

Background: The nucleases hSNM1A and hSNM1B are implicated in DNA interstrand cross-link repair.
Results: hSNM1A and hSNM1B were biochemically characterized using undamaged and cross-linked DNA. A real-time assay for the nucleases suitable for inhibitor identification was developed.
Conclusion: Preferential hSNM1A activation by high molecular weight and cross-linked DNA was observed.
Significance: This work provides a basis for hSNM1A inhibitor development for improved cancer therapy.
Keywords: DNA Damage, DNA Enzymes, DNA Repair, DNase, Metalloproteins, Nucleic Acid Enzymology, DNA Interstrand Cross-link, Metallo-β-lactamase
Abstract
Human SNM1A and SNM1B/Apollo have both been implicated in the repair of DNA interstrand cross-links (ICLs) by cellular studies, and SNM1B is also required for telomere protection. Here, we describe studies on the biochemical characterization of the SNM1A and SNM1B proteins. The results reveal some fundamental differences in the mechanisms of the two proteins. Both SNM1A and SNM1B digest double-stranded and single-stranded DNA with a 5′-to-3′ directionality in a reaction that is stimulated by divalent cations, and both nucleases are inhibited by the zinc chelator o-phenanthroline. We find that SNM1A has greater affinity for single-stranded DNA over double-stranded DNA that is not observed with SNM1B. Although both proteins demonstrate a low level of processivity on low molecular weight DNA oligonucleotide substrates, when presented with high molecular weight DNA, SNM1A alone is rendered much more active, being capable of digesting kilobase-long stretches of DNA. Both proteins can digest past ICLs induced by the non-distorting minor groove cross-linking agent SJG-136, albeit with SNM1A showing a greater capacity to achieve this. This is consistent with the proposal that SNM1A and SNM1B might exhibit some redundancy in ICL repair. Together, our work establishes differences in the substrate selectivities of SNM1A and SNM1B that are likely to be relevant to their in vivo roles and which might be exploited in the development of selective inhibitors.
Introduction
Interstrand cross-links (ICLs)3 are an extremely cytotoxic form of DNA damage that covalently tethers the two strands of the double helix and which inhibits fundamental nuclear functions, including transcription and DNA replication (1–3). Accumulating evidence implicates spontaneously arising cellular ICLs, induced by metabolic intermediates and oxidized lipids, as a formidable threat to genomic integrity (4). Indeed, the inability to repair these ICLs might directly contribute to developmental defects and tumor formation, as exemplified in the inherited disorder Fanconi anemia (FA) (5, 6), where ICL repair is compromised (7). In addition, a variety of clinically important alkylating anticancer drugs induces cell death through ICL formation, and response to therapy has been directly linked to ICL repair capacity (8). It is, therefore, critical to gain an improved understanding of ICL repair from both cancer treatment and prevention viewpoints.
In mammalian cells ICLs are primarily detected during DNA replication, where fork stalling elicits the repair response, and proteins within the FA pathway coordinate several key stages during ICL repair (1, 7, 9–12). A number of recent studies implicate the XPF-ERCC1 nuclease in the initial incision of the ICLs, and the localization and targeting of this factor to ICL-damaged chromatin is dependent upon the Slx4/FANCP factor (10, 12–14). The incised ICL intermediates are then subject to further processing allowing fork restart through the combined actions of translesion synthesis polymerases and homologous recombination factors (15–17). Evidence suggests that a key step during processing of incised ICLs involves the trimming or degradation of the tethered oligonucleotide remaining after ICL incision. Indeed, this step has been directly visualized in a Xenopus in vitro ICL repair system that employs replication-competent cell extracts (17). We have recently shown that the hSNM1A exonuclease might play an important role during this trimming step, as this protein acts in concert with XPF-ERCC1 to initiate ICL repair in vivo and displays biochemical activities consistent with such a role in vitro (14).
Purified hSNM1A is able to digest DNA-bearing site-specific ICLs produced by the non-distorting minor groove binding agent SJG-136 past the site of the lesion, leaving a single nucleotide covalently attached to the opposing strand (14) (illustrated schematically in Fig. 1A). These tethered mononucleotides are established as good substrates for translesion synthesis polymerases to copy past (18–20), suggesting that hSNM1A facilitates ICL repair by providing a substrate for the later stages of ICL repair. hSNM1A is a member of the β-CASP structural family of metallo-β-lactamase (MBL)-fold-containing proteins that includes antibiotic resistance genes and proteins involved in nucleic acid processing. In Saccharomyces cerevisiae a single, possibly founder member of this family, Pso2 (formerly known as Snm1), is present and is critical for normal ICL resistance (21, 22). Vertebrates possess three orthologs of Pso2, hSNM1A, hSNM1B/Apollo, and hSNM1C/Artemis. Biochemically, the three human proteins have been shown to exhibit either 5′-3′ exonuclease (hSNM1A and hSNM1B) (14, 23–27) or structure-specific endonuclease (hSNM1C) (28, 29) activities; although Artemis was initially thought to have a constitutive exonuclease activity, this has more recently been shown to be due to a contaminating co-purified exonuclease (30). hSNM1B (also known as Apollo) has recently been suggested to play a role in DNA repair, possibly in both ICL repair within the FA pathway and in the ATM-mediated response to ionizing radiation-induced double-strand breaks (31, 32). Furthermore, hSNM1B was identified as a TRF2-interacting protein and, therefore, plays a role in telomere maintenance in association with the Shelterin complex, protecting leading strand telomeres and preventing non-homologous end-joining-mediated chromosome fusions (25–27, 33–35). Because disruption of non-homologous end-joining factors rescues the embryonic lethality in hSNM1B-disrupted mice, this suggests that the dysfunctional telomeres and other abnormal chromosomal structures that accumulate in hSNM1B-deficient cells prevent the development of viable mammals (36). Indeed, a splice variant in hSNM1B that leads to production of a form of hSNM1B that is unable to interact with TRF2 leads to a form of Hoyeraal-Hreidarsson syndrome where patients exhibit premature aging, bone marrow failure, and immunodeficiency (37).
FIGURE 1.

hSNM1A and hSNM1B hydrolysis of dsDNA is supported by a variety of divalent metal ion cofactors and inhibited by metal ion chelators. A, shown is a schematic representing the hSNM1A-catalyzed hydrolysis of cross-linked substrate described recently (14); hSNM1A, indicated in green, hydrolyzes DNA from the 5′-end, pausing at the cross-linked base and then continuing to digest DNA past the modified base, presumably leaving a single nucleotide attached to the opposite strand via the cross-link. B, shown is alignment of the Pso2/Snm1 family, indicating the conserved MBL and β-CASP domains in green and orange, respectively. The canonical MBL motifs 1–4 are shown in blue, and motifs A-C of the β-CASP family of MBLs are shaded in purple. The green hatching of the C-terminal part of the β-CASP domain containing motifs B and C indicates the possibility suggested by structural studies that B and C form part of a C-terminal extension of the MBL domain. C, shown is a model for the hSNM1A and hSNM1B active site arrangement based on the structure of human CPSF73 (43) and computational alignments (42). Human SNM1A residues are numbered in blue, and hSNM1B residues are in red, blue spheres indicate Zn2+ ions, the red sphere represents bridging water, and arrows indicate possible hydrolytic mechanism. The conserved motifs are shown in green. D–E, the extent of hydrolysis in 30 min after the addition of wild-type hSNM1A (0.002 μg; 4.1 nm) (D) or wild-type hSNM1B (0.02 μg; 43.8 nm) (E) to 1000 nm 3′ fluorescently labeled dsDNA in the presence of 0.01, 0.1, 1, 5, or 10 mm MgCl2 (lanes 1–5), 0.01, 0.1, 1, 5, or 10 mm MnCl2 (lanes 6–10), 0.01, 0.1, 1, 5, or 10 mm ZnCl2 (lanes 11–15), or 0.01, 0.1, 1, 5, or 10 mm CaCl2 (lanes 16–20). Lane UT, untreated sample of 3′-fluorescein-containing 21-mer DNA. F and G, shown is the extent of hydrolysis in a 30-min period after the addition of wild-type hSNM1A (0.007 μg; 14.3 nm) (F) or hSNM1B (0.045 μg; 98.6 nm) (G) to 1000 nm 3′-labeled substrate in the presence of 10 mm MgCl2 and 0, 0.1, 1, 5, 10, or 25 mm o-phenanthroline (lanes 1–6), 0, 0.1, 1, 5, 10, or 25 mm EGTA (lanes 7–12), or 0, 0.1, 1, 5, 10, or 25 mm EDTA (lanes 13–18). H, hSNM1B (0.002 μg; 4.1 nm) was incubated with 1000 nm 3′-fluorescently labeled dsDNA in the presence (lanes 12–20) or absence (lanes 2–10) of 1 mm MgCl2. Titrations of ZnCl2 were performed from 0 to 1 mm in each case (lanes 2–10 and 11–20). Lane NE is samples of substrate DNA where no enzyme was added, and either 0 mm (lane 1) or 1 mm MgCl2 was included (lane 11), respectively. For all panels, lane M indicates 3′-labeled marker oligonucleotides of the size indicated. Reactions were carried out in a volume of 10 μl, and gels are representative of multiple experiments, which show qualitatively similar results. nt, nucleotides.
