

Background: Elevated CO2 is toxic to mammalian cells.
Results: Molecular CO2 reduces cellular cAMP dependent on intracellular Ca2+.
Conclusion: CO2 can alter cellular physiological processes through IP3-mediated Ca2+ release.
Significance: Altered Ca2+ signaling mediated by CO2 might underpin the detrimental effects of CO2 on the cell.
Keywords: Adenylate Cyclase (Adenylyl Cyclase); Carbon Dioxide; Cyclic AMP (cAMP); Inositol 1,4,5-Trisphosphate; Parathyroid Hormone
Abstract
Elevated CO2 is generally detrimental to animal cells, suggesting an interaction with core processes in cell biology. We demonstrate that elevated CO2 blunts G protein-activated cAMP signaling. The effect of CO2 is independent of changes in intracellular and extracellular pH, independent of the mechanism used to activate the cAMP signaling pathway, and is independent of cell context. A combination of pharmacological and genetic tools demonstrated that the effect of elevated CO2 on cAMP levels required the activity of the IP3 receptor. Consistent with these findings, CO2 caused an increase in steady state cytoplasmic Ca2+ concentrations not observed in the absence of the IP3 receptor or under nonspecific acidotic conditions. We examined the well characterized cAMP-dependent inhibition of the isoform 3 Na+/H+ antiporter (NHE3) to demonstrate a functional relevance for CO2-mediated reductions in cellular cAMP. Consistent with the cellular biochemistry, elevated CO2 abrogated the inhibitory effect of cAMP on NHE3 function via an IP3 receptor-dependent mechanism.
Introduction
The importance of CO2 in biology is paramount. CO2 is integral to all life as the substrate for the CO2-fixing enzyme ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco)2 in photosynthetic organisms and is a substrate/product for many other metabolic enzymes. The pH-dependent CO2/bicarbonate equilibrium is fundamental to physiology and is intimately associated with homeostatic mechanisms, including pH regulation, volume control, and fluid secretion.
All life on Earth has continued to flourish despite being subjected to large fluctuations in the levels of CO2 in both the atmosphere and aquatic environments (1). Photosynthetic organisms are able to acclimate to large changes in atmospheric CO2 (2). Fluctuations in CO2 can also apply stress to unicellular and multicellular organisms over much shorter time scales. Aquatic environments can show both diurnal and long-term seasonal variations in CO2 with consequent effects on photosynthetic organisms (3). Increased respiration during exercise can cause the partial pressure of CO2 rise from 35–45 mm Hg to over 120 mm Hg. Specific mechanisms exist to detect elevated CO2 and enable appropriate responses, but CO2 can also have relatively nonspecific deleterious effects on the cell (4).
CO2 is proposed to enter cells through aquaporin, Amt, and Rhesus channels (5–7) and have direct effects on protein through carbamate formation, for example on Rubisco and hemoglobin (8). About 20 Protein Data Bank structures have CO2 as a ligand with a variety of modes of interaction but primarily through interactions with basic side chains (9). CO2 and HCO3− also influence a number of cell signaling processes. CO2 activates fungal pathogenesis through AC, and additional ACs from prokaryotes and mammals also respond directly to CO2 and HCO3− (10–12). HCO3− activates guanylyl cyclase types D and G to enable CO2 olfaction (13–15). The role of the cGMP pathway in CO2 chemosensing has also been conserved in Caenorhabditis elegans avoidance behavior (16–18). Chemosensing of CO2 in Drosophila melanogaster is mediated through Gr21a and Gr63a, two receptors of the seven-transmembrane domain class (19, 20). In mammals, ATP release is key to chemosensory control of respiration and increased CO2 permits ATP release from the medulla oblongata through a mechanism that requires connexin 26 (21, 22). CO2 is also chemosensed by specific taste receptor cells that express carbonic anhydrase type 4 and has evolutionarily conserved inhibitory effects on innate immunity through inhibition of NF-κB signaling (23–26). In acute acid-base disturbance, the proximal tubule cells of the mammalian kidney respond directly to CO2 to stimulate H+ secretion through a mechanism involving a tyrosine kinase of the epidermal growth factor receptor family and intracellular Ca2+ (27, 28). It is clear that specific mechanisms exist, through which CO2 can interact with biological systems.
The examples provided are mechanisms by which CO2 is detected specifically to initiate adaptive physiological responses, but CO2 can have generally detrimental effects on animal cellular processes. In C. elegans, increased CO2 causes slowed development, reduced fertility, and causes deterioration of body musculature (29). In Drosophila, elevated CO2 causes defects in embryonic development and egg laying and hatching (25). Elevated CO2 in rats stimulates renal phosphate excretion that is independent of other physiological factors, including pH (30). CO2 will also impair alveolar fluid reabsorption in alveolar type II epithelial cells by inducing Na+,K+-ATPase endocytosis (31, 32). CO2 can also chronically decrease cell proliferation through increasing levels of the miR-183 microRNA (33). Generally speaking, elevated CO2 is tolerated in humans, although toxic effects on the central nervous system, cardiovascular, renal, metabolic, and respiratory systems are evident. Despite this, some individuals may show a greater sensitivity to the adverse effects of CO2, for example, in the presence of an increased intracranial pressure. The significance of this is that ventilation strategies in patients that induce hypercapnia, so-called “permissive hypercapnia,” improves prognosis in models of acute lung injury, ischemia-reperfusion injury and acute respiratory distress syndrome (34, 35). The protective effect of permissive hypercapnia is explained to a large extent by the anti-inflammatory influence of CO2, but such immune suppression may be detrimental in clinical settings where infection or wounding is present (26, 36). There is a requirement, therefore, to understand the molecular basis of CO2 interactions with the core processes of the cell to understand how CO2 can be detrimental to cell function across the animal kingdom and also to inform clinical decisions regarding the use of permissive hypercapnia. In direct contrast to the current paradigm where CO2 can activate specific physiological processes through accumulation of cyclic nucleotides, we demonstrate that CO2 blunts cellular activities regulated by cAMP. This effect is independent of pH and requires Ca2+ release via the IP3 receptor. Given the ubiquity of cAMP and Ca2+ signaling in mammalian cells, this work suggests a key mechanism by which CO2 can have a broad spectrum of effects on cell physiology independent of pH.
EXPERIMENTAL PROCEDURES
Cell Culture
OK cells (gift of Heini Murer, University of Zurich) and HEK-PR1 cells (gift of Colin Taylor, University of Cambridge) were cultured in Dulbecco's modified Eagle's medium (DMEM)/Ham's Nutrient Mixture F12 (1:1 volume), 15 mm HEPES, 14 mm NaHCO3, 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin-streptomycin, 2% (v/v) non-essential amino acids, 1% (v/v) l-glutamine, and 500 μg/ml G418 (HEK-PR1 cells only). UMR-106 cells (gift of James Gallagher, University of Liverpool) were cultured in DMEM, 15 mm HEPES, 14 mm NaHCO3, 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin, 2% (v/v) non-essential amino acids, and 1% (v/v) l-glutamine. DT40KO and DT40-IP3R1 cells (gift of Colin Taylor) were cultured in RPMI 1640 medium, 15 mm HEPES, 14 mm NaHCO3, 10% (v/v) FBS, 1% (v/v) heat-inactivated chicken serum, 1% (v/v) penicillin-streptomycin, 2 mm glutamine, and 50 μm 2-mercaptoethanol.
