

Abstract
Variable number tandem repeats (VNTRs) constitute a relatively under-examined class of genomic variants in the context of complex disease because of their sequence complexity and the challenges in assaying them. Recent large-scale genome-wide copy number variant mapping and association efforts have highlighted the need for improved methodology for association studies using these complex polymorphisms. Here we describe the in-depth investigation of a complex region on chromosome 8p21.2 encompassing the dedicator of cytokinesis 5 (DOCK5) gene. The region includes two VNTRs of complex sequence composition which flank a common 3975 bp deletion, all three of which were genotyped by polymerase chain reaction and fragment analysis in a total of 2744 subjects. We have developed a novel VNTR association method named VNTRtest, suitable for association analysis of multi-allelic loci with binary and quantitative outcomes, and have used this approach to show significant association of the DOCK5 VNTRs with childhood and adult severe obesity (Pempirical= 8.9 × 10−8 and P= 3.1 × 10−3, respectively) which we estimate explains ∼0.8% of the phenotypic variance. We also identified an independent association between the 3975 base pair (bp) deletion and obesity, explaining a further 0.46% of the variance (Pcombined= 1.6 × 10−3). Evidence for association between DOCK5 transcript levels and the 3975 bp deletion (P= 0.027) and both VNTRs (Pempirical= 0.015) was also identified in adipose tissue from a Swedish family sample, providing support for a functional effect of the DOCK5 deletion and VNTRs. These findings highlight the potential role of DOCK5 in human obesity and illustrate a novel approach for analysis of the contribution of VNTRs to disease susceptibility through association studies.
INTRODUCTION
The contribution of copy number variants (CNVs) to complex disease susceptibility has been the subject of much debate in recent years. Although rare CNVs are responsible for severe highly penetrant forms of obesity (1,2) and other complex conditions (3,4), the impact of common CNVs is unclear. Associations between common CNVs and disease have been reported (5–8), but a recent large-scale array-based study from Craddock et al. (9) revealed little evidence of association between a subset of common CNVs and eight common diseases. Although it has been suggested that complex multi-allelic loci such as VNTRs are among the most likely structural variants to be enriched for functional impact (9,10), Craddock et al. (9) acknowledged the refractory nature of these more complex genomic structural variants in their analyses, highlighting in particular the challenge faced in investigating VNTRs. The involvement of VNTRs in complex disease has been previously evidenced by the association of a VNTR in the 5′ region of the insulin (INS) gene with type-1 diabetes (11), but little is known of their wider contribution to obesity. There is, therefore, a need for the development of novel methodology to analyse these relatively under-studied regions in the context of complex disease association.
We have developed a novel approach for the analysis of VNTRs, and have applied this method to identify a novel association of two VNTRs on chromosome 8p21.2 within the DOCK5 region with severe common obesity.
RESULTS
We first used signal intensity data from Illumina SNP arrays in a childhood obesity case–control study (12,13) consisting of 646 cases and 589 controls from France to carry out genome-wide CNV prediction using three different copy number prediction algorithms: CNVPartition (14), PennCNV (15) and cnvHap (16). All three algorithms identified the presence of a copy number variable region (CNVR) on chromosome 8p21.2 encompassing multiple intensity-only probes.
Initial association analysis of cnvHap copy number predictions identified two independent clusters of probes in this region showing association with obesity in the childhood obesity case–control study (Supplementary Material, Table S1). The most significant association in the region was observed at cnv12003p2 [β (SE) = 1.03 (0.09); P = 4.29 × 10−33], which was ranked 22nd most significant among the 27 942 included in this analysis. The copy number–obesity associations at each of the two clusters of probes in this region were subsequently reproduced in silico in an independent adult obesity case–control cohort (13,17,18), also from France, for which Illumina SNP genotyping data were available for 709 cases and 197 controls. In the adult sample, the most significant association in this region was again detected at the same probe cnv12003p2 [β (SE) = 1.98 (0.23); P = 1.88 × 10−18] that had shown the strongest signal of association in the child obesity sample (Supplementary Materials, Table S1).
The initial association results from the child and adult obesity case–control studies carried out using CNV predictions from Illumina SNP array data were then combined by meta-analysis using METAL, with genomic control inflation correction (19). In this combined association analysis, the highest ranking probe in the DOCK5 region (cnv12003p2) was the 32nd most significant of 27 942 probes tested in our association analysis (Supplementary Material, Fig. S1; P = 5.41 × 10−5), and all 9 intensity-only probes within the region were ranked within the top 1.1% of associations in this analysis (Supplementary Material, Fig. S1 and Table S2).
These probes were located within two distinct hypervariable regions, referred to here as VNTR A (chr8: 25 085 372–25 085 875) and VNTR B (chr8: 25 129 579–25 130 501) (Fig. 1A). The two VNTRs flank a simple, common, previously identified 3975 bp deletion (20) (chr8: 25 122 602–25 126 576; deletion frequency 82–83%), with stable breakpoints across unrelated individuals (Fig. 1C and Supplementary Material, Fig. S2). Despite the high frequency of the deletion, it is indeed the mutant allele, as syntenic alignments from ENSEMBL indicate that orthologous sequence exists in all eutherian mammals (21). VNTR A lies ∼12 kb upstream of the dedicator of cytokinesis 5 (DOCK5) gene, whereas both VNTR B and the common deletion are located within the first intron of DOCK5. In our cnvHap analysis using the Illumina SNP array data for the child and adult obesity case–control studies, DOCK5 was the only gene region containing two separate structurally variable regions associated with obesity; probes within both of which were ranked within the top ∼1% of associations. Association signals at both VNTRs were replicated between the child and adult obesity case–control studies, and the presence of a known deletion between these two hypervariable regions suggested this locus to be highly complex. As a result, the region was prioritized for further in-depth investigation.
Figure 1.

The DOCK5 region. (A) Schematic overview of the DOCK5 CNV region. Position of DOCK5 is shown by the black arrow, with exons shown as black boxes. The position of probe rs6997760 (chr8: 25 101 829) is indicated relative to VNTRs A and B and a 3975 bp deletion on chr8p21.2 in intron 1–2 of DOCK5. (B and D) Cluster plots of the first (x-axis) versus second (y-axis) principal components of the LRR across three and six intensity-only probes within each of VNTRs A and B, respectively. Probe positions located between chr8: 25 085 709–25 085 826 (VNTR A) and chr8: 25 129 632–25 130 278 (VNTR B). Red closed circles: obesity cases; black closed circles: normal-weight controls. (C) CGH analytics (Agilent Technologies) view of the 3975 bp CNV on chr8p21.2. Array CGH was carried out on 9 child obese cases, 10 adult obese cases and 9 child controls, using Agilent 8 × 15 k custom arrays. Log2 ratios are ∼0 for homozygous deleted samples, and ∼2 for heterozygous samples, as the reference sample appears to have a homozygous deletion at this locus. Samples homozygous for the deletion are shown in black, whereas samples with two copies are shown in red. Both the 3975 bp deletion and VNTR B lie within intron 1–2 of the DOCK5 gene (represented by the black arrow in A). Exons are represented as black boxes in (A).
