318 [DOI] [PubMed] [Google Scholar]
- 41. Lewis B. A., Engelman D. M. (1983) Bacteriorhodopsin remains dispersed in fluid phospholipid bilayers over a wide range of bilayer thicknesses. J. Mol. Biol. 166, 203–210 [DOI] [PubMed] [Google Scholar]
- 42. Fernandes F., Loura L. M., Prieto M., Koehorst R., Spruijt R. B., Hemminga M. A. (2003) Dependence of M13 major coat protein oligomerization and lateral segregation on bilayer composition. Biophys. J. 85, 2430–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smeijers A. F., Pieterse K., Markvoort A. J., Hilbers P. A. (2006) Coarse-grained transmembrane proteins: hydrophobic matching, aggregation, and their effect on fusion. J. Phys. Chem. B 110, 13614–13623 [DOI] [PubMed] [Google Scholar]
- 44. Matveev S. V., Smart E. J. (2002) Heterologous desensitization of EGF receptors and PDGF receptors by sequestration in caveolae. Am. J. Physiol. Cell Physiol. 282, C935–946 [DOI] [PubMed] [Google Scholar]
- 45. Pike L. J., Casey L. (2002) Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking. Biochemistry 41, 10315–10322 [DOI] [PubMed] [Google Scholar]
- 46. Saffarian S., Li Y., Elson E. L., Pike L. J. (2007) Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 93, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Coskun Ü., Grzybek M., Drechsel D., Simons K. (2011) Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. U.S.A. 108, 9044–9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.