Measurement of Intracellular pH
Cells attached to glass coverslips were loaded with 5 μm (HEK-PR1 cells) or 7.5 μm (OK cells) 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) for 30 min at 37 °C and 5% (v/v) CO2. pHi was measured using a microspectrofluorometric system (excitation, 490/440 nm; emission, 535 nm). pHi calibration was performed using high K+ nigericin solutions (37).
cAMP Accumulation
Cells were starved overnight in 0.2% (w/v) BSA in serum-free medium and labeled for 2 h with 0.75 μCi ml−1 [3H]adenine. Cells were washed with phosphate-buffered saline and incubated for 30 min at 37 °C at the desired CO2 concentration in 990 μl of pre-incubation media (DMEM/F12 1:1 or DMEM depending on cell type, 15 mm HEPES, 1% (v/v) penicillin-streptomycin, 1 mm 3-isobutyl-1-methylxanthine) pre-gassed with the appropriate CO2 concentration and with the pH adjusted. Assays were initiated with 10 μl of agonist. After 10 min at 37 °C, medium was removed, and cells were lysed with 1 ml 5% (w/v) trichloroacetic acid containing 1 mm ATP and 1 mm cAMP (OK, HEK-PR1, UMR-106 cells). cAMP was quantified by twin column chromatography (38). DT40KO and DT40-IP3R1 cell cAMP was assayed using the Biotrak cAMP enzyme immunoassay (GE Healthcare) according to the manufacturers instructions. Antagonists were added to the pre-incubation media.
In Vitro Adenylyl Cyclase Assay
Cell monolayers were washed with phosphate-buffered saline and suspended in lysis buffer (10 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 5 mm CaCl2) for 20 min. The cell suspension was pelleted, re-suspended in lysis buffer, and incubated for a further 20 min. The cell suspension was pelleted and resuspended in 20 mm Tris-HCl, pH 7.5, 5 mm NaCl, 1 mm DTT, 1 mm 3-isobutyl-1-methylxanthine, 20% (v/v) glycerol, and homogenized through a 21-gauge needle. Adenylyl cyclase assays were performed at 37 °C in a final volume of 100 μl and contained 100 mm Tris-HCl, 100 mm NaCl, 1 mm DTT, 2 mm MgCl2, 1 mm 3-isobutyl-1-methylxanthine, 5 units of creatine phosphokinase, 5 μm creatine phosphate, and 1 mm [α32P]ATP (25 kBq). Reactions were stopped by the addition of 150 μl of 50 mm Tris-HCl, pH 7.5, 5% (w/v) SDS. A further 650 μl of H2O and 100 μl of 1 mm ATP, 1 mm [2,8-3H]cAMP (150 Bq) were added prior to separation of product [α32P]cAMP by the twin column method (38).
Measurement of NHE3 Activity
NHE3 activity was monitored by measuring pHi recovery after a NH4Cl pulse using BCECF-AM. OK cells were grown to 100% confluence on glass coverslips and starved overnight in 0.2% (v/v) BSA in serum-free media. 3-min NH4Cl pulses (110 mm NaCl, 25 mm glucose, 20 mm NH4Cl, 20 mm HEPES, 14 mm NaHCO3, 5 mm KCl, 1 mm CaCl2, 1 mm MgSO4, pH 7.4) were followed by at least 5 min of perfusion in the same solution with NaCl replacing NH4Cl.
Ca2+ Imaging
Cells were loaded with 10 μm of the Ca2+-sensitive fluorescent dye Fura 2-AM in serum-free media for 30 min at 37 °C in 5% (v/v) CO2 in air. Cells were washed and resuspended in Krebs-Ringer-HEPES solution (130 mm NaCl, 25 mm glucose, 20 mm HEPES, 14 mm NaHCO3, 5 mm KCl, 1 mm CaCl2, 1 mm MgSO4, pH 7.4) for 30 min at 37 °C in 5% (v/v) CO2 in air. CaCl2 was omitted when examining the effect of extracellular Ca2+. Cells were transferred to fresh Krebs-Ringer-HEPES pre-gassed with the appropriate CO2 concentration and the pH adjusted. Fura 2 emission was measured using a spectrofluorometer with simultaneous excitation at 340 and 380 nm and emission at 510 nm.
Statistical Analysis
Error bars represent the S.E. Statistical significance was determined by using Student's t test between indicated groups, unless otherwise indicated, and a 95% confidence interval was taken as p < 0.05.
RESULTS
The study of the effects of molecular CO2 in vivo are confused by delineating CO2 effects from those due to the associated acidosis and in differentiating between CO2 effects on the tissue of interest from those secondary to changes in the endocrine and autonomic nervous systems. As elevated CO2 influences renal processes regulated by cAMP (39), we studied a renal proximal tubule-derived cell line (OK cells (40)) as a model to investigate the impact of CO2 upon cAMP signaling. A previous study had revealed that elevated (10%) CO2 had no apparent influence on cAMP accumulation but a drop in cAMP-response element-binding protein phosphorylation suggested that elevated CO2 might be inhibitory for cAMP signaling (12). Methodology was therefore developed on the basis of this study to investigate the influence of elevated CO2 on cAMP signaling.