Direct sequencing of the VNTRs revealed a complex internal structure (Supplementary Material, Fig. S3). Each VNTR was shown to be highly polymorphic, varying in both size and sequence composition, with alleles of the same size also showing variability in repeat block composition (Supplementary Material, Fig. S3). Figure 1B and D provide summarized illustrations of signal intensity in obesity cases and controls at these two VNTRs.
Since we considered that the repetitive multi-allelic nature of the sequence at both VNTR loci, along with the challenges presented by batch effects on intensity-only probes, might have rendered in silico copy number estimation unreliable, we sought to confirm the putative association of the DOCK5 region with obesity using alternative methods. Local single nucleotide polymorphism (SNP) association analysis in the DOCK5 region in the pooled French childhood and adult obesity data sets showed the SNP rs6997760 (chr8: 25 101 829) to be significantly associated with obesity [odds ratio (OR) = 0.57; P= 5.68 × 10−4; Pcorrected = 0.025]. We also examined publicly available data from a meta-analysis conducted on 249 796 subjects for evidence of SNP association with body mass index (BMI) in the DOCK5 region (22). Although these results did not show any significant association of the SNP rs6997760 with BMI (P> 0.05), it is not uncommon for associated variants identified in study samples consisting of early-onset and/or extreme obesity cases not to show association with BMI in population samples (22).
In order to identify the source of the association signals, we directly genotyped the 3975 bp deletion and both flanking VNTRs in a larger subset of the French child and adult obesity samples, consisting of 706 child obesity cases and 644 child controls, as well as 714 adult obesity cases and 680 controls. Genotyping of the deletion was carried out using a direct polymerase chain reaction (PCR)-based assay (Table 1 and Supplementary Material, Figs. S4 and S5). The deletion was found to be in moderate linkage disequilibrium (LD) with the associated SNP (rs6997760, r2 = 0.45, P < 1 × 10−16); and both the SNP and homozygous non-deletion of this region showed similar protective effects against obesity in the combined adult and child cohorts [OR = 0.46 (95% confidence interval 0.29–0.78), Pcombined = 1.6 × 10−3].
Table 1.
Multivariate association analysis of fragment-length alleles within both VNTRs and the common 3975 bp deletion in the DOCK5 region
| Allele frequency |
β (SE) | Association with obesity |
||||
|---|---|---|---|---|---|---|
| Child | Adult | Child | Adult | Child | Adult | |
| 3975 bp deletion | 17%a | 18%a | −1.2 (0.5) | −0.5 (0.3) | 1.1 × 10−2 | 5.2 × 10−2 |
| VNTR-selected variables | ||||||
| VNTR A: 590–640 bp | 1.3% | 1.4% | 0.8 (0.3) | 1.1 (0.4) | 4.9 × 10−3 | |
| VNTR B: 944–1022 bp | 20% | 16% | −0.4 (0.1) | 0.1 (0.1) | 0.27 | |
| VNTR B: 1112–1127 bp | 1.9% | 1.2% | −1.5 (0.4) | 0.1 (0.4) | 0.68 | |
| VNTR B: 1073–1084 bp | 3.9% | 2.0% | −1.5 (0.3) | −0.6 (0.3) | 6.4 × 10−2 | |
| VNTR B: 1099–1103 bp | 1.6% | 1.2% | −0.9 (0.4) | 0.8 (0.4) | 8.3 × 10−2 | |
| VNTR A + B multivariate model | 8.9 × 10−8 | 3.1 × 10−3 | ||||
Associations between the 3,975 bp deletion and obesity were assessed under a recessive model in the child and adult obesity case-control samples. Multivariate models were trained in the child obesity case–control study and replicated in the adult obesity study. Models were built for VNTR A and subsequently VNTR B. Effect sizes and standard errors, as well as replication (adult) P-values were calculated using a logistic regression model. The VNTRA + VNTRB model significance is calculated using a likelihood ratio test between a model that includes all variables and a model including only the 3975 bp deletion and gender. The model significance presented for the child training data set is the empirical P-value (10 000 permutations).
aNote that the allele frequency presented in this table for the 3975 bp deletion in DOCK5 is that of the rarer undeleted allele at this locus.
PCR followed by fragment analysis of fluorescent PCR fragments for each of the VNTR loci produced a quasi-continuous distribution of PCR fragment sizes ranging from 502 to 770 bp in VNTR A and 631 to 1202 bp in VNTR B. Comparison of sequence lengths obtained from PCR fragment analysis by capillary electrophoresis and those obtained from Sanger sequencing for VNTR B revealed that fragment analysis provided total length estimates 3 bp shorter than those determined using sequence data. As this was consistent across the samples examined, it did not affect between-sample comparisons. Once this was taken into account, sample size estimates derived from sequencing and PCR fragment analysis were perfectly correlated.
We first used the software CLUMP (23) to carry out association analyses at the DOCK5 VNTR A and VNTR B loci using our fragment analysis data in the child and adult obesity samples. However, although significant association with obesity was clearly detected for both VNTRs, a subset of susceptibility alleles shared between the two study samples could not be identified using a binning method that operated independently of allele size during clustering (such as the one implemented in CLUMP, Supplementary Material, Table S3). Therefore, we implemented a novel algorithm called VNTRtest, which only considers bins comprising contiguous fragment lengths. VNTRtest calculates the significance of each possible contiguous bin (defined by a lower and upper fragment length bound) by regressing the phenotype on the number of instances of the alleles within the bin. In order to identify all independent signals of association in the data, VNTRtest iterates this procedure and subtracts the component of disease log-odds that can be attributed to previously selected VNTR bins at each iteration, including also relevant covariates and/or risk factors (such as the 3975 bp deletion in this specific study). At each stage, the P-value of the selected bin as well as the P-value of the full model up to that point is recorded, and the iteration terminates when the P-value is >0.05. To correct for testing multiple hypotheses, VNTRtest repeats this procedure on 10 000 data sets in which phenotype and covariates are permuted to infer a parametric form of the null distribution from which it calculates approximate empirical P-values (see Materials and Methods for further details).
We first tested the performance of VNTRtest by carrying out simulations using the empirical distribution of VNTRs observed in this study. In simulations conducted under the null hypothesis, VNTRtest showed a type-I error of 0.051. Under an alternative hypothesis of one associated VNTR bin, simulations showed that VNTRtest had >80% power to detect a single associated VNTR bin with simulated odds ratio of <0.5 or >2.5 (Supplementary Material, Fig. S7a and Table S4), with power <80% otherwise. In the scenario of two associated VNTR bins, VNTRtest exhibited >80% power with one moderately associated bin (OR = 1.2) provided the second bin had OR < 0.6 or > 1.7 (Supplementary Material, Fig. S7b and c and Table S4). Under the alternative hypothesis of three associated VNTR bins, VNTRtest had a power of >80% to detect all three bins when two were in the same direction and fixed, provided the third bin had OR < 0.7 or OR >1.4 (Supplementary Material, Fig. S7d and Table S4). Finally, in the case of four associated VNTR bins, VNTRtest showed >80% power to detect all four associated bins when three were fixed and followed the same direction, regardless of the effect size of the fourth (Supplementary Material, Fig. S7e and Table S4). These results demonstrated the increased power obtained from having multiple associated allele bins.