Despite their importance in maintaining human genomic integrity, no detailed biochemical analysis of the human SNM1A and SNM1B factors has been carried out. Here we present a detailed characterization of the enzymatic properties of hSNM1A and hSNM1B/Apollo, identifying metal ion requirements, substrate preferences, and a striking, differential capacity for processing high molecular weight and damaged DNA.
EXPERIMENTAL PROCEDURES
Purification of hSNM1A and hSNM1B
N-terminal-truncated hSNM1A (hSNM1A-(608–1040)) was purified as in a previous study (14). Briefly, hSNM1A-(608–1040) was purified from 24 liters of bacterial culture using expression from the pNH-TrxT plasmid containing an N-terminal His6 and thioredoxin TEV protease cleavable tag (accession number GU269914, (38)). After cell lysis (50 mm HEPES, pH 7.5, 150 mm NaCl, 5% glycerol, 0.5 mm TCEP, supplemented with a protease inhibitor mixture (Roche Applied Science) using a Emulsiflex-C5 cell breaker (Avestin) followed by sonication (2 × 15 min, 15 s on/15 s off) and centrifugation (38,000 × g, 1 h)), the lysate was separated using phosphocellulose P11 resin (Whatman), loaded at 150 mm NaCl, and eluted with 20 mm HEPES, pH 7.5, 600 mm NaCl, 0.5 mm TCEP, 10% glycerol. The eluate was further purified on a His-Trap column (50 mm HEPES, pH 7.5, 300 mm NaCl, 10 mm imidazole, 0.5 mm TCEP, elution gradient 10–300 mm imidazole) and in a S75 gel filtration step (10 mm HEPES, pH 7.5, 300 mm NaCl, 0.5 mm TCEP, 5% glycerol). After removal of the tag with tobacco etch virus protease (1:20 ratio) at 4 °C overnight, the protein was rebound to a His-Trap column and eluted as above to remove cleaved tag followed by further purification using a heparin column (20 mm HEPES, pH 7.5, 150 mm NaCl, 0.5 mm TCEP, 5% glycerol, elution gradient 150 mm-2 M NaCl) and an SP column (20 mm HEPES, pH 7.5, 50 mm NaCl, 0.5 mm TCEP, 5% glycerol, elution gradient 50 mm 2 m NaCl). At each stage the presence of protein was confirmed on an InstantBlue-stained SDS-PAGE gel, and the identity of the final preparation was confirmed using electrospray ionization-TOF mass spectrometry or by tandem mass spectrometry of tryptic peptides.
A truncated hSNM1B-(6–328) protein was purified from recombinant Escherichia coli. The protein was cloned into the expression vector pNIC-Zb, which uses a combination of a His10 sequence and the Z-basic tag (GenBankTM accession number GU452710) to promote solubility and efficient purification. The plasmid was introduced into BL21(DE3)-R3-pRARE2, the cells were grown in Terrific Broth (TB) medium containing kanamycin, and expression was induced with isopropyl 1-thio-β-d-galactopyranoside. The proteins were extracted by sonication in binding buffer (20 mm HEPES, pH 8.0, 1 m NaCl, 5% glycerol, 10 mm imidazole, 1 mm TCEP) and loaded on a 5-ml HisTrap column at 4 ml/min. The column was washed with 20 volumes of wash buffer (20 m HEPES, pH 8.0, 250 mm NaCl, 5% glycerol, 30 mm imidazole, 0.5 mm TCEP) and eluted with Elution buffer (20 mm HEPES, pH 8.0, 250 mm NaCl, 5% glycerol, 300 mm imidazole, 0.5 mm TCEP). The eluted fraction was further purified by size exclusion chromatography on a Superdex S75 HR 16/60 column in a buffer containing 20 mm HEPES, pH 8.0, 150 mm NaCl, 20 mm glycine, 5% glycerol, 20 μm ZnCl2, 1 mm TCEP, at 1.2 ml/min. An inactive mutant version of hSNM1B-(6–328, D35A/H36A) in the vector pNIC-Zb was expressed and purified using an identical procedure. Antibodies used to confirm the identity of proteins were the anti-hSNM1A antibody from Abcam, Cambridge, UK (goat-polyclonal, catalogue number ab14805, used at 1:1000 dilution) and the anti-hSNM1B from Abgent Europe, Oxford, UK (rabbit-polyclonal, catalogue number ap5426c, used at 1:500 dilution).
Substrate Preparation
The substrates were constructed as shown in supplemental Table S1 using the sequences listed in supplemental Table S2. All oligonucleotides, including the fluorescein- and/or BHQ1-containing substrates were synthesized by Eurofins MWG Operon (Ebersberg, Germany). To make double-stranded DNA (dsDNA), RNA/DNA hybrids, or double-stranded RNA (dsRNA) and to ensure hairpin substrates were folded back, complementary strands were annealed by heating to 100 °C for 5 min followed by stepwise cooling to room temperature (20 °C). The methodology used to prepare and purify DNA oligonucleotide duplexes containing a single site-directed ICL has recently been published in detail (14, 39). If required, single-stranded DNA substrates were labeled at the 3′-end with [α-32P]dATP (1 μl; 0.37 MBq) and terminal deoxynucleotidyltransferase (20 units; Fermentas), and dsDNA substrates, after annealing where necessary, were labeled with [α-32P]dATP and Klenow fragment lacking 3′-exonuclease activity (5 units; New England Biolabs).
Nuclease Assays
Standard exonuclease assays were carried out in 10-μl reactions containing 20 mm HEPES-KOH, pH 7.5, 50 mm KCl, 0.5 mm DTT, 10 mm MgCl2, 0.05% Triton-X, 0.1 mg/ml BSA, 5% glycerol, and the indicated amounts of enzyme. The reactions were started by the addition of substrate (amount indicated) and incubated at 37 °C for 30 min followed by the addition of 2 μl of stop mixture (95% formamide, 10 mm EDTA, 0.25% xylene cyanol, 0.25% bromphenol blue) and heating to 95 °C for 3 min. The samples were analyzed using denaturing 20% polyacrylamide gels containing 0.5× Tris borate EDTA buffer and 7 m urea. Gels were run at 525 V for 1.5 h. With radioactive substrate, gels were dried and exposed to a Kodak phosphorimaging screen and scanned using a Typhoon 9400 instrument (GE Healthcare); with fluorescently labeled substrates, gels were imaged using the fluorescence mode of the Typhoon 9400 instrument, with blue laser (488 nm) excitation and an emission filter set to 520 nm band-pass 40 (detecting light between 500–540 nm).
Metal ion dependence experiments were carried out in buffer containing 0.01, 0.1, 1, 5, or 10 mm MgCl2, MnCl2, ZnCl2, or CaCl2 instead of the 10 mm MgCl2 of the standard reactions, keeping ionic strength constant by varying potassium chloride concentration. The substrate concentration chosen for comparing metal ion dependence of the two enzymes was 1000 nm, which exceeds the Km (10-fold) for the hSNM1A-catalyzed hydrolysis (see Fig. 3A). It is worth noting that at this substrate concentration the hSNM1B-catalyzed hydrolysis of dsDNA is not maximal, but an equivalent level of substrate was used for comparison purposes after a normalization experiment was carried out to determine the amounts of hSNM1A and hSNM1B that would result in an approximately equivalent level of hydrolysis (supplemental Fig. S2A). The chelator experiments were carried out as in the standard assay but supplemented with 0.1, 1, 5, 10, or 25 mm o-phenanthroline, EGTA, or EDTA.
FIGURE 3.

hSNM1A and hSNM1B multiple-turnover catalytic parameters for the hydrolysis of double-stranded or single-stranded 21-mer DNA (dsDNA or ssDNA). Initial rates of reaction normalized to enzyme concentration (v/[E]) were measured in at least triplicate at substrate concentrations above and below the Km values. The mean of v/[E] is shown, with error bars representing the standard error. Non-linear regression fitting was used to fit data to the Michaelis-Menten equation (Equation 1) and generate the catalytic parameters and associated standard errors, as shown in the inset. The parameters were measured at pH 7.5 in reaction buffer containing 10 mm MgCl2 for hSNM1A-catalyzed hydrolysis of dsDNA (A), hSNM1A-catalyzed hydrolysis of ssDNA (the inset shows data between 0–100 nm substrate for clarity), hSNM1B-catalyzed hydrolysis of dsDNA (C), and hSNM1B-catalyzed hydrolysis of ssDNA (D). Substrate concentrations ranged from 10 to 1200 nm (hSNM1A on dsDNA), 1.25–1200 nm (hSNM1A on ssDNA), 10–12,000 nm (hSNM1B on dsDNA), and 1.25–18,000 nm (hSNM1B on ssDNA). In hSNM1B hydrolysis of ssDNA, it was not possible to reach saturation even at the highest substrate concentration (18,000 nm).