PTH couples to the cAMP generating enzyme AC through its cognate receptor and the G protein subunit, Gαs. cAMP accumulation in OK cells was reduced at 10% compared with 5% CO2 at a PTH concentration (5 nm) of similar magnitude to that used for previous analysis of the influence of CO2 on OK cell physiology (Fig. 1A) (41). Batch to batch variation is known to influence the sensitivity of OK cells to PTH (42), but the response to CO2 was independent of cell batch or passage number. The reduction in cAMP was independent of extracellular pH, as reduction of the medium pH from 7.5 to 7.0 did not affect the response (Fig. 1B). The EC50 for the response was unchanged as medium pH was dropped from 7.5 to 7.0 and cAMP accumulation actually increased (supplemental Fig. S1). The drop in cAMP in response to elevated CO2 is therefore not explained by any potential acidification of medium pH on the assay. We measured final assay pH to assess whether changes in pHe at elevated CO2 might still influence the observed response through an effect on the potency and efficacy of PTH stimulation of AC. Final assay pHe in assays performed at a starting pHe of 7.5 at 5% CO2 was 7.5 ± 0.1 (mean ± S.D.) and at 10% CO2 was 7.4 ± 0.1 (mean ± S.D.). The observed drop in cAMP accumulation, caused by elevated CO2, was therefore not unduly affected by any influence of pHe on signaling. cAMP levels were depressed by elevated CO2 when AC was directly activated by 10 μm forskolin (FSK), indicating that the effect of CO2 is independent of the mechanism used to stimulate AC (Fig. 1C). The FSK response was insensitive to a drop in medium pH from 7.5 to 7.0 (supplemental Fig. S2) indicating, together with the relative stability of assay pH at elevated CO2, that the response to CO2 is not confused by any undue influence on pHe. To differentiate between effects of molecular CO2 and effects due to intracellular pH (pHi), we examined the transient intracellular acidification caused by CO2 (ΔpHi = −0.72 ± 0.17; Fig. 1D). We approximated the extent of intracellular acidification with media containing 2 mm propionic acid (ΔpHi = −0.60 ± 0.35; Fig. 1E). Transient intracellular acidification by propionic acid had no influence on cAMP indicating that the effect of CO2 on cAMP is not mediated through pHi (Fig. 1F). Furthermore, propionic acid did not influence the response of cAMP to CO2, indicating that propionic acid does not influence the cAMP pathway such that it cannot respond to inhibitory signals (Fig. 1G). The effect of CO2 on cAMP levels was fully reversible. Cells grown at 5% CO2 and then exposed to 10% CO2 prior to assay at 5% CO2 with FSK demonstrated cAMP accumulation indistinguishable from cells maintained and assayed at 5% CO2 (Fig. 1H). We examined whether the effect of CO2 on AC activity was due to protein degradation. The in vitro AC activity of OK cell crude membrane preparations exposed to 5 or 10% CO2 was similar (the specific activity at 10% CO2 was 115 ± 8% (S.E., n = 6) that at 5% CO2), indicating similar protein levels and consistent with the reversibility of the response to CO2.
FIGURE 1.
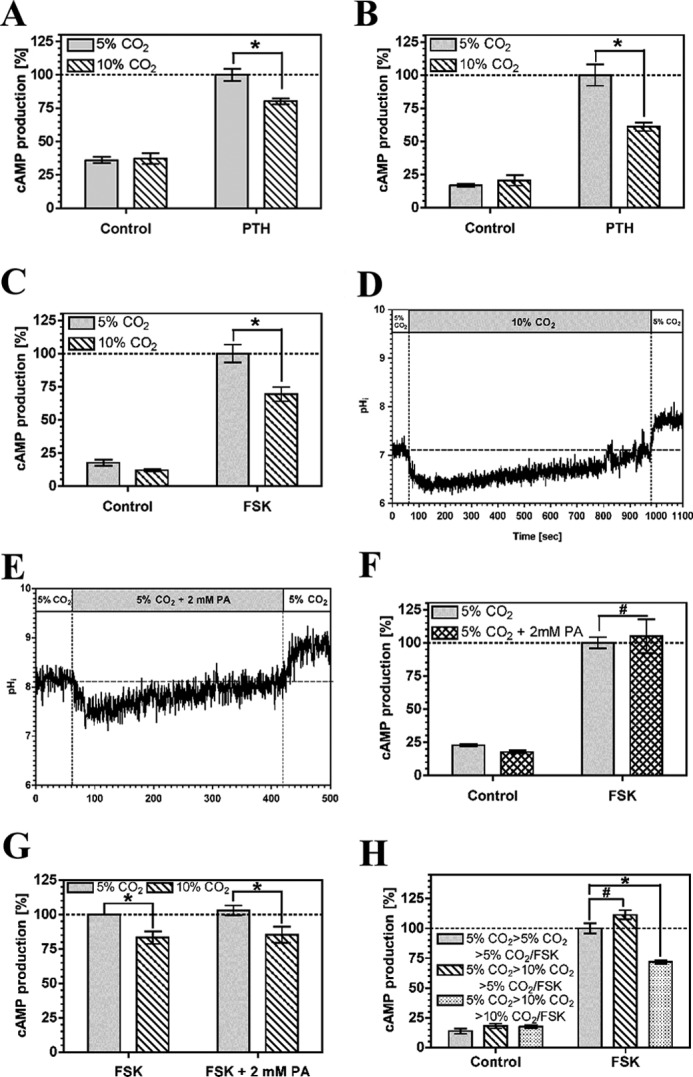
Elevated CO2 reduces cAMP accumulation in OK cells. A, 5 nm PTH stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.5. B, 5 nm PTH stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.0. C, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.0. D, pHi in OK cells in response to elevated CO2. E, pHi in OK cells in response to 2 mm propionic acid (PA). F, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 with or without 2 mm propionic acid at pH 7.5. G, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 with 2 mm propionic acid at pH 7.5. H, OK cells at 5% (v/v) CO2 were transferred to 5% (v/v) CO2 or 10% (v/v) CO2 for 30 min before assay at 5% (v/v) CO2 or 10% (v/v) CO2 with 10 μm FSK (pH 7.5). cAMP accumulation in each graph is normalized to the value in the presence of agonist at 5% (v/v) CO2 (n > 3; *, p < 0.05; #, not significant).
To determine whether the CO2 effect was cell context-specific, we examined the response of HEK 293 cells stably transfected with the human type 1 PTH receptor (HEK-PR1 cells) (43). cAMP accumulation stimulated by 5 nm PTH was reduced at elevated CO2 independent of extracellular pH (Fig. 2, A and B, supplemental Fig. S1) and any specific pathway used to stimulate AC (10 μm FSK; Fig. 2C and supplemental Fig. S2). 2 mm propionic acid gave a drop in pHi (ΔpHi = −0.61 ± 0.32; Fig. 2E) greater than that for elevated CO2 (ΔpHi = −0.21 ± 0.14; Fig. 2D) but had no influence on cAMP accumulation (Fig. 2F). Similar to OK cells, propionic acid did not influence the response of cAMP to CO2 (Fig. 2G). As the experimental process of elevating CO2 in air causes a small hypoxic effect (from 19.9% to 18.9% O2 (v/v)) we examined whether a shift from 20% to 18% O2 at constant CO2 influenced cAMP levels as an additional control. The mild hypoxia had no influence on cAMP indicating that the effect is mediated through CO2 (Fig. 2H). Similar to OK cells, the in vitro AC activity of HEK-PR1 crude membrane preparations exposed to 5 or 10% CO2 was similar (the specific activity at 10% CO2 was 99 ± 5% (S.E., n = 5) that at 5% CO2) indicating similar protein levels and cells grown at 5% CO2 and then exposed to 10% CO2 prior to assay at 5% CO2 with FSK demonstrated cAMP accumulation indistinguishable from cells maintained and assayed at 5% CO2 (Fig. 2I). To confirm that the effect of elevated CO2 on cAMP is a broadly applicable phenomenon we also demonstrated that the PTH-responsive rat osteosarcoma cell line, UMR-106 (44), showed an identical response to CO2 (supplemental Fig. S3).
FIGURE 2.