We then applied VNTRtest to the DOCK5 VNTR data from the child and adult obesity cases and controls. Application of VNTRtest to the childhood cohort resulted in the selection of a multivariate model which included a VNTR A bin of 590–640 bp and VNTR B bins of sizes 944–1022, 1112–1127, 1073–1084 and 1099–1103 bp, respectively (Table 1 and Figs 2 and 3). This model was significantly associated with obesity after permutation in the childhood cohort used for training (Pempirical = 8.9 × 10−8), and association of this model with obesity was replicated in the adult obesity cohort (P= 3.1 ×1 0−3). Figure 2 shows the significance levels of all possible VNTR bins during the first iteration of VNTRtest, highlighting the reproducibility of VNTR A bin associations between the two cohorts.
Figure 2.

Association of DOCK5 VNTR bins with obesity. The heat map indicates the level of association (−log10 P-value) between VNTR bins and obesity in a logistic regression model, after regressing out gender and the DOCK5 3795 bp deletion (see Materials and Methods). (A) VNTR A bins; (B) VNTR B bins. Top left triangle: association in child cohort; middle triangle: association in adult cohort; lower triangle: association in combined child and adult cohort.
Figure 3.

Log odds ratios of variants significantly associated with obesity in both the child and adult obesity samples. For the 3975 bp DOCK5 deletion, the log odds of disease risk for homozygote undeleted relative to homozygote deleted is shown. For VNTR bins, the log odds of disease risk for heterozygote relative to homozygote is shown.
We calculated the proportion of heritability of obesity explained by the three variants at the DOCK5 locus using a multi-factorial liability threshold model (24). The model assumes an underlying continuous liability to a disease determined by the sum of genetics and environmental liabilities and the disease occurs when an individual's total liability exceeds a certain threshold. Genotypic configurations at a susceptibility locus show different mean liabilities. Based on a lifetime risk of obesity of 16.85% (25), the DOCK5 VNTRs and 3975 bp deletion explain a total of 1.24% of the variance in liability to obesity (0.42, 0.36 and 0.46% for VNTR A, VNTR B and the 3975 bp deletion, respectively).
A widely recognized challenge encountered in association studies is the tendency to overestimate the effect size of an identified variant, known as the ‘winner's curse’ (26). A 1000-fold 80%-training-to-20%-testing cross-validation analysis (Supplementary Material, Table S6) on the child obesity cohort demonstrated that although the VNTR B bins show limited inflation (up to 20%), the VNTR A 590–640 bin shows 140% inflation in children. Nevertheless, we still see a stronger effect size in adults for this particular bin. Further studies will be required to provide additional confirmation of the effect sizes of all three variants at the DOCK5 locus investigated in this study.
We next investigated the population history of the 3975 bp deletion. The sequence comprising the deletion is shared with other eutherian but not metatherian mammals or non-mammalian vertebrates, although the DOCK5 gene itself is shared within Euteleostomi (21) and is thus older (Supplementary Material, Figs S10 and S11). CNV genotypes for the deletion [CNVR3831.1 in Conrad et al. (10)] were available for 450 Hapmap samples (10). Allele frequencies for the deletion in the European, Han Chinese + Japanese and Yoruban African population samples are shown in Supplementary Material, Figure S8. SNP frequencies for rs6997760, which we found to be in moderate LD with the deletion, are presented in Supplementary Material, Figure S9. We observed a large population differentiation in the 3975 bp deletion allele as well as the SNP rs6997760, with the deletion showing an ∼60% increase in frequency between the Yoruban African and the European population samples (Supplementary Material, Fig. S8 and 9), indicating that this deletion has been under on-going positive selection in the migration out of Africa.
We then sought to assess functional effects of variation at the DOCK5 VNTRs in an independent Swedish sib-pair cohort discordant for BMI (27), for which SNP genotyping data and gene expression data from subcutaneous adipose tissue were available for 149 families (28). Structural variation at both DOCK5 VNTRs was modelled indirectly using the total signal intensity (LRR, log R ratio) at intensity-only probes located within VNTRs A and B, and the association of signal intensity at these probes with DOCK5 transcript levels was assessed using the famCNV (29) program. Copy number at DOCK5 VNTRs A and B was significantly associated with DOCK5 transcript levels (minimum Pempirical = 0.015 at probe cnvi0018215) (Table 2).
Table 2.
Association of LRR signal intensity with DOCK5 transcript levels in a Swedish sib-pair cohort
| Illumina probe | Chromosome 8 position | Empirical P-value | β (SE) | |
|---|---|---|---|---|
| VNTR A | cnvi0001691 | 25085709 | 0.175 | — |
| cnvi0001692 | 25085821 | 0.072 | — | |
| cnvi0001690 | 25085826 | 0.036 | 0.069 (0.025) | |
| VNTR B | cnvi0018215 | 25129632 | 0.015 | 0.071 (0.024) |
| cnvi0001693 | 25129709 | 0.116 | — | |
| cnvi0001694 | 25129964 | 0.100 | — | |
| cnvi0001695 | 25130171 | 0.215 | — | |
| cnvi0001697 | 25130231 | 0.143 | — | |
| cnvi0001696 | 25130278 | 0.024 | 0.066 (0.023) |
Empirical P-values were calculated by permutation of transcript levels, with P-values significant after correction shown in bold. Note that probes differ in name between Table 2 and Supplementary Material, Tables S1 and S2 due to the fact that a different probe nomenclature for intensity-only probes was introduced by Illumina between the Illumina Human CNV370-Duo and Illumina Human 610-Quad arrays.
We also noted that the region encompassing the 3975 bp deletion region is predicted to be transcriptionally repressive (Supplementary Material, Fig. S12) (30,31). Thus, a deletion of this region may result in an increase in transcription levels. To test this hypothesis, we genotyped the 3975 bp deletion in the Swedish discordant sib-pair cohort, for whom gene expression data were available (28). The deletion was found to be associated with increased DOCK5 expression in this sample (β = −0.46; P = 0.027) (Supplementary Material, Fig. S13).
Finally, analysis of publicly available gene expression profiles from subcutaneous adipose tissue from overweight subjects (32) revealed significantly increased expression of DOCK5 (Supplementary Material, Fig. S14) following exposure to a controlled-feeding high-saturated fatty acid diet (P= 3.9 × 10−3, exact permutation paired t-test; median paired fold-change = 1.03), thus suggesting diet-induced variation in DOCK5 expression levels.