Oligonucleotide competition experiments were carried out in 10-μl reactions as for the standard assay, with three reactions started concurrently by the addition of dsDNA or single-stranded DNA (ssDNA) substrate (1000 nm). Reaction 1 was stopped after 5 min by the addition of stop mixture and boiling, reaction 2 was supplemented with an excess (16.67 μm) of unlabeled substrate and subsequently stopped at 60 min, and reaction 3 was stopped at 60 min, with all samples analyzed as described above.
Plasmid digestion experiments were carried out in two stages. First, plasmid (325 ng, supplemental Fig. S7C) was either left untreated, gapped (nicked at five sites within a 52 base pair region) with Nb.BbvCI (5 units; New England Biolabs), or linearized with HindIII (10 units; New England Biolabs) in 10 μl for 90 min at 37 °C in 1× New England Biolabs reaction buffer 2. Second, reaction buffer (final composition 20 mm HEPES-KOH, pH 7.5, 50 mm KCl, 0.5 mm DTT, 10 mm MgCl2, 0.05% Triton X, 0.1 mg/ml BSA, 5% glycerol) and, in competition experiments unlabeled 21-mer dsDNA, were added and hSNM1A or hSNM1B were allowed to digest the plasmid for 1 h. Products were analyzed on 1% agarose gels containing 1× Tris borate EDTA (130 V, 150 min).
Kinetic Characterization of hSNM1A and hSNM1B
Reactions were carried out with varying concentrations of substrate and enzyme in the reaction buffer as described above. Reactions, prewarmed to 37 °C, were initiated by the addition of radiolabeled substrate, sampled at 4 appropriate time intervals, and quenched with stop mixture. The amount of product was determined after gels were dried and scanned by quantification using the ImageQuant (GE Healthcare) software package. The gels were quantified, and the products were analyzed assuming that the product of length n − 1 (where substrate is n nucleotides long) is the result of one hydrolysis reaction cycle, the product of length n − 2 is the result of two hydrolysis cycles, and the product of length n − m is the result of m hydrolysis (40, 41). The substrate concentrations were chosen to range from Km/8 to 8-fold Km where possible, and the enzyme concentrations were adjusted appropriately to measure a linear part of the initial reaction rate. The final substrate concentrations ranged from 10 to 1,200 and from 1.25 to 1,200 nm to analyze hSNM1A hydrolysis of ds- and ssDNA, respectively, and from 10–12,000 nm and from 1.25–18,000 nm to analyze hSNM1B hydrolysis of ds and ssDNA. Enzyme concentrations ranged from 0.5 to 4.1 nm (hSNM1A on dsDNA and ssDNA), 6 to 88 nm (hSNM1B on dsDNA), and 6 to 131 nm (hSNM1B on ssDNA). Initial rates of reaction at the range of substrate concentrations, normalized to enzyme concentration, were determined, and kinetic parameters were determined by nonlinear regression fitting of the data to the Michaelis-Menten equation (Equation 1),
 |
Curve-fitting was performed using Kaleidagraph software (Synergy Software).
Real-time Measurements
All measurements were carried out in 96-well plates using a SpectraMax M2e fluorescent plate reader (Molecular Devices) in fluorescence top read mode using SoftMaxPro software (Molecular Devices) to control the settings. Emission spectra were measured with 6 readings taken from 500 to 600 nm (2 nm steps) with excitation set to 495 nm and cutoff at 515 nm. Time courses were measured, with 6 readings taken at 7-s intervals for 12 min, with excitation at 495 nm, emission at 525 nm, and cutoff set to 515 nm.
pH Rate Profile of kcat for the hSNM1A WT and H994A Reactions
Reactions were carried out with 1000 nm dsDNA substrate when hydrolysis of dsDNA by hSNM1A WT reached saturation (see Fig. 3A) and when initial rate could be reasonably assumed to be an estimate of the kcat of the reaction. Reactions, prewarmed to 37 °C, were initiated by the addition of radiolabeled substrate, sampled at four appropriate time intervals to measure initial reaction rate, and quenched with stop mixture followed by determination of amount of product as described above for kinetic characterization. The experiments were carried out as described for the standard nuclease assays but used HEPES buffer (pH 7.0–8.8) or potassium glycinate (pH 8.6–11.0) to maintain constant pH. The real-time measurements were carried out as described above, with the initial rate determined during the linear part of the reaction and quoted as fluorescence units/nm enzyme/min.
Due to the bell-shaped curve obtained, the pH dependence of kcat was fitted to a model whereby the enzyme-substrate complex is subject to two possible ionizations but is active in the singly ionized form (Equation 2).
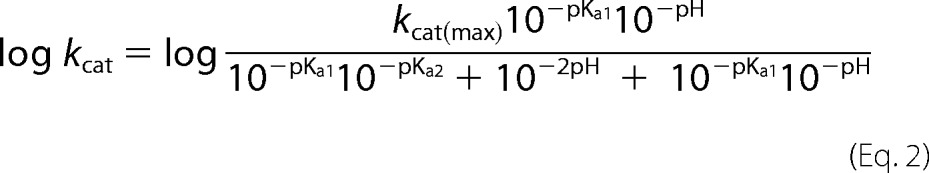 |
where kcat(max) is the maximal turnover number of the enzyme, and Ka1 and Ka2 are the acid dissociation constants of the enzyme-substrate complex. Curve fitting was carried out using logged data in Kaleidagraph software (Synergy Software).
RESULTS
hSNM1A and hSNM1B Purification
hSNM1A was overproduced and purified from E. coli as an N-terminal-truncated form containing all hSNM1 conserved regions (hSNM1A-(608–1040)) as described elsewhere (14). The protein, purified close to homogeneity, is shown on an InstantBlue-stained gel, and its identity was confirmed by an immunoblot probed with anti-hSNM1A antibody (supplemental Fig. S1, A–B) as well as by electrospray ionization-TOF mass spectrometry (data not shown). hSNM1B was also purified from E. coli using a C-terminal-truncated construct (hSNM1B-(6–328)) containing all conserved domains (shown in Fig. 1B), and the purified proteins are shown on an InstantBlue-stained gel in supplemental Fig. S1C, with their identity confirmed by immunoblot with anti-hSNM1B antibodies (supplemental Fig. S1D) as well as by mass spectrometry (data not shown). The relative domain architecture of the human members of the Snm1 family of proteins is shown compared with the related yeast interstrand cross-link repair protein Pso2 in Fig. 1B.
hSNM1A and hSNM1B Are 5′-to-3′ Exonucleases That Require Divalent Metal Cations and Are Inhibited by Metal Chelation
Previously published data suggested that hSNM1A requires the presence of magnesium divalent ions as a cofactor and is inhibited in the presence of zinc and iron(III) (23, 24), whereas the 5′-exonuclease activity of hSNM1B was studied only in the presence of magnesium (25, 26). Here, we initially assessed the protein activity in the presence of a broad range of concentrations of several divalent cations over a 30-min time course.