Elevated CO2 reduces cAMP accumulation in HEK-PR1 cells. A, 5 nm PTH stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.5. B, 5 nm PTH stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.0. C, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.0. D, pHi in HEK-PR1 cells in response to elevated CO2. E, pHi in HEK-PR1 cells in response to 2 mm propionic acid. F, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 with or without 2 mm propionic acid at pH 7.5. G, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2 or 10% (v/v) CO2 with 2 mm propionic acid (PA) at pH 7.5. H, 10 μm FSK stimulated cAMP accumulation at 5% (v/v) CO2/20% (v/v) O2, 5% (v/v) CO2/18% (v/v) O2, or 10% (v/v) CO2/18% (v/v) O2 at pH 7.5. I, HEK-PR1 cells at 5% (v/v) CO2 were transferred to 5% (v/v) CO2 or 10% (v/v) CO2 for 30 min before assay at 5% (v/v) CO2 or 10% (v/v) CO2 with 10 μm FSK (pH 7.5). cAMP accumulation in each graph is normalized to the value in the presence of agonist at 5% (v/v) CO2 (n > 3; * p < 0.05; #, not significant).
The activity of the nine identified mammalian G protein-responsive AC isoforms can be modulated by a number of other signaling processes, and we investigated whether any of these pathways was responsible for the reduction in cAMP accumulation in response to CO2. We used antagonists whose broad target range enabled us to simultaneously inhibit multiple cAMP interacting signaling pathways (Fig. 3A). The effect of CO2 on cAMP accumulation in HEK-PR1 cells did not require the activity of cAMP phosphodiesterase (1 mm 3-isobutyl-1-methylxanthine), soluble adenylyl cyclase (10 μm KH7), cAMP-dependent protein kinase (PKA) (10 μm H-89), calcium-calmodulin-dependent protein kinase II (100 nm autocamtide II), or protein kinase C (1 μm staurosporine/1 mm Gö 6983). The lack of an effect of CO2 with the AC inhibitor SQ 22,536 (200 μm) demonstrated the requirement for a G protein-responsive AC (45) as opposed to soluble AC, which is unresponsive to SQ 22,536 (46, 47). Carbonic anhydrase inhibition (100 μm acetazolamide) had no effect, indicating no requirement for conversion of CO2 to HCO3− (14). 1 mm extracellular ethylene glycol tetraacetic acid (EGTA) had no effect on the CO2 response, whereas it was ablated by the acetoxymethyl ester of 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA-AM; 1 mm), indicating a requirement for intracellular but not extracellular Ca2+. The influence of BAPTA-AM on the CO2 response was confirmed in OK cells, demonstrating a likely common mechanism of action (Fig. 3B). We used further inhibitors to investigate the source of the intracellular Ca2+. The CO2 effect was insensitive to 100 μm nifedipine (L- and T-type voltage-dependent Ca2+ channel blocker) in both OK and HEK-PR1 cells and 5 μm rotenone (a mitochondrial inhibitor) in HEK-PR1 cells (Fig. 4, A and B). Rotenone ablated the response of cAMP to CO2 in OK cells; however, we noted significant toxicity and cell death in response to rotenone in this cell line, indicating that this might be a nonspecific effect. The cAMP response was ablated by 10 μm thapsigargin (endoplasmic reticulum Ca2+-ATPase inhibitor) in both HEK-PR1 and OK cells (Fig. 4, A and B). This indicated a likely role for CO2 mediated Ca2+ release from a thapsigargin-sensitive store, most likely via the IP3 receptor and the endoplasmic reticulum. Further evidence for this was obtained in HEK-PR1 cells using the IP3R inhibitor 100 μm 2-APB (Fig. 4C). To eliminate the possibility of off target effects with thapsigargin and 2-APB, particularly as significant variability was observed with the latter due to toxicity, we investigated more specific evidence for the involvement of the IP3 receptor in Ca2+ release. We examined the effect of elevated CO2 on the DT40KO cell line (48). DT40KO cells are a chicken B lymphocyte-derived cell line genetically ablated for type 1, 2, and 3 IP3 receptors and are a null background for IP3 receptor studies. We examined cAMP accumulation in response to elevated CO2 in DT40KO cells compared with DT40-IP3R1 cells that have had the rat IP3 type 1 receptor introduced (49). Elevated CO2 did not blunt cAMP accumulation in DT40KO cells (Fig. 4D) but did in DT40-IP3R1 cells (Fig. 4E), proving that the type 1 IP3 receptor is required for the response to CO2. Intracellular acidification through 2 mm propionic acid had no influence on cellular cAMP in DT40-IP3R1 cells as observed for both OK and HEK-PR1 cells.
FIGURE 3.
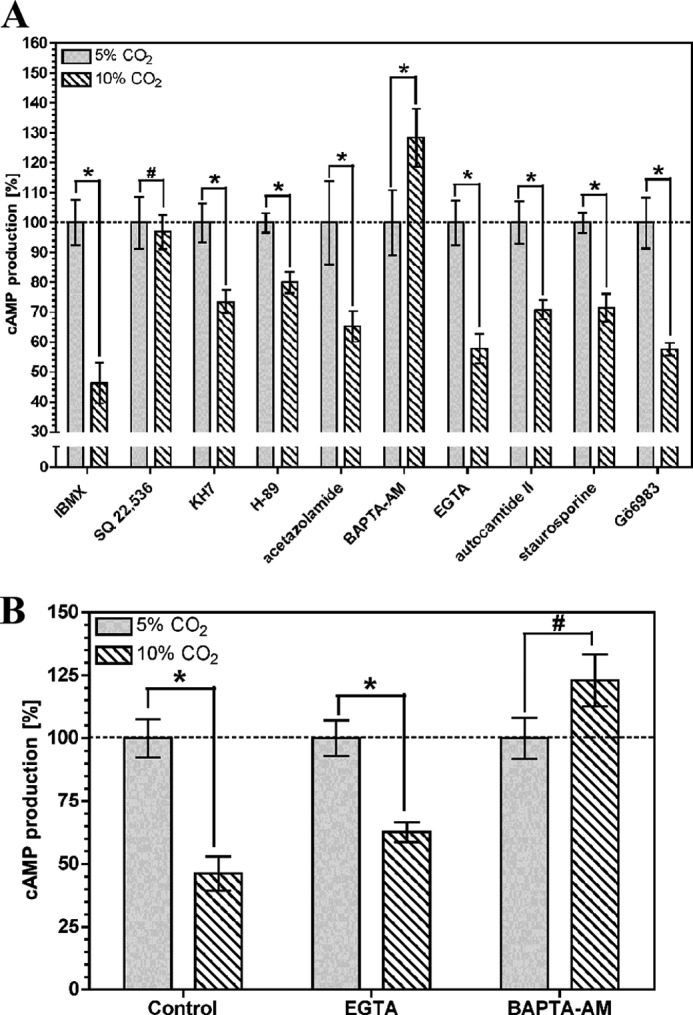
Intracellular Ca2+ is required for CO2 to reduce cellular cAMP. cAMP accumulation in HEK-PR1 (A) or OK cells (B) exposed to 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.5 in the presence of 10 μm FSK and various antagonists. cAMP accumulation for each antagonist is normalized to the value at 5% (v/v) CO2 (n > 4; *, p < 0.05; #, not significant). IBMX, 3-isobutyl-1-methylxanthine.