Taken together, these results suggest putative links between copy number variation and dietary fat intake and expression levels of DOCK5 in adipose tissue that may lead to the subsequent development of obesity.
DISCUSSION
Association analyses between VNTR loci and complex phenotypes are greatly complicated by the large number of different alleles that may be found at such loci, varying in both size and sequence composition. By applying our novel VNTR allele bin refinement method implemented in VNTRtest, we have shown that specific VNTR alleles at DOCK5 are significantly associated with childhood and adult obesity. VNTRtest models the association between complex multi-allelic VNTRs and both quantitative and binary phenotypes through the identification of bin configurations that favour clustering of allele combinations of similar size which maximize the observed association, with significance of association determined empirically. In this study, we show that VNTRtest outperforms size-independent binning of alleles by identifying associations replicating between the two data sets. The program allows the inclusion of covariates and confounders in association analyses, and the generation of multivariate models encompassing multiple CNVs in regions under study, which is particularly useful in the case of CNVRs containing multiple multi-allelic markers, as in the DOCK5 region.
These data support the association of VNTR variants at the DOCK5 locus with adult and childhood obesity. We also showed that structural variation at the DOCK5 VNTRs and deletion of a 3975 bp region within intron 1–2 of DOCK5 were independently associated with increased expression levels of the gene transcript. In addition, analysis of publicly available expression data indicated that DOCK5 expression levels are increased relative to baseline in individuals exposed to a diet high in saturated fatty acids. Taken together with previously reported associations between high saturated fatty acid diets and fat accumulation (33,34), we hypothesize that increased expression of DOCK5, mediated either through copy number changes resulting in increased expression, or through environmentally induced increases in expression levels, may result in an increased risk of obesity.
Relatively little is known about the DOCK5 gene, which is a member of the DOCK family of guanine-nucleotide exchange factors that activate Rho-family GTPases by exchanging bound GDP for free guanosine triphosphate (GTP) (35). Phylogenetic reconstruction shows that DOCK5 has arisen from two duplication events in Euteleostomi (Supplementary Material, Fig. S11), also giving rise to its closest paralogues DOCK1 and DOCK2 (21). DOCK5 is expressed in multiple tissues, including adipose tissue, brain and pancreas (36), and interacts with the regulatory and catalytic subunits of protein phosphatase 2, encoded by PPP2R1A, PPP2R1B and PPP2CA, respectively (37). Protein phosphatase 2 has been shown to inactivate v-akt murine thymoma viral oncogene homolog (Akt) proteins (38,39) as well as mitogen-activated protein kinase 1 and 3 (MAPK1 and 3, also known as ERK2 and ERK1, respectively) (40). The well-established involvement of Akt and MAPK in body weight regulation in humans, with both pathways having been shown to mediate the anorectic effects of leptin, provides a possible potential mechanistic basis for the involvement of DOCK5 in obesity (41,42). Interestingly, DOCK5 has recently been identified as a regulator of osteoclast function, playing an essential role in bone resorption (43–45). Taken together with the association of complex CNVRs within the DOCK5 region with obesity presented here, these findings suggest that DOCK5 may play an important role in the well-established relationship between adiposity and bone density (46).
In the present study, we suggest that multiple allele size classes within DOCK5 VNTRs, as well as an intervening common 3975 bp deletion, are significantly associated with obesity in childhood and adult case–control cohorts, and that they explain >1% of the variance in liability. We also propose a novel powerful approach to carry out association studies using these complex structural polymorphisms, thus enabling more systematic investigation of the role of VNTRs in complex diseases and the relatively unexplored contribution of these loci to the ‘missing heritability’ (47) of these disorders.
MATERIALS AND METHODS
Study subjects
Informed consent and ethical approval were obtained for all subjects included in this study.
Childhood obesity cohort
A previously described childhood obesity case–control sample set from France consisting of 706 cases and 644 controls was used for this study (13). Child obesity cases were below the age of 18 with BMI z-score ≥97th percentile for their age and gender, with a minimum of one obese first-degree relative. Controls included in this study were selected from the STANISLAS cohort (12). Selected controls had age- and sex-corrected BMI z-score <90th percentile, and were all below 18 years of age (13).
Adult obesity cohort
The adult obesity study consisted of a case–control sample from France comprising 714 cases with BMI ≥ 40 kg/m2 and 680 normal-weight controls, described in a previous study (13). Informed consent from all participants and ethical approval were obtained as detailed previously (12,48).
Swedish sib-pair cohort
For expression analysis of DOCK5 transcripts, data were taken from a Swedish discordant sibling cohort, consisting of 154 nuclear families with BMI discordant sibling pairs (BMI difference >10 kg/m2), giving a total of 732 subjects (27). Average family size was 4.75. Expression data were available for 342 subjects and Illumina SNP genotyping data for 349 subjects.
In silico CNV prediction
Data quality control
Childhood and adult obesity case–control cohorts were genotyped on the Illumina Human CNV370-Duo chip as previously described (13). Illumina genotyping data were available for 646 childhood obesity cases and 589 child controls, 709 adult obesity cases and 197 adult controls. Illumina genotyping data were also available for 349 siblings from the Swedish sib-pair cohort, who were genotyped on the Illumina Human610-Quad chip.
Illumina raw data were uploaded into Beadstudio 2.0 (Illumina) and SNP genotype re-clustering carried out for each of the cohorts, using the Beadstudio re-clustering algorithm (Illumina). Samples with a call rate <0.95 were excluded from further analyses.
Sample genotype calls, LRR and B-allele frequency (BAF) were exported for CNV analysis.
Data normalization
For each sample, LRR median subtraction was carried out on a per-chromosome basis. In order to correct for additional known sources of CNV discovery artefacts, LRR data were corrected using a probe-by-probe regression on GC content, as well as a localized Loess function within a 500 kb window to correct for wave effects.
CNV prediction
LRR and B allele frequency were used for the prediction of copy number state in the child and adult obesity case–control cohorts, using cnvHap (16), which employs a hidden Markov model to predict CNVs at the haplotype level, integrating joint CNV and SNP haplotype structure to refine copy number predictions. Model parameters were empirically optimized for each data set.
PennCNV, an algorithm that also employs a hidden Markov model for CNV prediction, and CNV Partition, which models CNVs, using a Gaussian mixture model, were also employed for CNV prediction in these data sets (14,15).
Principal component analysis
Principal component analysis (PCA) was carried out independently at each of VNTR A and VNTR B using three and six intensity-only probes within each of the respective VNTRs in the DOCK5 region, using Illumina LRR data in the child and adult obesity cases and controls. PCA was carried out using the svd function in R version 2.11 (49).