Both enzymes catalyzed the hydrolysis of dsDNA in the 5′-to-3′ direction in the presence of magnesium (Fig. 1, D and E, lanes 1–5) or manganese ions (Fig. 1, D and E, lanes 6–10) in a dose-dependent manner, with the reaction rate being significantly higher in the presence of manganese for hSNM1B, but not for hSNM1A, where the reaction rate throughout the tested manganese concentration range (0.01–10 mm) was approximately equivalent to that observed in the presence of 1 mm magnesium. This pattern has been consistently reproduced with multiple enzyme preparations obtained using a variety of procedures (data not shown), so is unlikely, at least solely, to be a function of metal ions co-purified with the enzyme. The hSNM1A and hSNM1B reaction is stimulated to a lesser extent by calcium (Fig. 1, D and E, lanes 16–20) and for hSNM1B occurs in the presence of a low concentration of zinc (0.01 mm) (Fig. 1E, lane 11) while being apparently inhibited at concentrations of zinc above 0.1 mm (Fig. 1E, lanes 12–15). For hSNM1B, we therefore explored the effect of varying zinc concentrations in the presence and absence of a fixed concentration of magnesium (Fig. 1H). This revealed that in the presence of magnesium, increasing zinc concentrations above 0.002 mm is stimulatory but becomes inhibitory above 0.1 mm zinc, as confirmed by the reactions performed in the absence of magnesium. For hSNM1A (Fig. 1D, lane 11, as compared with lanes 12–15), activity was not apparent even at low zinc concentrations as confirmed by titrating zinc in the absence of magnesium ions (data not shown). We saw no significant activity in the presence of a variety of concentrations of CoCl2 or NiCl2, although these experiments are complicated by the fact that the cofactors precipitate quickly due to their reduction in the dithiothreitol containing reaction buffer (data not shown). Finally, by substituting, on the basis of a homology model (42, 43), two predicted active site residues, D736A/H737A in hSNM1A and D35A/H36A in hSNM1B as indicated in Fig. 1C, we showed that the nuclease activity of the protein is intrinsic to the protein rather than due to contaminating exonucleases because substitution of these residues hindered catalysis (supplemental Fig. 2, A and B). Some residual nuclease activity was apparent with the double-mutated hSNM1A protein in the presence of manganese ions (supplemental Fig. S2A, lanes 6–10), which might be due to the tighter binding of manganese in the active site as compared with magnesium that could lead to retention of manganese cofactor in the active site even in the absence of the D736A/H737A residues (44, 45).
Because the identity of the physiological active site metal ion is still unclear, we addressed this issue by carrying out the reaction in the presence of a variety of metal ion chelating compounds. We used o-phenanthroline, which is relatively specific toward zinc (46, 47), EDTA, a general metal chelator, and EGTA, which has a higher affinity for calcium as compared with magnesium (48, 49). The activity of both hSNM1A and hSNM1B was effectively inhibited in the presence of the zinc chelator o-phenanthroline (Fig. 1, F and G, lanes 1–6), with a lower concentration required to inhibit hSNM1B (compare lanes 3 in Fig. 1, F and G). EGTA was not strongly inhibitory to the hSNM1A reaction, stimulating it at low concentrations up to 5 mm and still showing activity similar to that in the absence of chelator at 25 mm EGTA (Fig. 1F, lanes 7–12). In contrast, hSNM1B activity was inhibited at EGTA concentrations of 5 mm and higher, with some residual activity at 25 mm EGTA (Fig. 1G, lanes 7–12). EDTA, a nonspecific metal chelator, stimulated hSNM1A at low concentrations where it did not affect hSNM1B activity significantly (0.01–1 mm, Fig. 1, F and G, lanes 14 and 15). At higher concentrations (5–25 mm), EDTA was inhibitory to both hSNM1A and hSNM1B to a greater extent than EGTA but to a lesser extent than o-phenanthroline (Fig. 1, F and G, lane 16–18). We conclude that divalent cations are important for hSNM1A and hSNM1B catalysis, and due to the efficient inhibition conferred by o-phenanthroline, zinc is a very attractive candidate reaction co-factor, especially given its role in bacterial metallo-β-lactamase catalysis. However, further work is required before unequivocal assignment of the metal cofactor requirements of the SNM1 proteins.
hSNM1A and hSNM1B Digest DNA with a Similar Catalytic Efficiency but Show Distinct Substrate Binding Properties
hSNM1A and hSNM1B have previously been shown to be able to digest both dsDNA and ssDNA (14, 23–26). To investigate the relative efficiency of these reactions, we first ascertained whether or not the hSNM1A and hSNM1B reactions are processive; for a non-processive enzyme, the assumption can be made that each nucleotide removal is the result of an independent hydrolysis event. Unlike an endonuclease activity, which usually occurs at a single incision point, during an exonuclease reaction the substrate may be released and rebound by the enzyme between hydrolysis of subsequent nucleotides (non-processive), or further hydrolysis of the product may occur without product release (processive). We investigated the processivity of both hSNM1A and hSNM1B by adding an excess of unlabeled substrate to the reaction after a measured time delay and stopping the reaction after a fixed time period (Fig. 2). The amount of hSNM1A and hSNM1B used was such that the reaction progress in a 30-min time period was approximately similar, as defined by experiments normalizing for activity shown in supplemental Fig. S3. The results suggest that hSNM1A hydrolysis of both dsDNA (Fig. 2A, lanes 1–3) and ssDNA (Fig. 2A, lanes 4–6) is not strongly processive, because the addition of excess unlabeled DNA (lanes 2 and 5) has the same effect as stopping the reaction with stop buffer (lanes 1 and 4) as quantified in Fig. 2B. The hSNM1B reaction may be slightly more processive in character as hydrolysis of dsDNA (Fig. 2A, lanes 7–9) and ssDNA (Fig. 2A, lanes 10–12), quantified in Fig. 2C, is less sensitive to the addition of excess competitor (lanes 8 and 11 compared with lanes 7 and 10, respectively). The greater processivity of hSNM1B compared with hSNM1A could be related to the lower substrate binding efficiency of hSNM1B, as discussed below. Nevertheless, overall processivity of hSNM1B was low and likely does not invalidate the assumptions used in analyzing our data.
FIGURE 2.

hSNM1A and hSNM1B processivity in dsDNA and ssDNA hydrolysis and ability to digest DNA in the context of a plasmid substrate. A, experiments were carried out in 10-μl reaction volumes with 1 μm substrate (3′ fluorescently labeled dsDNA or ssDNA) and 0.006 μg (12.3 nm) of hSNM1A or 0.06 μg (131 nm) of hSNM1B containing dsDNA with hSNM1A (lanes 1–3), ssDNA with hSNM1A (lanes 4–6), dsDNA with hSNM1B (lanes 7–9), and ssDNA with hSNM1B (lanes 10–12). Lane UT, untreated samples of 3′-fluorescein-containing 21-mer DNA. Competition experiments were carried out as follows. The reaction was either stopped after 5 min by the addition of 2 μl of stop buffer and boiling (lanes 1, 4, 7, and 10), the reaction had excess unlabeled substrate (16.67 μm) added to it after 5 min followed by stop buffer at 60 min (lanes 2, 5, 8, 11), or the reaction was stopped after 60 min (lanes 3, 6, 9, 12). Gels are representative of at least three different experiments, and the same results were obtained qualitatively when the timing of the delay in adding unlabeled competitor DNA was varied. B and C, data from three independent experiments from A were quantified for hSNM1A (B) and hSNM1B (C). nt, nucleotides.
We were then able to measure the initial rate of reaction on both ssDNA and dsDNA substrates, normalized against enzyme concentration, at varying substrate concentrations such that the Michaelis-Menten plots in Fig. 3 could be constructed and the kinetic parameters kcat and Km obtained through non-linear regression fitting of the data. The kinetic parameters, tabulated as an inset of Fig. 3, suggest that hSNM1A and hSNM1B digest dsDNA with comparable catalytic efficiency (kcat ∼ 25 min−1), although substrate binding, reflected in the Km value, appeared to be stronger for the hSNM1A/dsDNA interaction (Fig. 3A) as compared with the hSNM1B/dsDNA interaction (Fig. 3C). When hydrolysis of ssDNA was assessed, the catalytic efficiency of the hSNM1A reaction was comparable with dsDNA hydrolysis (14 min−1 compared with 26 min−1), but ssDNA appeared to be bound more tightly, with saturation reached at low nanomolar substrate concentrations (Fig. 3B). This observation explains why hSNM1A was previously shown to hydrolyze ssDNA with greater efficiency than dsDNA (14, 23) because in these studies substrate concentration (100 nm) was at subsaturation levels for dsDNA hydrolysis and in excess of saturation for ssDNA hydrolysis. Finally, when ssDNA hydrolysis by hSNM1B was investigated, it was impossible to reach saturation even at the highest concentration of substrate used (18 μm) (Fig. 3D), and although the turnover rate appeared relatively high (in excess of 55 min−1), it is unlikely to be reached at physiological DNA concentrations.
Another human 5′-3′ exonuclease, human exonuclease 1, has been shown to have a high preference for a nicked substrate compared with blunt-ended DNA (50). To ensure that the difference in substrate binding by hSNM1B and hSNM1A was not simply due to substrate preference, we measured the initial rate of hydrolysis by hSNM1A and hSNM1B using blunt-ended, recessed, and nicked dsDNA, constructed from the oligonucleotides shown in supplemental Table S1 (sequences listed in supplemental Table S2) and saw no significant preference for recessed or nicked dsDNA by either enzyme as shown qualitatively in supplemental Fig. S4, A–D and as quantified in supplemental Fig. S4E.
hSNM1A and hSNM1B Preferentially Hydrolyze DNA Over RNA Substrates
It has been previously shown that hSNM1A protein hydrolyzes RNA as well as DNA substrates (24). This result was confirmed here but with a strong preference for DNA shown (supplemental Fig. S5A). We found that hSNM1B can also hydrolyze both RNA and DNA and has a similar preference for DNA (supplemental Fig. S5B). An in silico study of the β-CASP family of MBL proteins has suggested that motif C (Fig. 1C) is a histidine in RNA-processing enzymes such as CPSF-73 but a valine in DNA-processing enzymes including yeast Pso2 and human SNM1A, SNM1B, and SNM1C (42). To test the proposed importance of this residue in determining DNA/RNA substrate specificity, we made the V1016H change in the context of hSNM1A (Fig. 1C). However, this substitution had no effect on the DNA/RNA substrate preference (supplemental Fig. S5C), i.e. it did not increase relative preference for RNA versus DNA and, therefore, a role for motif C in RNA specificity is unlikely.