FIGURE 4.

Ca2+ release via IP3R is required for CO2 to reduce cellular cAMP. A–C, cAMP accumulation in HEK-PR1 (A and C) or OK (B) cells exposed to 5% (v/v) CO2 or 10% (v/v) CO2 at pH 7.5 in the presence of 10 μm FSK and various antagonists. cAMP accumulation for each antagonist is normalized to the value at 5% (v/v) CO2 (n > 4; *, p < 0.05; #, not significant). D and E, 10 μm FSK stimulated cAMP accumulation in DT40KO (D), and DT40-IP3R1 cells (E) exposed to 5% (v/v) CO2, 10% (v/v) CO2, or 5% (v/v) CO2 with 2 mm propionic acid (PA) at pH 7.5. cAMP accumulation in each graph is normalized to the value at 5% (v/v) CO2 (n > 3; *, p < 0.05; #, not significant). F, ratio of cytosolic Ca2+ at a test condition versus 5% (v/v) CO2 at pH 7.5 in DT40-IP3R1 or DT40-KO cells in the presence of various antagonists (n > 6; *, p < 0.05; #, not significantly <10% (v/v) CO2 by one-way analysis of variance with post-hoc one-sided Dunnett test).
As the CO2/cAMP effect is sensitive to intracellular Ca2+ chelation and shows a requirement for the IP3 receptor, we investigated whether elevated CO2 altered steady state cytoplasmic [Ca2+] in both DT40KO cells and DT40-IP3R1 cells. Cytoplasmic [Ca2+] was elevated at 10% compared with 5% CO2 in DT40-IP3R1 but not DT40KO cells (Fig. 4F). Nonspecific intracellular acidification through propionic acid had no influence on cytoplasmic [Ca2+]. Elevated CO2 therefore mediates Ca2+ release from the endoplasmic reticulum via the IP3 receptor. To provide an independent validation for the role of the IP3 receptor in the DT40 cell response to CO2, we treated DT40-IP3R cells with either 1 mm EGTA, 100 μm 2-APB, or 500 nm xestospongin C (Fig. 4F). Cytoplasmic [Ca2+] was elevated at 10% compared with 5% CO2 in DT40-IP3R1 cells in the presence of EGTA, consistent with the absence of an effect of EGTA on CO2 modulation of cAMP. The Ca2+ release in response to CO2 was ablated by the IP3 receptor antagonists xestospongin C and 2-APB. This result supports the interpretation that these inhibitors are blocking IP3 receptor signaling in HEK-PR1 and OK cells (Figs. 4C and 5B). Inclusion of the AC inhibitor SQ 22,536 (200 μm) had no influence on CO2-mediated Ca2+ release. These data confirm that an increase in cytosolic Ca2+ is a prerequisite for the effect of CO2 on cAMP (Fig. 4, D and E) and that cAMP lies downstream of the increase in cytosolic Ca2+ (Fig. 4F).
FIGURE 5.

Elevated CO2 blunts cAMP inhibition of NHE3 in OK cells. A, monitoring of pHi in OK cells in response to NH4Cl. The inset shows a sample experiment monitoring post-alkalization pHi recovery analyzed to generate the graph below. B, ratio of slope of pHi recovery at 10% (v/v) CO2 versus 5% (v/v) CO2 after shift from NH4Cl to NaCl in the presence of 10 μm FSK or 5 nm PTH and various inhibitors (n > 3; *, p < 0.05; #, not significant compared with FSK by one-way analysis of variance with post-hoc Bonferroni test). C, model for the impact of CO2 on PTH signaling at normal and elevated CO2. ER, endoplasmic reticulum.
We investigated the functional consequences of CO2-mediated reductions in intracellular cAMP by assessing a cAMP-dependent physiological process in OK cells. Sodium-proton exchanger isoform 3 (NHE3) is an apical Na+-H+ antiporter of renal epithelial (and OK) cells with a crucial role in H+, Na+, and fluid homeostasis (50). NHE3 is inhibited by PKA phosphorylation at serine residues 552 and 605 (51), and we examined the effect of elevated CO2 on cAMP-mediated suppression of NHE3 activity. OK cells exposed to NH4Cl alkalize due to H+ buffering by NH3, but pH regulatory mechanisms returns pHi to normal (Fig. 5A). On exchange of NH4Cl for NaCl, pHi drops as the accumulated intracellular NH4+ releases H+. The alkalization to restore pHi is due to NHE3 (52), and we analyzed this phase of the response.
Comparison of control pHi recoveries at 5 and 10% CO2 demonstrated a cAMP-independent suppression of recovery at elevated CO2 (Fig. 5, A and B; note the ratio of recovery at 10% compared with 5% CO2 < 1). Inhibition of NHE3-mediated pHi recovery by FSK or PTH was greater in 5% compared with 10% CO2 consistent with the effect of CO2 on cAMP levels (note the ratio of recovery at 10% CO2 compared with 5% CO2 > 1). The effect of CO2 on cAMP inhibition of pHi recovery was reduced by H-89 and BAPTA-AM, demonstrating a requirement for both PKA and intracellular Ca2+. 10 μm dantrolene (a ryanodine receptor antagonist) had no influence on CO2 suppression of cAMP signaling. The IP3 receptor antagonists xestospongin C (500 nm) and 2-APB (100 μm) eliminated the effect of CO2 on cAMP-dependent NHE3 inhibition. CO2 therefore suppresses the activity of the cAMP signaling pathway through Ca2+ release via the IP3 receptor with functional consequences for cAMP-dependent cellular processes (Fig. 5C).
DISCUSSION
In this work, we make two original claims. The first is that molecular CO2 reduces levels of cellular cAMP when the G-protein responsive cAMP signaling pathway is activated and this has functional consequences for downstream processes. This is in direct contrast to the current paradigm where cyclic nucleotide levels, where they are observed to respond to CO2, increase. The second is that molecular CO2 increases steady state cytoplasmic Ca2+ concentrations dependent on the IP3 receptor. The effect of CO2 on cAMP is a consequence of this altered Ca2+. These findings significantly advance our understanding of the effects of CO2 on the cell.