Custom array CGH
Custom-designed Agilent 8 × 15 k arrays were used to validate CNVs predicted from obesity genome-wide association study (GWAS) data (unpublished data). Array CGH was carried out on 9 childhood obesity cases, 10 adult obesity cases and 9 child controls, using the manufacturer's recommended protocol (Agilent Technologies).
DNA sequencing
Purification of PCR products was carried out using the Cambio Ultra-Clean PCR Clean-Up Kit following the manufacturer's recommended conditions. Purified products were sequenced in both forward and reverse directions on an ABI3730xl DNA analyser (Applied Biosystems) (primer sequences are available upon request). Analysis of sequence data was carried out using the Chromas 2.01 software (Technelysium Pty Ltd). Human reference sequences were retrieved from the UCSC Genome Browser (hg18) (50), and ClustalW was used for multiple alignment analysis of sample and reference sequences (51). For the 3975 bp deletion, the upstream breakpoint was determined by aligning the reference sequence with sequences generated by the forward primer in the sequencing reaction. The downstream breakpoint was determined by aligning the reference sequence with the reverse complement of sequences generated using the reverse primer.
Genotyping of VNTRs A and B in child and adult cohorts
High-throughput PCR-based assays were designed to permit genotyping of two loci within the DOCK5 region found to encompass variable number tandem repeats (referred to as VNTRs A and B) in the French child and adult obesity case–control cohorts. VNTR A was genotyped by PCR amplification using a fluorescently labelled forward primer, followed by fragment analysis on the ABI 3730xl DNA analyser and sizing with reference to the ABI LIZ1200 size standard (Applied Biosystems). A semi-nested PCR strategy followed by fragment analysis was employed to permit genotyping of VNTR B (Supplementary Material, Fig. S5, primer sequences and detailed reaction conditions are available upon request). Data analysis for both VNTRs was carried out using the GeneMarker software (SoftGenetics). To determine the reproducibility of the PCR and fragment analysis assay used for VNTR genotyping, 93 samples were genotyped in duplicate. Reproducibility of VNTR fragment size calling was found to be >98% for both VNTR A and VNTR B.
Genotyping of 3975 bp deletion in French child and adult obesity cohorts and the Swedish obesity-discordant sib-pair sample
Further characterization of an additional CNV detected upstream of region B, established to be a 3975 bp deletion, was carried out by PCR amplification, using primers designed as shown in Supplementary Material, Figure S2 (primer sequences are available upon request). This PCR-based assay was employed to genotype the DOCK5 3975 bp CNV in the previously described child and adult obesity case-control cohorts from northern France and the siblings from the Swedish discordant sib-pair cohort. PCR primers were designed using the Primer 3 software (52). All PCR amplification steps were carried out using the Clontech Advantage 2 PCR kit under the manufacturer's recommended conditions.
Genotype PCA
PCA was carried out on 25 190 autosomal SNP genotypes from the Illumina GWAS data for the child and adult obesity cases and controls, using singular value decomposition in R version 2.13.2 (53). The top principal components for the child cases and controls showed no association with obesity (P > 0.05), and therefore were not included in the association analyses. The first principal component in the adult obesity case–control study showed significant association with obesity (P = 1.07 × 10−7), and was included as a covariate in our association analyses.
Association analyses using in silico CNV predictions
CNV predictions at the 8p21.2 locus by cnvHap were tested for association with obesity using logistic regression, with gender and sample LRR variance included as covariates in association analyses. Only probes at which cnvHap predicted a CNV with a frequency >5% (27 942 probes) in both the child and adult obesity studies were included in the association analyses.
Meta-analysis of association analyses using in silico CNV predictions
Meta-analysis of the genome-wide CNV association results from the child and adult obesity Illumina array data was carried out using the software package METAL (19), combining the effect size estimates from both studies, weighted using the inverse of the corresponding standard errors and correcting for inflation using genomic control. Only probes at which cnvHap predicted a CNV with a frequency >5% (27 942 probes) were included in the association analyses.
SNP association analyses in the DOCK5 region
After quality control, a total of 44 SNPs within the DOCK5 gene and the surrounding 5′ and 3′ intergenic regions, extending to the nearest genes upstream and downstream, were tested for association in the pooled French childhood and adult obesity data sets using logistic regression under a recessive model.
VNTR association analysis using the VNTRtest algorithm
We developed a novel algorithm for the assessment of the association of VNTR fragment-length genotypes (G) with a phenotype (D), called VNTRtest.
VNTRtest is an iterative, greedy forward selection algorithm. Let k denote the iteration. We define the set of variables selected up to iteration k as Vk and initialize this set as the set of covariates: V0 = {cj}, where cj denotes the jth covariate, such as gender. At each iteration, VNTRtest looks for the VNTR bin, defined by lower and upper fragment-length bounds (lk, uk) for which VNTR bin genotypes Bk are most strongly associated with the phenotype in a regression model after regressing out the previously selected variables Vk−1. We define the VNTR bin genotypes Bk as the count of fragment-length alleles x within the range lk ≤ x < uk. We consider bins only which either (i) do not overlap with previously selected bins; (ii) are completely nested within previously selected bins; or (iii) completely contain previously selected bins.
We define an initial logistic regression model LM0 as log(π/(1 − π)) ∼ V0 and record the regression coefficients of this model as β0. In the iteration step, we then search for the most significant bin Bk under the regression models 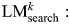 log(π/(1−π)) − βk−1 × Vk−1 ∼ Bk. We then update the list of selected variables: Vk = {Vk−1, Bk}, build a new regression model LMk as log(π/(1 − π)) ∼ Vk and use this to calculate a new set of regression coefficients βk. The significance of the variable Bk within the regression model
log(π/(1−π)) − βk−1 × Vk−1 ∼ Bk. We then update the list of selected variables: Vk = {Vk−1, Bk}, build a new regression model LMk as log(π/(1 − π)) ∼ Vk and use this to calculate a new set of regression coefficients βk. The significance of the variable Bk within the regression model 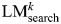 is denoted as pk. The iteration terminates once pk> 0.05. We also calculate the significance of the model up to and including variable Bk (pmodelk) as the significance of twice the log-likelihood ratio 2 × log(P(D|G, Vk)/P(D|G,V0)) under the χ2 distribution with k degrees of freedom.
is denoted as pk. The iteration terminates once pk> 0.05. We also calculate the significance of the model up to and including variable Bk (pmodelk) as the significance of twice the log-likelihood ratio 2 × log(P(D|G, Vk)/P(D|G,V0)) under the χ2 distribution with k degrees of freedom.
In order to make this algorithm more computationally efficient, we have derived a heuristic strategy for efficiently searching the space of bin boundaries (lk, uk) at each iteration k. First, we restrict our search to a grid consisting of all the observed fragment lengths in the data set. Second, at the beginning of our search strategy, we further restrict our set of predictors to a coarse grid of (lk, uk) consisting of every fifth potential fragment length. The most significant coarse bin is subsequently refined by considering all possible fragment lengths in the surrounding window.