The pH Profile of the hSNM1A Protein Does Not Support the Role of Conserved Histidine 994 as a General Acid Catalyst
In a structural study of CPSF-73, an mRNA processing factor, which is a member of the same protein family as hSNM1 factors, the co-crystallization of a sulfate ion as a structural mimic of the scissile phosphate in the active site of the protein hinted at a possible mechanism for phosphodiester bond hydrolysis (see Fig. 1C). The study suggested that a hydroxide bridging two zinc ions is well positioned for an in-line attack of the phosphate (sulfate) and that a histidine residue (residue 396 in CPSF-73, residue 994 in hSNM1A) is a good candidate for acting as a general acid catalyst (43). We tested this hypothesis by comparing the pH profile of log(kcat) for wild-type hSNM1A with that for the H994A mutant. No large shift in pH optimum values was observed (compare supplemental Fig. S6, A with B), and our data thus do not indicate a role for hSNM1A histidine 994 as a general acid catalyst. This discrepancy with the structural study may be explained by the fact that the mechanism for leaving group stabilization could differ in the RNA- and DNA-processing enzymes of the β-CASP family of metallo-β-lactamase type enzymes. This result highlights the need for further structural studies of hSNM1A/B and other β-CASP MBL family members. However, it is also possible that another step is rate-limiting the nuclease reaction such that changing the rate of the leaving group departure has little effect on the actual observed reaction rate. A recent study of yeast Pso2 biochemical activities proposes that the equivalent residue in Pso2, His-611, is important for catalysis because its mutation abrogates nucleolytic activities of Pso2 (51). This is interesting because, in contrast, in our work we saw very little effect of the H994A mutation on activity.
hSNM1A, but Not hSNM1B, Digests Higher Molecular Weight DNA Substrates with Increased Efficiency
Because the major physiological substrate of both hSNM1A and hSNM1B is likely to be high molecular weight chromosomal DNA, in contrast to the oligonucleotide substrates used to date, we addressed the issue of whether the exonucleolytic behavior of the hSNM1 proteins is altered on high molecular weight substrates. An excess of hSNM1B and hSNM1A was incubated with an unmodified (circular), gapped or linearized plasmid, a modified version of pUC18 (pUC18-shortgap4) illustrated in supplemental Fig. S7C. Stimulation of the hSNM1B by gapped or linearized plasmid was not observed for any version of this plasmid substrate, as no significant digestion could be resolved when the products were separated on relatively low resolution agarose gels (Fig. 4A, compare lanes 2 and 8 and lane 3 with 9). By contrast, gapped and linearized plasmid substrates were hydrolyzed efficiently by hSNM1A (Fig. 4A, comparing lane 2 with 5, and lane 3 with 6) with an apparent digestion of at least 0.5 kb of plasmid substrate in 1 h; the reaction proceeded much farther than on oligonucleotide substrates in the same amount of time (Fig. 4A, lane 6, compared with Fig. 2A, lane 3). To address whether activity of hSNM1A becomes more processive on high molecular weight substrates, we digested gapped or linearized plasmid in the presence of a large excess of unlabeled double-stranded oligonucleotide substrate. Titrating in competitor substrate to a hydrolysis by hSNM1A of gapped (Fig. 4B, lanes 1–9) or linearized (Fig. 4B, lanes 10–18) plasmid, we found that a large excess (more than 25-fold) of competitor dsDNA was required to inhibit the hSNM1A reaction, suggesting that the hydrolysis of higher molecular weight substrate becomes processive.
FIGURE 4.

hSNM1A and hSNM1B are both able to hydrolyze dsDNA past a blockage in the hydrolyzed as well as in the complementary strand, but only hSNM1A hydrolyzes plasmid-based gapped and linearized DNA, a reaction that is only inhibited by the presence of a large excess (more than 25-fold) of oligonucleotide substrate. A, hydrolysis of plasmid substrate, either intact, gapped, or linearized, by hSNM1A or hSNM1B is shown. Lane M, GeneRuler 1kb DNA ladder, plasmid substrate (pUC18-shortGAP46, 325 ng; 16 nm free ends) in the absence of enzyme (lanes 1–3), in the presence of 21 nm hSNM1A (lanes 4–6), or in the presence of 219 nm hSNM1B (lanes 7–9). Plasmid substrate was either left untreated (lanes 1, 4, and 7), gapped with NbBbvCI (lanes 2, 5, and 8), or linearized with HindIII (lanes 3, 6, and 9). B, hydrolysis of plasmid substrate either gapped or linearized by hSNM1A in the presence of competitor dsDNA. Lane M, GeneRuler 1-kb DNA ladder, plasmid substrate (pUC18-shortGAP46, 325 ng; 16 nm free ends) that has been gapped by NbBbvCI (lanes 1–9) or linearized by HindIII (lanes 10–18) and treated with by 21 nm hSNM1A in the presence of an increasing concentration of 21mer dsDNA. C, shown are time courses (0, 5, 30, 60, 120 min) of hydrolysis of cross-linked substrate (lanes 1–10) and a non-cross-linked control (lanes 11–20) by 0.001 μg (2.0 nm) hSNM1A (lanes 1–5 and 11–15) or 0.195 μg (427 nm) hSNM1B (lanes 6–10 and 16–20). nt, nucleotides.
A recent study proposed that one of the functions of yeast Pso2 (51), aside from its role in interstrand cross-link repair, may be the opening of hairpin substrates in analogy with the hSNM1C/Artemis protein in mammals (28, 30, 52). To exclude the possibility that the plasmid substrate was hydrolyzed by hSNM1A endonucleolytically and in the light of this hairpin opening activity proposed for yeast Pso2 protein, we used gel-purified hairpin substrates blocked at the 5′-end to confirm that neither hSNM1A nor hSNM1B exhibit any endonuclease activity (supplemental Fig. S7, A and B). Specifically, a fold-back hairpin substrate with a 5′-OH moiety (supplemental Fig. S7, A and B, lanes 1–6) or blocked with a 5′-biotin (supplemental Fig. S7, A and B, lanes 7–12) was not degraded by an excess of hSNM1A or hSNM1B within 120 min of enzyme addition, in contrast with a 5′-phosphate containing hairpin, which was degraded very efficiently (supplemental Fig. Fig. S7, A and B, lanes 13–18). The same activity was observed using an alternative preparation of the enzymes overexpressed in insect cells, and moreover, a 10-fold increase of enzyme concentration, which led to complete hydrolysis of the 5′-phosphate hairpin within 5 min, did not hydrolyze the 5′-OH and 5′-biotin hairpin substrates (data not shown). Thus we conclude that neither hSNM1A nor hSNM1B, at least in the absence of other proteins, displays any hairpin opening activity.
Both hSNM1A and hSNM1B Digest Past ICL Lesions in Vitro
We recently demonstrated that hSNM1A is a nuclease with ability to digest DNA past a site-specific cross-link (14). Given that both hSNM1A and hSNM1B have been proposed to participate in ICL repair (14, 53, 54), possibly redundantly, we investigated the relative activity of hSNM1A and hSNM1B on a substrate containing an SJG-136 ICL lesion between the 5′-terminal guanosine and the complementary strand (0 nt, shown in supplemental Table 1 and described in detail elsewhere (39)). When such a substrate containing a 5′-terminal ICL was hydrolyzed in a time-dependent manner, both hSNM1A and hSNM1B were able to hydrolyze past a cross-linked base on the hydrolyzed strand within 30 min (Fig. 4C, lanes 3 and 8, for hSNM1A and hSNM1B, respectively). However, an extended time course (up to 120 min) suggested that hSNM1A may be better at hydrolyzing DNA past a cross-linked base on the complementary strand (Fig. 4C, compare lanes 4 and 5 with lanes 9 and 10). This result was consistently observed with multiple batches of both enzymes, with a larger proportion of 11-mer product visible after an extended time period in the hSNM1B reaction as compared with the hSNM1A reactions.