CO2 toxicity is not straightforward to study due to the effects of the associated acidosis and secondary effects on the endocrine and autonomic nervous systems. We circumvented these problems through the use of cultured cells to demonstrate that elevated CO2 blunts cellular cAMP production independent of pH. The use of sub-maximally activating concentrations of cAMP stimulating agonists enabled us to detect either activation or down-regulation of the cAMP signaling pathway under any given experimental condition. Submaximal 5 nm PTH gave only a very small increase in cAMP in experiments to examine the influence of pHe in OK cells (supplemental Fig. S1). As the assay procedure used has minimal effects on pHe, this issue with this batch of cells does not affect the main findings. Cell batches used to examine the influence of CO2 gave robust responses at submaximal 5 nm PTH (Figs. 1 and 5). In addition to a failure of intracellular acidification to modulate cAMP, two further lines of evidence make it unlikely that the effects of CO2 are mediated by pHi. First, pHi is allowed to normalize after CO2 elevation requiring a hypothesized acid signal to persist long after pH homeostasis. Second, FSK (activating all AC isoforms) and PTH (coupling to AC6 in HEKPR1 cells (53)) give identical responses to CO2 arguing against a localized acid signal communicating with a distinct signaling enzyme. Extracellular pH is also unlikely to be responsible as medium acidification (as might be proposed to occur mid-assay were buffering insufficient) does not alter the EC50 for PTH and actually gives an increase in cAMP. Medium acidification would therefore cause an underestimation of the decrease in cAMP. An increase in pCO2 causes a small decrease in pO2, but the decrease in pO2 did not explain the effects of pCO2 on cAMP. We conclude, therefore, that the effect of hypercapnia is mediated by molecular CO2 and not pH or any other variable.
The influence of CO2 on cAMP signaling might be due to a direct interaction with AC or with an alternative signaling pathway, which impacts on cAMP levels. Pharmacological inhibitors offered the best initial route to investigate CO2 responsive pathways that influence cAMP as the experiment could exploit the broad spectrum of targets antagonised under moderate concentrations. This method identified a role for intracellular Ca2+ in the response to CO2. The Ca2+ was most likely derived from an intracellular store as external EGTA did not influence the response. Further support came from the use of inhibitors previously used to study the role of Ca2+ in cellular responses to CO2 (27). Rotenone antagonized the cellular response to CO2 in OK cells, but this is most likely due to its observed toxicity, particularly as the effect was not reproduced in HEK-PR1 cells. Thapsigargin ablated the cAMP response to CO2 in both OK and HEK-PR1 cells. The use of 2-APB, which inhibits both the IP3 receptor and transient receptor potential channels, supported this finding. The lack of a role for extracellular Ca2+ eliminated the transient receptor potential channels and supported a role for Ca2+ release from the endoplasmic reticulum, in particular via the IP3 receptor. A role for the IP3 receptor was proven through the use of a cell line genetically ablated for all three IP3 receptor isoforms. The most likely interpretation of the pharmacological analysis in HEK-PR1 and OK cells is therefore supported unambiguously through genetics in an independent cell line. It is formally possible that the underlying signaling response to CO2 in HEK-PR1 and OK cells does not involve the IP3 receptor. This possibility is unlikely, however, as the IP3 receptor antagonists used in this study were validated by demonstrating that their effect on CO2-mediated Ca2+ release in DT40-IP3R1 cells was identical to the effect of the genetic ablation. A combination of the subtlety of the effects of CO2 and the incomplete knockdown of the IP3 receptor using siRNA (53) makes a genetic validation for the role of the IP3 receptor in these cells currently unfeasible. Future experiments using knock-outs generated via zinc finger nucleases will permit a genetic approach to be adopted in these cells.
Despite this, a coherent picture does emerge and supports what is known of the toxicity of CO2. A role for altered Ca2+ signaling in the response to CO2 is consistent with generic toxicity as Ca2+ signaling is a core physiological process evident in all mammalian cell types and elevated cytosolic Ca2+ is associated with cell toxicity (54). The approximate 20% increase in steady state cytoplasmic Ca2+ and 10–50% drop in cellular cAMP are relatively subtle effects and are consistent with the reasonable tolerance of mammals to elevated CO2 (55). A role for Ca2+ in CO2 responses has been investigated previously. Vadász and co-workers (32) noted a transient increase in cytoplasmic Ca2+ in response to hypercapnia, whereas Bouyer and co-workers (27) identified a rise in intracellular Ca2+ proposed as originating from an unconventional source. Our work provides significant clarity in its use of genetics to identify the Ca2+ source, but we cannot eliminate the possibility of context-dependent Ca2+ sources. It will be instructive in the future to investigate whether the effects of Ca2+ on AC are direct (56) or occur through a Ca2+-sensitive signaling pathway (57).
A crucial discovery is that elevated CO2 suppresses a cAMP-dependent cellular process. The effect of CO2 on a cAMP-responsive Na+-H+ antiporter is consistent with the observed biochemistry of the influence of CO2 on cAMP. We propose, therefore, that altered cAMP and/or Ca2+ signaling can be investigated further as a key mechanism by which the toxic effects of CO2 are manifested. For example, there is growing evidence that chronic daytime hypercapnia associated with obstructive sleep disorders predisposes individuals to cardiovascular disease (58). The central role of cAMP and Ca2+ in cardiac and circulatory physiology (57) suggests a key route to understanding this pathophysiology and possibilities for therapeutic intervention.
Ca2+ efflux through the IP3 receptor is modulated both by signaling pathways that regulate phospholipase C and by the local cellular environment (59–61). A major challenge for the future will be to determine how CO2 causes Ca2+ release through the IP3 receptor and its direct cellular target. Identification of such a CO2 target will further inform our understanding of the cell biology of CO2, in particular other cell processes that are influenced by CO2 to mediate its toxicity.
Supplementary Material
Acknowledgment
We thank Phil Townsend for helpful discussion and critical comments on the manuscript.
This work was supported by Leverhulme Trust Grant F/00 128/AU, Biotechnology and Biological Sciences Research Council Grant BB/I011994/1, and Wellcome Trust Grant GR083381MA (to M. J. C.).

This article contains supplemental Figs. S1–S3.
- Rubisco
- ribulose 1,5-bisphosphate carboxylase/oxygenase
- AC
- adenylyl cyclase
- 2-APB
- 2-aminoethoxydiphenyl borate
- Fsk
- forskolin
- Fura 2-AM
- Fura-2 pentakis-acetoxymethyl ester
- IP3
- inositol 1,4,5-triphosphate
- IP3R
- IP3 receptor
- PTH
- parathyroid hormone
- BCECF-AM
- 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- OK
- opossum kidney.