We use a permutation procedure to address the problem of multiple hypothesis testing (in this case, multiple bins). We generate 10 000 permuted data sets, in which both phenotype and initial covariates are permuted relative to the fragment-length genotypes, thus generating 10 000 sets of {pmodelk} values under the null distribution. For each value of k, we fit a parametric distribution to the −log 10(p) values, and we use this distribution to estimate empirical P-values.
VNTRtest first builds a model for VNTR A which is subsequently expanded to include VNTR B. VNTRtest builds a model using one cohort at a time, which is then assessed on the remaining cohort. The algorithm for assessing significance is the same as above, except that we use pre-defined Bk, and as such there is no requirement for a permutation procedure to correct for multiple hypothesis testing.
A prototype of our algorithm VNTRtest is available at: http://www1.imperial.ac.uk/medicine/people/l.coin/.
VNTRtest simulations
We generated 1000 simulated data sets under the null hypothesis of no association, in order to assess the type-I error. Each simulation consisted of generating fragment-length genotypes for X = 646 cases and Y = 589 controls, the same sample size and case:control ratio as the child study. For each simulation, we simulated fragment-length genotypes assuming Hardy–Weinberg equilibrium and using the per base-pair fragment-length allele frequency distribution observed at the VNTR B locus in the child study. We simulated 2000 samples, which is more than the total number of the original samples in order to be able to subsample at least X cases and Y controls.
For simulations under the null hypothesis of no association, we randomly chose X cases and Y controls, irrespective of genotype. The type-I error was calculated as the fraction of data sets simulated under the null for which the VNTRtest-reported permutation model P-value was <0.05. We also simulated various scenarios under the alternative hypothesis, where we varied both the number and the effect size of the simulated, associated VNTRs. For each simulation under the alternative hypothesis, we randomly selected one, two, three or four bins which were causative, which did not overlap and which each showed an allele frequency >5%. This was achieved by randomly selecting a lower bound for the bin, and then incrementing the upper bound for the bin until the allele frequency was at least 5%, followed by confirmation that the bin did not overlap a previously selected bin.
Once a set of bins was selected, we calculated the bin genotypes for bin k (see the section on VNTRtest) Bk and simulated case–control status for each individual according to log(p/(1 − p)) ≈ Σkβk × Bk. When only one associated VNTR was simulated, we varied the OR between OR = 0.2 to OR = 3.6 to include the values of the discovered ORs in the original study. Under the scenario of two associated VNTRs, we fixed one effect being OR = 0.2 (protective) and we varied the other. We repeated with fixing one effect being OR = 1.2 (predisposing). In the scenario of three VNTRs being associated, we fixed the two effects being protective and we varied the third. The same was applied for the four effects. The simulated ORs were chosen so as to resemble and include the ones detected in the original study.
The power of each scenario was calculated as the fraction of simulations for which the VNTRtest-reported permutation P-value was <0.05.
VNTR association analysis using CLUMP
VNTR alleles at both DOCK5 VNTRs A and B were tested for association with obesity using the CLUMP T4 χ2 statistic, generated by grouping the VNTR alleles into a 2 × 2 table to maximize the χ2 value (23). The analysis was carried out using the child obesity data set for training and the adult obesity data set for replication, as well as vice versa. Empirical distributions of the T4 χ2 statistic were generated for each data set using 100 000 Monte Carlo simulations. A χ2 value for VNTR allele clusters identified by CLUMP as significantly associated with obesity (Pempirical < 0.05) in the training data set (either child or adult obesity) was then calculated in the replication data set. Empirical P-values for the replication data set were then calculated as P= (r + 1)/(n + 1), where r is the number of simulations where a χ2 value higher than the test χ2 value was obtained, and n is the total number of Monte Carlo simulations carried out.
Estimation of the variance in liability (Vg) explained by the DOCK5 variants
We calculated the variance explained for the 3975 bp deletion using the method described by So et al. (24), in which we used the odds ratios and frequencies of each genotype in the adult (replication) cohort. In order to apply the same approach to the VNTR loci, we first calculated for each individual the ‘extended’ genotype over all associated bins as the vector consisting of the number of copies the individual had in each bin listed in order. We calculated the odds ratio (OR) and frequency of each extended genotype that was observed in at least 15 individuals in the adult cohort. These OR and frequency values were then used to evaluate the total variance explained by each VNTR using the same method. In order to check that the variance explained calculated by these methods was independent, we re-calculated Vg for VNTR A, excluding those samples with the 3975 bp protective non-deletion homozygote genotype, and also for VNTR B, excluding both those samples as well as samples with the protective heterozygote 590–640 bp VNTR A genotype. In each case, the variance explained increased slightly, indicating that the original variance explained estimates were unlikely to be inflated by LD. The lifetime risk of obesity was taken to be 16.85% (25).
RNA extraction and microarray analysis
Subcutaneous adipose tissue biopsies were obtained from all participants in the Swedish sib-pair cohort as described previously (27). Gene expression was measured for 376 siblings, using the Affymetrix Human Genome U133 Plus 2.0 gene expression arrays (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's recommendations. Gene expression levels were normalized using the robust multiarray average (RMA) method (54). Informed written consent was obtained from all participants. This study was approved by the Ethics Committee at Gothenburg University (55).
Association of copy number at the DOCK5 VNTRs with DOCK5 gene expression
Association of copy number at the DOCK5 VNTRs with DOCK5 expression in subcutaneous adipose tissue was carried out in the Swedish sib-pair sample set using famCNV (29). The program models LRR and BAF in a mixed linear model to control for phenotypic resemblance among family members due to polygenic effects. The association test between intensity signal at a particular probe and gene expression level is evaluated by comparing the full model where LRR is included in the fixed effects and a null model where its effect is constrained to zero. famCNV is available at: http://www1.imperial.ac.uk/medicine/people/m.falchi.
Association of copy number at the DOCK5 3975 bp with DOCK5 gene expression
A linear mixed effects model, implemented in the lmekin function from the R kinship package (56), was used to carry out association analysis between copy number at the DOCK5 3975 bp deletion and DOCK5 expression in subcutaneous adipose tissue in the Swedish sib-pair sample. Sex and age were included as covariates in this analysis.
Paired-sample analysis of dietary-fat-induced variation in DOCK5 expression levels
Gene expression analysis from subcutaneous adipose tissue of moderately overweight individuals (n = 20) at risk of metabolic syndrome subjected to a 2-week run-in saturated fat diet followed by a 8-week diet intervention was obtained from gene expression omnibus data set GDS3678 (32). Individuals were assigned to two independent groups and subjected to a saturated (n = 10) and mono-unsaturated (n = 10) fat-supplemented diet. Gene expression after the 2-week run-in and at the end of the program was collected for both groups. A paired comparison of the individual expression profiles before and after intervention was performed for the two groups independently. Quantile normalization was applied to all microarrays and paired samples t-test and exact permutation t-test were consequentially applied using R (49) with package exactRankTests (57).