Digestion by hSNM1A and hSNM1B of Fluorescein-containing DNA Enables the Development of a Real-time Assay for Measuring Their Activity
Expanding on the result obtained above, we assessed the ability of hSNM1A and hSNM1B to digest substrates containing other bulky lesions, specifically DNA blocked with fluorescein, by the introduction of a fluorescein-dT phosphoramidite during the standard synthesis cycle either in the digested (top) or complementary (bottom) strand. Qualitatively, the data suggested that blocking the 5′-terminus of the dsDNA by the introduction of a dT-fluorescein base did not affect the time course of the nuclease digestion catalyzed by hSNM1A (Fig. 5A) or hSNM1B (Fig. 5B), as the extent of reaction was similar with the top strand-modified substrate (Fig. 5, A and B, lanes 1–6), the bottom strand-modified substrate (Fig. 5, A and B, lanes 7–12), and the unmodified substrate (Fig. 5, A and B, lanes 12–18). In addition to the qualitative assessment of the hSNM1A and hSNM1B activity on blocked substrates, we measured the turnover rate for the removal of a 5′ fluorescein-T base and found this to be in the same range as the rate measured with unmodified dsDNA (data not shown), indicating that the proteins hydrolyzed lesion-containing substrates with a similar efficiency as unmodified DNA.
FIGURE 5.

hSNM1A and hSNM1B hydrolyze dsDNA past a fluorescein blockage, which enables the establishment of a real-time assay to monitor the reaction of hSNM1A (and hSNM1B). A and B, time courses (0, 2, 5, 10, 30, 60 min) of hSNM1A (A) or hSNM1B (B) catalyzed hydrolysis of dsDNA blocked with a base-attached fluorescein on the first nucleotide of the top strand (lanes 1–6), the first nucleotide of the bottom strand (lanes 7–12), or not blocked at all (lanes 13–18). Lane M, 3′-labeled marker oligonucleotides of the size indicated. Reactions were carried out in a volume of 10 μl containing 1000 nm 3′ radiolabeled substrate and 0.01 μg (20.4 nm) of hSNM1A or 0.1 μg (219 nm) of hSNM1B. The gels shown are representative of multiple experiments and show qualitatively similar results. C, D, and E, hSNM1A hydrolysis of fluorescein and BHQ1 double-labeled oligonucleotide, ds- or ssDNA, is accompanied by an increase in fluorescence at 525 nm. C, shown is ds- and ssDNA substrate containing a fluorescein-T (green) and BHQ1-T (black) base. D, shown is an emission spectrum of fluorescein before and after the addition of hSNM1A (excitation at 495 nm, 515 nm cut-off). E, shown is the time course of fluorescein emission at 525 nm immediately after the addition of hSNM1A (excitation 495 nm, cut-off 515 nm) to 1000 nm ssDNA (open circles) or dsDNA (closed circles).
Although the gel-based assay was useful in determining the kinetic parameters and making basic comparisons between the hSNM1A and hSNM1B proteins, it is not very quantitative and is a relatively low throughput method. Recently, fluorescence and fluorescence resonance energy transfer (FRET) based assays have become prevalent in the assaying of a number of biological systems, including nucleases (55, 56). Fluorescence-based methods used to study nucleases involved in DNA metabolism include, for example, an assay to measure the endonuclease activity of archaeal XPF and Fen1 and the subsequent analysis of substrate distortion as a result of protein binding (57, 58), a high throughput assay to identify inhibitors of human flap endonuclease 1 (59), and a FRET-based way of identifying multiple modes of nucleolytic processing of DNA by lambda exonuclease (60).
We aimed to design an assay that could measure hSNM1A and hSNM1B activity in real time, exploiting the observation that both proteins were able to digest DNA past the bulky lesion (Fig. 5, A and B). In analogy with the previous studies (57–59), we tested the usefulness of a substrate with a 5′-acceptor (Cy3) and an internal donor (fluorescein) but saw no increase in fluorescence because the exonuclease reaction was blocked by the 5′-modification of substrate (data not shown). However, we were able to show that a real-time assay based on double labeling oligonucleotides internally with fluorescein and BHQ1 (black hole quencher) enabled us to measure the activity of hSNM1A and hSNM1B by following the increase in emission at 525 nm associated with the separation of fluorophore and quencher molecules (Fig. 5, C–E). Using ssDNA, we revisited the results obtained using the gel-based assay and confirmed that for reactions carried out in the presence of a variety of metal ion cofactors and inhibited by a range of metal chelators, qualitatively similar results could be obtained (supplemental Fig. S8). Thus, as a comparison of supplemental Fig. S8, A and B with Fig. 1, D and E, shows, it is clear that the same dependence of reactivity on magnesium or manganese concentration was observed, with activity of hSNM1A and hSNM1B increasing with increasing magnesium concentration (0.01–10 mm) and manganese being a better cofactor for hSNM1B than hSNM1A. Similarly, the hydrolysis rates measured in the presence of o-phenanthroline, EGTA, or EDTA in the real-time assay (supplemental Fig. S8, C and D) correspond to the extent of reaction after 30 min in a single time point gel-based experiment (Fig. 1, F and G, respectively). The reaction rate of wild-type and H994A hSNM1A was also measured at varied pH using the real-time assay, and the pattern observed and pKa values obtained did not differ significantly from those measured in a gel-based assay (compare supplemental Fig. S6, A and B with supplemental Fig. S6, C and D, and values in the inset table). This assay is potentially of great value in further directing structure-function relationships of this family of nucleases as well as identifying inhibitors in a high throughput screening effort.
DISCUSSION
Previous biochemical studies, including our own, of the SNM1A and SNM1B proteins have been qualitative (23, 24) and demonstrated that both proteins are 5′-to-3′ exonucleases that require a terminal 5′-phosphate for the digestion of ds- and ssDNA. The yeast orthologue of the human SNM1 factors, Pso2, has also been shown to have 5′-exonuclease activity on 5′-phosphate-containing ssDNA and dsDNA (23, 61) and, perhaps surprisingly, an endonucleolytic activity on hairpin substrates (51).
We have conducted a detailed kinetic analysis of the recombinant hSNM1A and hSNM1B proteins using both ds- and ssDNA to ascertain their binding characteristics and catalytic efficiency. We show that catalytic efficiency is comparable for the two proteins (Fig. 3), which is about an order of magnitude higher than the kcat values measured for yeast Pso2 recently (51). This could be related to the relative protein solubility as it has proven difficult to express and purify soluble full-length Pso2 in large quantities (Refs. 23 and 61 and data not shown). Although the catalytic efficiency of hSNM1A and hSNM1B is similar, with short oligonucleotide substrates, hSNM1A binds substrate more tightly than the related hSNM1B, as indicated by a Km value in the nanomolar as compared with the micromolar range. The Km values measured indicated that hSNM1A bound an ssDNA oligonucleotide substrate (21-mer) more efficiently than dsDNA (compare Fig. 3, A and B). Conversely, the Michaelis-Menten plot for hSNM1B hydrolysis of ssDNA (Fig. 3D) never reached saturation in the range of substrate concentrations studied (up to 18 μm), suggesting that hSNM1B bound dsDNA more tightly albeit still much more weakly than hSNM1A. The reactions were carried out with short oligonucleotide substrates, raising the possibility that the activity of these factors might be modified on high molecular weight substrates more akin to genomic DNA. Using a gapped and linearized plasmid substrate, we were unable to show a stimulation of hSNM1B by high molecular weight DNA, whereas hSNM1A efficiently degraded these substrates, digesting up to a kilobase during the 60-min incubation. This suggests that the activity of hSNM1A is altered upon incubation with such high molecular weight DNA. Much higher levels of competitor were required to inhibit the reaction of hSNM1A on high Mr DNA as compared with oligonucleotide substrates (at least 25-fold fold as compared with 5 fold), suggesting that hSNM1A becomes more processive or much more active on high molecular weight substrates.
In terms of the catalytic mechanism of SNM1A and SNM1B, we built upon previous work regarding substrate specificity and cofactor requirement. It was already known that the hSNM1A exonuclease activity is dependent on magnesium and is inactive in excess zinc, EDTA, or iron(III) (24). We have shown that the activity of both hSNM1A and hSNM1B is supported by magnesium and manganese and to a lesser extent by calcium (Fig. 1, D and E); the hSNM1B activity was qualitatively higher in the presence of manganese, whereas hSNM1A preferentially cleaves DNA in the presence of magnesium, a result that was consistently observed with multiple batches of the purified enzymes. In agreement with a previous study (24), zinc at high concentrations was inhibitory to the reactions, but surprisingly, we observed some hSNM1B activity in the presence of low (below 0.02 mm) concentrations of added zinc. Because the physiological metal ion cofactor of β-CASP MBLs is still unknown, an explanation for observation of activity at low but not high zinc concentrations may be the need for a zinc cofactor in one of the two active site metal ion sites and a second metal such as magnesium in the other metal ion site where binding of a second zinc ion may be inhibitory as is the case for some, but not all, metallo-β-lactamases. Indeed, further titration experiments exploring the effect of titrating zinc in the presence of a fixed amount of magnesium (Fig. 1H) suggested that two cation binding sites might be present and that occupancy of zinc in one site is required for activity, but occupation of both sites by zinc is inhibitory. The need for zinc in the active site may be further supported by the fact that activity of both hSNM1A and hSNM1B is more sensitive to attenuation by o-phenanthroline, a zinc-selective chelator, than by either EGTA or EDTA.