REFERENCES
- 1. Beerling D. J., Lomax B. H., Royer D. L., Upchurch G. R., Jr., Kump L. R. (2002) An atmospheric pCO2 reconstruction across the Cretaceous-Tertiary boundary from leaf megafossils. Proc. Natl. Acad. Sci. U.S.A. 99, 7836–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaplan A., Helman Y., Tchernov D., Reinhold L. (2001) Acclimation of photosynthetic microorganisms to changing ambient CO2 concentration. Proc. Natl. Acad. Sci. U.S.A. 98, 4817–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson M. S., Billett M. F., Dinsmore K. J., Wallin M., Dyson K. E., Jassal R. S. (2010) Direct and continuous measurement of dissolved carbon dioxide in freshwater aquatic systems: Method and applications. Ecohydrology 3, 68–78 [Google Scholar]
- 4. Sharabi K., Lecuona E., Helenius I. T., Beitel G. J., Sznajder J. I., Gruenbaum Y. (2009) Sensing, physiological effects, and molecular response to elevated CO2 levels in eukaryotes. J. Cell Mol. Med. 13, 4304–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soupene E., Inwood W., Kustu S. (2004) Lack of the Rhesus protein Rh1 impairs growth of the green alga Chlamydomonas reinhardtii at high CO2. Proc. Natl. Acad. Sci. U.S.A. 101, 7787–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musa-Aziz R., Chen L. M., Pelletier M. F., Boron W. F. (2009) Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc. Natl. Acad. Sci. U.S.A. 106, 5406–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakhoul N. L., Davis B. A., Romero M. F., Boron W. F. (1998) Effect of expressing the water channel aquaporin-1 on the CO2 permeability of Xenopus oocytes. Am. J. Physiol. 274, C543–548 [DOI] [PubMed] [Google Scholar]
- 8. Lorimer G. H. (1983) Carbon dioxide and carbamate formation: The makings of a biochemical control system. Trends Biochem. Sci. 8, 65–68 [Google Scholar]
- 9. Cundari T. R., Wilson A. K., Drummond M. L., Gonzalez H. E., Jorgensen K. R., Payne S., Braunfeld J., De Jesus M., Johnson V. M. (2009) CO2 Formatics: How do proteins bind carbon dioxide? J. Chem. Inf. Model 49, 2111–2115 [DOI] [PubMed] [Google Scholar]
- 10. Hammer A., Hodgson D. R., Cann M. J. (2006) Regulation of prokaryotic adenylyl cyclases by CO2. Biochem. J. 396, 215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klengel T., Liang W. J., Chaloupka J., Ruoff C., Schröppel K., Naglik J. R., Eckert S. E., Mogensen E. G., Haynes K., Tuite M. F., Levin L. R., Buck J., Mühlschlegel F. A. (2005) Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr. Biol. 15, 2021–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Townsend P. D., Holliday P. M., Fenyk S., Hess K. C., Gray M. A., Hodgson D. R., Cann M. J. (2009) Stimulation of mammalian G-protein-responsive adenylyl cyclases by carbon dioxide. J. Biol. Chem. 284, 784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chao Y. C., Cheng C. J., Hsieh H. T., Lin C. C., Chen C. C., Yang R. B. (2010) Guanylate cyclase-G, expressed in the Grueneberg ganglion olfactory subsystem, is activated by bicarbonate. Biochem. J. 432, 267–273 [DOI] [PubMed] [Google Scholar]
- 14. Guo D., Zhang J. J., Huang X. Y. (2009) Stimulation of guanylyl cyclase-D by bicarbonate. Biochemistry 48, 4417–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun L., Wang H., Hu J., Han J., Matsunami H., Luo M. (2009) Guanylyl cyclase-D in the olfactory CO2 neurons is activated by bicarbonate. Proc. Natl. Acad. Sci. U.S.A. 106, 2041–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bretscher A. J., Busch K. E., de Bono M. (2008) A carbon dioxide avoidance behavior is integrated with responses to ambient oxygen and food in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 105, 8044–8049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hallem E. A., Spencer W. C., McWhirter R. D., Zeller G., Henz S. R., Rätsch G., Miller D. M., 3rd, Horvitz H. R., Sternberg P. W., Ringstad N. (2011) Receptor-type guanylate cyclase is required for carbon dioxide sensation by Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 108, 254–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hallem E. A., Sternberg P. W. (2008) Acute carbon dioxide avoidance in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 105, 8038–8043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jones W. D., Cayirlioglu P., Kadow I. G., Vosshall L. B. (2007) Two chemosensory receptors together mediate carbon dioxide detection in Drosophila. Nature 445, 86–90 [DOI] [PubMed] [Google Scholar]
- 20. Kwon J. Y., Dahanukar A., Weiss L. A., Carlson J. R. (2007) The molecular basis of CO2 reception in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 104, 3574–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gourine A. V., Llaudet E., Dale N., Spyer K. M. (2005) ATP is a mediator of chemosensory transduction in the central nervous system. Nature 436, 108–111 [DOI] [PubMed] [Google Scholar]
- 22. Huckstepp R. T., id Bihi R., Eason R., Spyer K. M., Dicke N., Willecke K., Marina N., Gourine A. V., Dale N. (2010) Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J. Physiol. 588, 3901–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chandrashekar J., Yarmolinsky D., von Buchholtz L., Oka Y., Sly W., Ryba N. J., Zuker C. S. (2009) The taste of carbonation. Science 326, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cummins E. P., Oliver K. M., Lenihan C. R., Fitzpatrick S. F., Bruning U., Scholz C. C., Slattery C., Leonard M. O., McLoughlin P., Taylor C. T. (2010) NF-κB links CO2 sensing to innate immunity and inflammation in mammalian cells. J. Immunol. 185, 4439–4445 [DOI] [PubMed] [Google Scholar]
- 25. Helenius I. T., Krupinski T., Turnbull D. W., Gruenbaum Y., Silverman N., Johnson E. A., Sporn P. H., Sznajder J. I., Beitel G. J. (2009) Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 106, 18710–18715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Toole D., Hassett P., Contreras M., Higgins B. D., McKeown S. T., McAuley D. F., O'Brien T., Laffey J. G. (2009) Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-κB-dependent mechanism. Thorax 64, 976–982 [DOI] [PubMed] [Google Scholar]
- 27. Bouyer P., Zhou Y., Boron W. F. (2003) An increase in intracellular calcium concentration that is induced by basolateral CO2 in rabbit renal proximal tubule. Am. J. Physiol. Renal Physiol. 285, F674–687 [DOI] [PubMed] [Google Scholar]
- 28. Zhou Y., Bouyer P., Boron W. F. (2006) Role of a tyrosine kinase in the CO2-induced stimulation of HCO3 reabsorption by rabbit S2 proximal tubules. Am. J. Physiol. Renal Physiol. 291, F358–367 [DOI] [PubMed] [Google Scholar]