Genomic coordinates
Except where indicated, all genomic coordinates in this manuscript correspond to the human genome sequence build NCBI36/hg18.
SUPPLEMENTARY MATERIAL
FUNDING
J.S.El-S.M. is supported by an Imperial College Department of Medicine PhD studentship. J.L.B. is supported by Wellcome Trust fellowship grant number WT088431MA. A.C.A. and L.J.M.C. acknowledge support from the European Commission, Framework 7, grant number 223367. L.B. acknowledges the support of the Wellcome Trust Value-in-People award. A.I.F.B. acknowledges support from Diabetes UK. The STANISLAS Family Study was supported by the Caisse Nationale d'Assurance Maladies des Travailleurs Salariés (CNAM), the Institut National de la Santé et de la Recherche Médicale (INSERM), the Région Lorraine, the Communauté Urbaine du Grand Nancy, the Université de Lorraine and the BioIntelligence project. This work was supported by grants from the Wellcome Trust (grant ref. 079534/z/06/z), Swedish Research Council (K2010-55X-11285-13), the Swedish Foundation for Strategic Research to Sahlgrenska Center for Cardiovascular and Metabolic Research, the Swedish Diabetes Foundation and the Swedish federal government under the LUA/ALF agreement.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Simon Burbidge and Matt Harvey of the Imperial College High Performance Computing Service for their assistance (http://www.imperial.ac.uk/ict/services/teachingandresearchservices/highperformancecomputing). The authors acknowledge Ivan Andrew and the MRC core sequencing facility at Imperial College London for their support. We also thank Enrico Petretto for his advice and comments on the manuscript, and Kim Sung for technical assistance. The authors would also like to thank the study participants in each of the studies, as well as the field investigators for the recruitment and examinations of study subjects.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Walters R.G., Jacquemont S., Valsesia A., de Smith A.J., Martinet D., Andersson J., Falchi M., Chen F., Andrieux J., Lobbens S., et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. doi:10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glessner J.T., Bradfield J.P., Wang K., Takahashi N., Zhang H., Sleiman P.M., Mentch F.D., Kim C.E., Hou C., Thomas K.A., et al. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am. J. Hum. Genet. 2010;87:661–666. doi: 10.1016/j.ajhg.2010.09.014. doi:10.1016/j.ajhg.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. doi:10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 4.Williams N.M., Zaharieva I., Martin A., Langley K., Mantripragada K., Fossdal R., Stefansson H., Stefansson K., Magnusson P., Gudmundsson O.O., et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376:1401–1408. doi: 10.1016/S0140-6736(10)61109-9. doi:10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanciulli M., Norsworthy P.J., Petretto E., Dong R., Harper L., Kamesh L., Heward J.M., Gough S.C., de Smith A., Blakemore A.I., et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 2007;39:721–723. doi: 10.1038/ng2046. doi:10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollox E.J., Huffmeier U., Zeeuwen P.L., Palla R., Lascorz J., Rodijk-Olthuis D., van de Kerkhof P.C., Traupe H., de Jongh G., den Heijer M., et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. doi:10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fellermann K., Stange D.E., Schaeffeler E., Schmalzl H., Wehkamp J., Bevins C.L., Reinisch W., Teml A., Schwab M., Lichter P., et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 2006;79:439–448. doi: 10.1086/505915. doi:10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sha B.Y., Yang T.L., Zhao L.J., Chen X.D., Guo Y., Chen Y., Pan F., Zhang Z.X., Dong S.S., Xu X.H., et al. Genome-wide association study suggested copy number variation may be associated with body mass index in the Chinese population. J. Hum. Genet. 2009;54:199–202. doi: 10.1038/jhg.2009.10. doi:10.1038/jhg.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., et al. Genome-wide association study of CNVs in 16 000 cases of eight common diseases and 3000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. doi:10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. doi:10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Julier C., Hyer R.N., Davies J., Merlin F., Soularue P., Briant L., Cathelineau G., Deschamps I., Rotter J.I., Froguel P., et al. Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature. 1991;354:155–159. doi: 10.1038/354155a0. doi:10.1038/354155a0. [DOI] [PubMed] [Google Scholar]
- 12.Visvikis-Siest S., Siest G. The STANISLAS Cohort: a 10-year follow-up of supposed healthy families. Gene-environment interactions, reference values and evaluation of biomarkers in prevention of cardiovascular diseases. Clin. Chem. Lab. Med. 2008;46:733–747. doi: 10.1515/CCLM.2008.178. doi:10.1515/CCLM.2008.178. [DOI] [PubMed] [Google Scholar]
- 13.Meyre D., Delplanque J., Chevre J.C., Lecoeur C., Lobbens S., Gallina S., Durand E., Vatin V., Degraeve F., Proenca C., et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 2009;41:157–159. doi: 10.1038/ng.301. doi:10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- 14.Peiffer D.A., Le J.M., Steemers F.J., Chang W., Jenniges T., Garcia F., Haden K., Li J., Shaw C.A., Belmont J., et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. doi:10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. doi:10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coin L.J., Asher J.E., Walters R.G., Moustafa J.S., de Smith A.J., Sladek R., Balding D.J., Froguel P., Blakemore A.I. cnvHap: an integrative population and haplotype-based multiplatform model of SNPs and CNVs. Nat. Methods. 2010;7:541–546. doi: 10.1038/nmeth.1466. doi:10.1038/nmeth.1466. [DOI] [PubMed] [Google Scholar]
- 17.Balkau B., Eschwege E., Tichet J., Marre M. Proposed criteria for the diagnosis of diabetes: evidence from a French epidemiological study (D.E.S.I.R.) Diabetes Metab. 1997;23:428–434. [PubMed] [Google Scholar]
- 18.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S., et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. doi:10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 19.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. doi:10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T., et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. doi:10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 21.Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. doi:10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Speliotes E.K., Willer C.J., Berndt S.I., Monda K.L., Thorleifsson G., Jackson A.U., Allen H.L., Lindgren C.M., Luan J., Magi R., et al. Association analyses of 249 796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010;42:937–948. doi: 10.1038/ng.686. doi:10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sham P.C., Curtis D. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann. Hum. Genet. 1995;59:97–105. doi: 10.1111/j.1469-1809.1995.tb01608.x. doi:10.1111/j.1469-1809.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- 24.So H.C., Gui A.H., Cherny S.S., Sham P.C. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet. Epidemiol. 2011;35:310–317. doi: 10.1002/gepi.20579. doi:10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 25.Widen E., Lehto M., Kanninen T., Walston J., Shuldiner A.R., Groop L.C. Association of a polymorphism in the beta 3-adrenergic-receptor gene with features of the insulin resistance syndrome in Finns. N. Engl. J. Med. 1995;333:348–351. doi: 10.1056/NEJM199508103330604. doi:10.1056/NEJM199508103330604. [DOI] [PubMed] [Google Scholar]