The role and identity of metal ions in the active site will in the future be supported by structural studies, but the structural data available on β-CASP metallo-β-lactamases is still scarce. One of the RNA-hydrolyzing members of the protein family, CPSF-73, has been solved structurally in the presence of a sulfate ion mimicking a scissile phosphate (43), and the authors propose a general acid catalysis mechanism involving a histidine residue. We substituted the equivalent histidine residue in hSNM1A to test its role but found no biochemical evidence that the residue acts as a general catalyst in hSNM1A (supplemental Fig. S6). Although the sulfate ion may have cocrystallized with CPSF-73 in a location distinct from the scissile phosphate binding pocket, such as in the 5′-phosphate binding pocket, the mechanism suggested by the structural study remains an attractive one due to the position of the two metal ions, apparently well located to deprotonate an attacking water nucleophile. As implied elsewhere with relation to Artemis (42), a possible explanation for the discrepancy is that the nucleolytic mechanism differs between RNA- and DNA-processing enzymes because of the differing intrinsic stabilities of the scissile phosphate. However, it is of note that a very recent publication discussing the biochemical activities of Pso2, the yeast homologue of hSNM1A, suggested that mutation of the equivalent histidine in yeast Pso2 (histidine 611) abolishes its nuclease activity completely (51), which contrasts with our data that suggest that the hSNM1A H994A mutant protein exhibits near wild-type activity (supplemental Fig. S6). Intriguingly also, the Pso2 biochemistry study suggested a hairpin opening endonucleolytic activity for Pso2 (51), in contrast with hSNM1A, where such activity has not been observed (Ref. 24 and data not shown). We confirmed that neither hSNM1A nor hSNM1B, at least in the absence of other protein cofactors, shows hairpin opening endonuclease activity (supplemental Fig. S7, A and B). It is thus possible that both functional and mechanistic differences between the hSNM1A and Pso2 proteins may become apparent, particularly because in yeast, Pso2 is the only factor of this protein family so far identified, whereas human cells contain three homologues, hSNM1A, hSNM1B/Apollo, and hSNM1C/Artemis.
Taking advantage of the near-normal hydrolysis activity of both SNM1A and SNM1B on DNA containing fluorescein moieties, we were able to develop a higher throughput assay to replace the gel-based method of kinetic parameter measurement. Using the assay to assess metal ion dependence and inhibition by metal chelators, we obtained results that were qualitatively similar to those obtained in a gel-based assay, and the pH profiles of wild-type and H994A hSNM1A hydrolysis were similar whether measured in a gel-based or real-time fluorescence based assay. The higher throughput nature of the real-time assay compared with a gel-based system clearly opens up the possibility for advanced mechanistic studies of the SNM1 family of proteins, and after optimization and scaling of the assay, our study provides a good starting point for this process. In addition to being a rapid platform to further characterize SNM1-family biochemical characteristics, the real-time assay can be used in screens for small molecule inhibitors. This is of relevance particularly because the development of small molecule inhibitors of the human SNM1-family proteins might suggest new strategies to therapeutically sensitize tumors to ICL-inducing agents as well as providing valuable research reagents. Importantly, due to the similarity of the hSNM1A active site and bacterial MBLs (43, 62, 63), candidate MBL inhibitors are already available and may be a good starting point for identifying potential hSNM1A and hSNM1B inhibitors.
Supplementary Material
This work was supported by a Cancer Research UK Programme Grant to PMcH.

This article contains supplemental Tables S1 and S2 and Figs. S1–S8.
- ICL
- interstrand cross-link
- FA
- Fanconi anemia
- MBL
- metallo-β-lactamase
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1. Deans A. J., West S. C. (2011) DNA interstrand cross-link repair and cancer. Nat. Rev. Cancer 11, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dronkert M. L., Kanaar R. (2001) Repair of DNA interstrand cross-links. Mutat. Res. 486, 217–247 [DOI] [PubMed] [Google Scholar]
- 3. McHugh P. J., Spanswick V. J., Hartley J. A. (2001) Repair of DNA interstrand cross-links. Molecular mechanisms and clinical relevance. Lancet Oncol. 2, 483–490 [DOI] [PubMed] [Google Scholar]
- 4. Niedernhofer L. J., Daniels J. S., Rouzer C. A., Greene R. E., Marnett L. J. (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 278, 31426–31433 [DOI] [PubMed] [Google Scholar]
- 5. Langevin F., Crossan G. P., Rosado I. V., Arends M. J., Patel K. J. (2011) Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 [DOI] [PubMed] [Google Scholar]
- 6. Rosado I. V., Langevin F., Crossan G. P., Takata M., Patel K. J. (2011) Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 [DOI] [PubMed] [Google Scholar]
- 7. Moldovan G. L., D'Andrea A. D. (2009) How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 43, 223–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spanswick V. J., Craddock C., Sekhar M., Mahendra P., Shankaranarayana P., Hughes R. G., Hochhauser D., Hartley J. A. (2002) Repair of DNA interstrand cross-links as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 100, 224–229 [DOI] [PubMed] [Google Scholar]
- 9. Akkari Y. M., Bateman R. L., Reifsteck C. A., Olson S. B., Grompe M. (2000) DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol. Cell. Biol. 20, 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Silva I. U., McHugh P. J., Clingen P. H., Hartley J. A. (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol. 20, 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knipscheer P., Räschle M., Smogorzewska A., Enoiu M., Ho T. V., Schärer O. D., Elledge S. J., Walter J. C. (2009) The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Niedernhofer L. J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A. F., de Wit J., Jaspers N. G., Beverloo H. B., Hoeijmakers J. H., Kanaar R. (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 24, 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crossan G. P., van der Weyden L., Rosado I. V., Langevin F., Gaillard P. H., McIntyre R. E., Sanger Mouse Genetics Project, Gallagher F., Kettunen M. I., Lewis D. Y., Brindle K., Arends M. J., Adams D. J., Patel K. J. (2011) Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 43, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang A. T., Sengerová B., Cattell E., Inagawa T., Hartley J. M., Kiakos K., Burgess-Brown N. A., Swift L. P., Enzlin J. H., Schofield C. J., Gileadi O., Hartley J. A., McHugh P. J. (2011) Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 25, 1859–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Long D. T., Räschle M., Joukov V., Walter J. C. (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niedzwiedz W., Mosedale G., Johnson M., Ong C. Y., Pace P., Patel K. J. (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell 15, 607–620 [DOI] [PubMed] [Google Scholar]
- 17. Räschle M., Knipscheer P., Knipsheer P., Enoiu M., Angelov T., Sun J., Griffith J. D., Ellenberger T. E., Schärer O. D., Walter J. C. (2008) Mechanism of replication-coupled DNA interstrand cross-link repair. Cell 134, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ho T. V., Guainazzi A., Derkunt S. B., Enoiu M., Schärer O. D. (2011) Structure-dependent bypass of DNA interstrand cross-links by translesion synthesis polymerases. Nucleic Acids Res. 39, 7455–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumari A., Minko I. G., Harbut M. B., Finkel S. E., Goodman M. F., Lloyd R. S. (2008) Replication bypass of interstrand cross-link intermediates by Escherichia coli DNA polymerase IV. J. Biol. Chem. 283, 27433–27437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minko I. G., Harbut M. B., Kozekov I. D., Kozekova A., Jakobs P. M., Olson S. B., Moses R. E., Harris T. M., Rizzo C. J., Lloyd R. S. (2008) Role for DNA polymerase κ in the processing of N2-N2-guanine interstrand cross-links. J. Biol. Chem. 283, 17075–17082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henriques J. A., Moustacchi E. (1980) Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics 95, 273–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruhland A., Kircher M., Wilborn F., Brendel M. (1981) A yeast mutant specifically sensitive to bifunctional alkylation. Mutat. Res. 91, 457–462 [DOI] [PubMed] [Google Scholar]