- 29. Sharabi K., Hurwitz A., Simon A. J., Beitel G. J., Morimoto R. I., Rechavi G., Sznajder J. I., Gruenbaum Y. (2009) Elevated CO2 levels affect development, motility, and fertility and extend life span in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 106, 4024–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guntupalli J., Matthews B., Carlin B., Bourke E. (1987) Effect of acute hypercapnia on PTH-stimulated phosphaturia in dietary Pi-deprived rat. Am. J. Physiol. Renal Physiol. 253, F34–40 [DOI] [PubMed] [Google Scholar]
- 31. Briva A., Vadász I., Lecuona E., Welch L. C., Chen J., Dada L. A., Trejo H. E., Dumasius V., Azzam Z. S., Myrianthefs P. M., Batlle D., Gruenbaum Y., Sznajder J. I. (2007) High CO2 levels impair alveolar epithelial function independently of pH. PLoS ONE 2, e1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vadász I., Dada L. A., Briva A., Trejo H. E., Welch L. C., Chen J., Tóth P. T., Lecuona E., Witters L. A., Schumacker P. T., Chandel N. S., Seeger W., Sznajder J. I. (2008) AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J. Clin. Invest. 118, 752–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vohwinkel C. U., Lecuona E., Sun H., Sommer N., Vadász I., Chandel N. S., Sznajder J. I. (2011) Elevated CO2 levels cause mitochondrial dysfunction and impair cell proliferation. J. Biol. Chem. 286, 37067–37076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laffey J. G., Kavanagh B. P. (1999) Carbon dioxide and the critically ill–too little of a good thing? Lancet 354, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 35. Taylor C. T., Cummins E. P. (2011) Regulation of gene expression by carbon dioxide. J. Physiol. 589, 797–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nichol A. D., O'Cronin D. F., Howell K., Naughton F., O'Brien S., Boylan J., O'Connor C., O'Toole D., Laffey J. G., McLoughlin P. (2009) Infection-induced lung injury is worsened after renal buffering of hypercapnic acidosis. Crit. Care Med. 37, 2953–2961 [DOI] [PubMed] [Google Scholar]
- 37. Hegyi P., Rakonczay Z., Jr., Gray M. A., Argent B. E. (2004) Measurement of intracellular pH in pancreatic duct cells: A new method for calibrating the fluorescence data. Pancreas 28, 427–434 [DOI] [PubMed] [Google Scholar]
- 38. Salomon Y., Londos C., Rodbell M. (1974) A highly sensitive adenylate cyclase assay. Anal. Biochem. 58, 541–548 [DOI] [PubMed] [Google Scholar]
- 39. Guntupalli J., Bourke E. (1985) Evidence that the phosphaturic effect of acute hypercapnia (HC) is not due to the low systemic pH of HC. Kidney Int. 27, 116 [Google Scholar]
- 40. Koyama H., Goodpasture C., Miller M. M., Teplitz R. L., Riggs A. D. (1978) Establishment and characterization of a cell line from the American opossum (Didelphys virginiana). In Vitro 14, 239–246 [DOI] [PubMed] [Google Scholar]
- 41. Jehle A. W., Hilfiker H., Pfister M. F., Biber J., Lederer E., Krapf R., Murer H. (1999) Type II Na-Pi cotransport is regulated transcriptionally by ambient bicarbonate/carbon dioxide tension in OK cells. Am. J. Physiol. 276, F46–53 [DOI] [PubMed] [Google Scholar]
- 42. Cole J. A., Forte L. R., Krause W. J., Thorne P. K. (1989) Clonal sublines that are morphologically and functionally distinct from parental OK cells. Am. J. Physiol. 256, F672–679 [DOI] [PubMed] [Google Scholar]
- 43. Short A. D., Taylor C. W. (2000) Parathyroid hormone controls the size of the intracellular Ca2+ stores available to receptors linked to inositol trisphosphate formation. J. Biol. Chem. 275, 1807–1813 [DOI] [PubMed] [Google Scholar]
- 44. Martin T. J., Ingleton P. M., Underwood J. C., Michelangeli V. P., Hunt N. H., Melick R. A. (1976) Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature 260, 436–438 [DOI] [PubMed] [Google Scholar]
- 45. Hurley J. H. (1999) Structure, mechanism, and regulation of mammalian adenylyl cyclase. J. Biol. Chem. 274, 7599–7602 [DOI] [PubMed] [Google Scholar]
- 46. Spehr M., Schwane K., Riffell J. A., Barbour J., Zimmer R. K., Neuhaus E. M., Hatt H. (2004) Particulate adenylate cyclase plays a key role in human sperm olfactory receptor-mediated chemotaxis. J. Biol. Chem. 279, 40194–40203 [DOI] [PubMed] [Google Scholar]
- 47. Leclerc P., Kopf G. S. (1999) Evidence for the role of heterotrimeric guanine nucleotide-binding regulatory proteins in the regulation of the mouse sperm adenylyl cyclase by the egg's zona pellucida. J Androl 20, 126–134 [PubMed] [Google Scholar]
- 48. Sugawara H., Kurosaki M., Takata M., Kurosaki T. (1997) Genetic evidence for involvement of type 1, type 2, and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laude A. J., Tovey S. C., Dedos S. G., Potter B. V., Lummis S. C., Taylor C. W. (2005) Rapid functional assays of recombinant IP3 receptors. Cell Calcium 38, 45–51 [DOI] [PubMed] [Google Scholar]
- 50. Alexander R. T., Grinstein S. (2009) Tethering, recycling, and activation of the epithelial sodium-proton exchanger, NHE3. J. Exp. Biol. 212, 1630–1637 [DOI] [PubMed] [Google Scholar]
- 51. Zhao H., Wiederkehr M. R., Fan L., Collazo R. L., Crowder L. A., Moe O. W. (1999) Acute inhibition of Na/H exchanger NHE-3 by cAMP. Role of protein kinase a and NHE-3 phosphoserines 552 and 605. J. Biol. Chem. 274, 3978–3987 [DOI] [PubMed] [Google Scholar]
- 52. Weinman E. J., Steplock D., Wade J. B., Shenolikar S. (2001) Ezrin binding domain-deficient NHERF attenuates cAMP-mediated inhibition of Na+/H+ exchange in OK cells. Am. J. Physiol. Renal Physiol. 281, F374–380 [DOI] [PubMed] [Google Scholar]
- 53. Tovey S. C., Dedos S. G., Taylor E. J., Church J. E., Taylor C. W. (2008) Selective coupling of type 6 adenylyl cyclase with type 2 IP3 receptors mediates direct sensitization of IP3 receptors by cAMP. J. Cell Biol. 183, 297–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johnson M. E., Gores G. J., Uhl C. B., Sill J. C. (1994) Cytosolic free calcium and cell death during metabolic inhibition in a neuronal cell line. J. Neurosci. 14, 4040–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adrogué H. E., Adrogué H. J. (2001) Acid-base physiology. Respir. Care 46, 328–341 [PubMed] [Google Scholar]
- 56. Mou T. C., Masada N., Cooper D. M., Sprang S. R. (2009) Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry 48, 3387–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Willoughby D., Cooper D. M. (2007) Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev. 87, 965–1010 [DOI] [PubMed] [Google Scholar]
- 58. Gopalakrishnan P., Tak T. (2011) Obstructive sleep apnea and cardiovascular disease. Cardiol. Rev. 19, 279–290 [DOI] [PubMed] [Google Scholar]
- 59. Rossi A. M., Tovey S. C., Rahman T., Prole D. L., Taylor C. W. (2011) Biochim. Biophys. Acta [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 60. Li G., Mongillo M., Chin K. T., Harding H., Ron D., Marks A. R., Tabas I. (2009) Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Higo T., Hattori M., Nakamura T., Natsume T., Michikawa T., Mikoshiba K. (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120, 85–98 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.