- 26.Zollner S., Pritchard J.K. Overcoming the winner's curse: estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 2007;80:605–615. doi: 10.1086/512821. doi:10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlsson L.M., Jacobson P., Walley A., Froguel P., Sjostrom L., Svensson P.A., Sjoholm K. ALK7 expression is specific for adipose tissue, reduced in obesity and correlates to factors implicated in metabolic disease. Biochem. Biophys. Res. Commun. 2009;382:309–314. doi: 10.1016/j.bbrc.2009.03.014. doi:10.1016/j.bbrc.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walley A.J., Jacobson P., Falchi M., Bottolo L., Andersson J.C., Petretto E., Bonnefond A., Vaillant E., Lecoeur C., Vatin V., et al. Differential coexpression analysis of obesity-associated networks in human subcutaneous adipose tissue. Int. J. Obes. (Lond.) 2012;36:137–147. doi: 10.1038/ijo.2011.22. doi:10.1038/ijo.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eleftherohorinou H., Andersson-Assarsson J.C., Walters R.G., El-Sayed Moustafa J.S., Coin L., Jacobson P., Carlsson L.M., Blakemore A.I., Froguel P., Walley A.J., et al. famCNV: copy number variant association for quantitative traits in families. Bioinformatics. 2011;27:1873–1875. doi: 10.1093/bioinformatics/btr264. doi:10.1093/bioinformatics/btr264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. doi:10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., Noble W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. doi:10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dijk S.J., Feskens E.J., Bos M.B., Hoelen D.W., Heijligenberg R., Bromhaar M.G., de Groot L.C., de Vries, J.H., Muller M., Afman L.A. A saturated fatty acid-rich diet induces an obesity-linked proinflammatory gene expression profile in adipose tissue of subjects at risk of metabolic syndrome. Am. J. Clin. Nutr. 2009;90:1656–1664. doi: 10.3945/ajcn.2009.27792. doi:10.3945/ajcn.2009.27792. [DOI] [PubMed] [Google Scholar]
- 33.Hariri N., Gougeon R., Thibault L. A highly saturated fat-rich diet is more obesogenic than diets with lower saturated fat content. Nutr. Res. 2010;30:632–643. doi: 10.1016/j.nutres.2010.09.003. doi:10.1016/j.nutres.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 34.DeLany J.P., Windhauser M.M., Champagne C.M., Bray G.A. Differential oxidation of individual dietary fatty acids in humans. Am. J. Clin. Nutr. 2000;72:905–911. doi: 10.1093/ajcn/72.4.905. [DOI] [PubMed] [Google Scholar]
- 35.Cote J.F., Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. doi:10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 36.Safran M., Dalah I., Alexander J., Rosen N., Iny Stein T., Shmoish M., Nativ N., Bahir I., Doniger T., Krug H., et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glatter T., Wepf A., Aebersold R., Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 2009;5:237. doi: 10.1038/msb.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andjelkovic M., Jakubowicz T., Cron P., Ming X.F., Han J.W., Hemmings B.A. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl Acad. Sci. USA. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. doi:10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ugi S., Imamura T., Maegawa H., Egawa K., Yoshizaki T., Shi K., Obata T., Ebina Y., Kashiwagi A., Olefsky J.M. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 2004;24:8778–8789. doi: 10.1128/MCB.24.19.8778-8789.2004. doi:10.1128/MCB.24.19.8778-8789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez N., Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353:170–173. doi: 10.1038/353170a0. doi:10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- 41.Niswender K.D., Morton G.J., Stearns W.H., Rhodes C.J., Myers M.G.,, Jr, Schwartz M.W. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. doi:10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 42.Rahmouni K., Sigmund C.D., Haynes W.G., Mark A.L. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes. 2009;58:536–542. doi: 10.2337/db08-0822. doi:10.2337/db08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brazier H., Pawlak G., Vives V., Blangy A. The Rho GTPase Wrch1 regulates osteoclast precursor adhesion and migration. Int. J. Biochem. Cell Biol. 2009;41:1391–1401. doi: 10.1016/j.biocel.2008.12.007. doi:10.1016/j.biocel.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Vives V., Laurin M., Cres G., Larrousse P., Morichaud Z., Noel D., Cote J.F., Blangy A. The Rac1 exchange factor Dock5 is essential for bone resorption by osteoclasts. J. Bone Miner. Res. 2010;26:1099–1110. doi: 10.1002/jbmr.282. doi:10.1002/jbmr.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kien C.L., Bunn J.Y., Ugrasbul F. Increasing dietary palmitic acid decreases fat oxidation and daily energy expenditure. Am. J. Clin. Nutr. 2005;82:320–326. doi: 10.1093/ajcn.82.2.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosen C.J., Bouxsein M.L. Mechanisms of disease: is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2006;2:35–43. doi: 10.1038/ncprheum0070. doi:10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- 47.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A., et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. doi:10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyre D., Boutin P., Tounian A., Deweirder M., Aout M., Jouret B., Heude B., Weill J., Tauber M., Tounian P., et al. Is glutamate decarboxylase 2 (GAD2) a genetic link between low birth weight and subsequent development of obesity in children? J. Clin. Endocrinol. Metab. 2005;90:2384–2390. doi: 10.1210/jc.2004-1468. doi:10.1210/jc.2004-1468. [DOI] [PubMed] [Google Scholar]
- 49.Large V., Hellstrom L., Reynisdottir S., Lonnqvist F., Eriksson P., Lannfelt L., Arner P. Human beta-2 adrenoceptor gene polymorphisms are highly frequent in obesity and associate with altered adipocyte beta-2 adrenoceptor function. J. Clin. Invest. 1997;100:3005–3013. doi: 10.1172/JCI119854. doi:10.1172/JCI119854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. doi:10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chenna R., Sugawara H., Koike T., Lopez R., Gibson T.J., Higgins D.G., Thompson J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. doi:10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 53.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. ISBN 3-900051-07-0 http://www.R-project.org/ [Google Scholar]
- 54.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 55.Jernas M., Olsson B., Sjoholm K., Sjogren A., Rudemo M., Nellgard B., Carlsson L.M., Sjostrom C.D. Changes in adipose tissue gene expression and plasma levels of adipokines and acute-phase proteins in patients with critical illness. Metabolism. 2009;58:102–108. doi: 10.1016/j.metabol.2008.08.012. doi:10.1016/j.metabol.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 56.Atkinson B., Therneau T.M. Kinship: Mixed-effects Cox Models, Sparse Matrices, and Modeling Data from Large Pedigrees. Rochester, MN: Mayo Foundation for Medical Education and Research; 2008. [Google Scholar]
- 57.Hothorn T., Hornik K. ExactRankTests: exact distributions for rank and permutation tests. R package version 0.8-19. http://CRAN.R-project.org/package=exactRankTests. 2010 doi:10.1038/sj.ijo.0801168. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.