- 23. Hazrati A., Ramis-Castelltort M., Sarkar S., Barber L. J., Schofield C. J., Hartley J. A., McHugh P. J. (2008) Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair 7, 230–238 [DOI] [PubMed] [Google Scholar]
- 24. Hejna J., Philip S., Ott J., Faulkner C., Moses R. (2007) The hSNM1 protein is a DNA 5′-exonuclease. Nucleic Acids Res. 35, 6115–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lenain C., Bauwens S., Amiard S., Brunori M., Giraud-Panis M. J., Gilson E. (2006) The Apollo 5′ exonuclease functions together with TRF2 to protect telomeres from DNA repair. Curr. Biol. 16, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 26. Ye J., Lenain C., Bauwens S., Rizzo A., Saint-Léger A., Poulet A., Benarroch D., Magdinier F., Morere J., Amiard S., Verhoeyen E., Britton S., Calsou P., Salles B., Bizard A., Nadal M., Salvati E., Sabatier L., Wu Y., Biroccio A., Londoño-Vallejo A., Giraud-Panis M. J., Gilson E. (2010) TRF2 and apollo cooperate with topoisomerase 2α to protect human telomeres from replicative damage. Cell 142, 230–242 [DOI] [PubMed] [Google Scholar]
- 27. Wu P., van Overbeek M., Rooney S., de Lange T. (2010) Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol. Cell 39, 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goodarzi A. A., Yu Y., Riballo E., Douglas P., Walker S. A., Ye R., Härer C., Marchetti C., Morrice N., Jeggo P. A., Lees-Miller S. P. (2006) DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 25, 3880–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pannicke U., Ma Y., Hopfner K. P., Niewolik D., Lieber M. R., Schwarz K. (2004) Functional and biochemical dissection of the structure-specific nuclease ARTEMIS. EMBO J. 23, 1987–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pawelczak K. S., Turchi J. J. (2010) Purification and characterization of exonuclease-free Artemis. Implications for DNA-PK-dependent processing of DNA termini in NHEJ-catalyzed DSB repair. DNA Repair 9, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Demuth I., Bradshaw P. S., Lindner A., Anders M., Heinrich S., Kallenbach J., Schmelz K., Digweed M., Meyn M. S., Concannon P. (2008) Endogenous hSNM1B/Apollo interacts with TRF2 and stimulates ATM in response to ionizing radiation. DNA Repair 7, 1192–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Demuth I., Digweed M., Concannon P. (2004) Human SNM1B is required for normal cellular response to both DNA interstrand cross-link-inducing agents and ionizing radiation. Oncogene 23, 8611–8618 [DOI] [PubMed] [Google Scholar]
- 33. Freibaum B. D., Counter C. M. (2006) hSnm1B is a novel telomere-associated protein. J. Biol. Chem. 281, 15033–15036 [DOI] [PubMed] [Google Scholar]
- 34. Lam Y. C., Akhter S., Gu P., Ye J., Poulet A., Giraud-Panis M. J., Bailey S. M., Gilson E., Legerski R. J., Chang S. (2010) SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 29, 2230–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Overbeek M., de Lange T. (2006) Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr. Biol. 16, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 36. Akhter S., Lam Y. C., Chang S., Legerski R. J. (2010) The telomeric protein SNM1B/Apollo is required for normal cell proliferation and embryonic development. Aging Cell 9, 1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Touzot F., Callebaut I., Soulier J., Gaillard L., Azerrad C., Durandy A., Fischer A., de Villartay J. P., Revy P. (2010) Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10097–10102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Savitsky P., Bray J., Cooper C. D., Marsden B. D., Mahajan P., Burgess-Brown N. A., Gileadi O. (2010) High throughput production of human proteins for crystallization. The SGC experience. J. Struct. Biol. 172, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kiakos K., Hartley J. M., Hartley J. A. (2010) Measurement of DNA interstrand cross-linking in naked DNA using gel-based methods. Methods Mol. Biol. 613, 283–302 [DOI] [PubMed] [Google Scholar]
- 40. Cheng C. H., Kuchta R. D. (1993) DNA polymerase ϵ. Aphidicolin inhibition and the relationship between polymerase and exonuclease activity. Biochemistry 32, 8568–8574 [DOI] [PubMed] [Google Scholar]
- 41. Elisseeva E., Mandal S. S., Reha-Krantz L. J. (1999) Mutational and pH studies of the 3′ → 5′ exonuclease activity of bacteriophage T4 DNA polymerase. J. Biol. Chem. 274, 25151–25158 [DOI] [PubMed] [Google Scholar]
- 42. de Villartay J. P., Shimazaki N., Charbonnier J. B., Fischer A., Mornon J. P., Lieber M. R., Callebaut I. (2009) A histidine in the β-CASP domain of Artemis is critical for its full in vitro and in vivo functions. DNA Repair 8, 202–208 [DOI] [PubMed] [Google Scholar]
- 43. Mandel C. R., Kaneko S., Zhang H., Gebauer D., Vethantham V., Manley J. L., Tong L. (2006) Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 444, 953–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Feng M., Patel D., Dervan J. J., Ceska T., Suck D., Haq I., Sayers J. R. (2004) Roles of divalent metal ions in flap endonuclease-substrate interactions. Nat. Struct. Mol. Biol. 11, 450–456 [DOI] [PubMed] [Google Scholar]
- 45. José T. J., Conlan L. H., Dupureur C. M. (1999) Quantitative evaluation of metal ion binding to PvuII restriction endonuclease. J. Biol. Inorg. Chem. 4, 814–823 [DOI] [PubMed] [Google Scholar]
- 46. Slater J. P., Mildvan A. S., Loeb L. A. (1971) Zinc in DNA polymerases. Biochem. Biophys. Res. Commun. 44, 37–43 [DOI] [PubMed] [Google Scholar]
- 47. Williams R. O., Loeb L. A. (1973) Zinc requirement for DNA replication in stimulated human lymphocytes. J. Cell Biol. 58, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Herbette S., Cochard H. (2010) Calcium is a major determinant of xylem vulnerability to cavitation. Plant Physiol. 153, 1932–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qin N., Olcese R., Bransby M., Lin T., Birnbaumer L. (1999) Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc. Natl. Acad. Sci. U.S.A. 96, 2435–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee Bi B. I., Nguyen L. H., Barsky D., Fernandes M., Wilson D. M., 3rd. (2002) Molecular interactions of human Exo1 with DNA. Nucleic Acids Res. 30, 942–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tiefenbach T., Junop M. (2012) Pso2 (SNM1) is a DNA structure-specific endonuclease. Nucleic Acids Res. 40, 2131–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma Y., Pannicke U., Schwarz K., Lieber M. R. (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108, 781–794 [DOI] [PubMed] [Google Scholar]
- 53. Bae J. B., Mukhopadhyay S. S., Liu L., Zhang N., Tan J., Akhter S., Liu X., Shen X., Li L., Legerski R. J. (2008) Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene 27, 5045–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mason J. M., Sekiguchi J. M. (2011) Snm1B/Apollo functions in the Fanconi anemia pathway in response to DNA interstrand cross-links. Hum. Mol. Genet. 20, 2549–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Biggins J. B., Prudent J. R., Marshall D. J., Thorson J. S. (2006) A continuous assay for DNA cleavage using molecular break lights. Methods Mol. Biol. 335, 83–92 [DOI] [PubMed] [Google Scholar]
- 56. Yang C. J., Li J. J., Tan W. (2006) Using molecular beacons for sensitive fluorescence assays of the enzymatic cleavage of nucleic acids. Methods Mol. Biol. 335, 71–81 [DOI] [PubMed] [Google Scholar]
- 57. Hutton R. D., Craggs T. D., White M. F., Penedo J. C. (2010) PCNA and XPF cooperate to distort DNA substrates. Nucleic Acids Res. 38, 1664–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hutton R. D., Roberts J. A., Penedo J. C., White M. F. (2008) PCNA stimulates catalysis by structure-specific nucleases using two distinct mechanisms. Substrate targeting and catalytic step. Nucleic Acids Res. 36, 6720–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dorjsuren D., Kim D., Maloney D. J., Wilson D. M., 3rd, Simeonov A. (2011) Complementary non-radioactive assays for investigation of human flap endonuclease 1 activity. Nucleic Acids Res. 39, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee G., Yoo J., Leslie B. J., Ha T. (2011) Single-molecule analysis reveals three phases of DNA degradation by an exonuclease. Nat. Chem. Biol. 7, 367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li X., Hejna J., Moses R. E. (2005) The yeast Snm1 protein is a DNA 5′-exonuclease. DNA Repair 4, 163–170 [DOI] [PubMed] [Google Scholar]
- 62. Bonatto D., Brendel M., Henriques J. A. (2005) In silico identification and analysis of new Artemis/Artemis-like sequences from fungal and metazoan species. Protein J. 24, 399–411 [DOI] [PubMed] [Google Scholar]
- 63. Daiyasu H., Osaka K., Ishino Y., Toh H. (2001) Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503, 1–6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.