

Abstract
Long QT syndrome (LQTS) is a genetic disease characterized by a prolonged QT interval in an electrocardiogram (ECG), leading to higher risk of sudden cardiac death. Among the 12 identified genes causal to heritable LQTS, ∼90% of affected individuals harbor mutations in either KCNQ1 or human ether-a-go-go related genes (hERG), which encode two repolarizing potassium currents known as IKs and IKr. The ability to quantitatively assess contributions of different current components is therefore important for investigating disease phenotypes and testing effectiveness of pharmacological modulation. Here we report a quantitative analysis by simulating cardiac action potentials of cultured human cardiomyocytes to match the experimental waveforms of both healthy control and LQT syndrome type 1 (LQT1) action potentials. The quantitative evaluation suggests that elevation of IKr by reducing voltage sensitivity of inactivation, not via slowing of deactivation, could more effectively restore normal QT duration if IKs is reduced. Using a unique specific chemical activator for IKr that has a primary effect of causing a right shift of V1/2 for inactivation, we then examined the duration changes of autonomous action potentials from differentiated human cardiomyocytes. Indeed, this activator causes dose-dependent shortening of the action potential durations and is able to normalize action potentials of cells of patients with LQT1. In contrast, an IKr chemical activator of primary effects in slowing channel deactivation was not effective in modulating action potential durations. Our studies provide both the theoretical basis and experimental support for compensatory normalization of action potential duration by a pharmacological agent.
Keywords: stem cells, drugs
The biophysical characteristics of an action potential (AP) are controlled by contributions of different ionic currents. In the human cardiac system, synchronized firing and conduction of APs of electrically coupled cardiomyocytes give rise to rhythmic contraction, which is commonly monitored by the surface electrocardiogram (ECG). Rapidly activating K+ current (IKr) and slow delayed rectifier K+ current (IKs) are major components of AP repolarization and hence have roles in determining AP duration (APD) (1, 2). Reducing their contributions pharmacologically or genetically is reflected in a lengthening of the Q-T interval between the QRS complex and the T wave in the ECG. Long QT syndrome (LQTS) caused by prolongation of APD has a prevalence of 1 in 5,000 among the general population. As a predisposed condition, patients with LQT have a high incidence of sudden cardiac death. Among 12 different genes causal to LQTS, remarkably, 90% of patients harbor mutations in KCNQ1 or KCNH2 [human Ether-a-go-go–Related Gene (hERG)] genes, causing a reduction in either IKs or IKr currents. Therefore, understanding the contributions of repolarizing ionic currents and potential compensatory effects between them is of considerable interest.
Reduction of the IKs current due to mutations in KCNQ1 has been reported in 40–55% of patients with LQT and represents the most frequent cause of congenital LQTS (3). In both rabbit and guinea pig, the IKs component is present in adult cardiomyocytes. Indeed, reduction of KCNQ1 expression resulted in prolongation of the QT interval (4). However, several important features of human cardiac function including diversity of genetic mutations of IKr or IKs and drug-induced LQT phenotype cannot readily be reproduced or modeled in animal systems (5). R-L3 is an active compound that was reported to potentiate IKs in guinea pig and had similar effects on KCNQ1. However, it was not effective on the oligomeric KCNQ1/KCNE1 channel, the molecular determinant for IKs (6). Phenylboronic acid (PBA) activates KCNQ1/KCNE1 complexes at millimolar effective concentrations (7).
Using induced pluripotent stem cells (iPSCs) derived from members of a family affected by LQT syndrome type 1 (LQT1) and harboring an R190Q heterozygous missense mutation in KCNQ1, we generated and investigated functional cardiomyocytes. The patient-derived cells recapitulated the electrophysiological features of the disorder with noticeably prolonged APs (8). By recording autonomous beating cardiomyocyte clusters displaying ventricular features, we investigated the feasibility of quantitatively analyzing both control and LQT1 action potentials. On the basis of this model system, we then tested whether the specific perturbation of hERG channel gating would effectively rescue the LQT1 disease phenotype.
Results
Quantitative Analyses of Compensatory Effects Between IKs and IKr.
A detailed characterization of different ionic components in differentiated human cardiomyocytes from pluripotent stem cells has not been reported. We compared the expression of genes generating the cardiac IKr (HERG1a and HERG1b) and IKs (KCNQ1) currents in cardiac myocytes obtained from control and LQT1 iPSC lines, as well as in adult and fetal human heart. Quantitative real-time PCR (qRT-PCR) analyses revealed that HERG transcript levels were similar between control and LQT1 cardiomyocytes (Fig. 1A), which is consistent with the earlier data of similar IKr current densities in control and LQT1 cells (8). Therefore, the subsequent analyses use the same IKr value for both the healthy control and LQT1 in modeling.
Fig. 1.

Comparison between simulated and recorded ventricular-like action potentials from iPSC-derived cardiomyocytes from a healthy control and patient with LQT1. (A) qRT-PCR analysis of HERG1a, HERG1b, and KCNQ1 in cardiomyocyte clusters from control (bar with light shading) and LQT1 (open bar) iPSCs at 4-mo maturation compared with human adult (AH, solid bar) and fetal (FH, bar with dark shading) heart. Expression values are relative to those of AH, normalized to GAPDH, and presented as mean ± SD, n = 3. (B) Representative traces of the ventricular-like AP recorded from cardiomyocytes derived from either a healthy control or a patient with LQT1 with R190Q mutation as indicated. The experimentally recorded traces are shown on the Left, whereas the simulated ventricular-like APs based on a modified ten Tusscher model are shown on the Right.
To assess the contributions of different current components in a typical ventricular-like AP, we recorded spontaneous electrical activity of human cardiomyocytes derived from iPSCs. We then used the model first developed by ten Tusscher, which describes a variety of electrophysiological behaviors of adult human ventricular cardiomyocytes (9). One unique feature in differentiated cardiomyocytes is the autonomous contraction absent in acutely isolated human cardiomyocytes, consistent with the notion that differentiated myocytes in culture are not mature (10). Some components in the ten Tusscher model were modified to match the recorded spontaneous APs. Inclusion of a late sodium current component (INaL) and a funny current component (If) afforded a better cross-examination between model-based simulation and the recorded action potential (SI Experimental Procedures). With the modifications outlined in Table S1, the simulated AP traces agree well with those of the representative APs recorded in control cell clusters (Fig. 1B). The key parameters include APD90 = 388.4 ms and action potential amplitude (APA) = 82.5 mV for simulated traces and APD90 = 377.3 ± 14.0 ms and APA = 79.6 ± 1.6 mV for recorded traces (Table 1). To model LQT1 APs, we reduced IKs by 30% of control, the level of IKs measured in LQT1 iPSC-derived myocytes with the R190Q-KCNQ1 mutation (11). This procedure resulted in a simulated action potential of APD90 = 523.6 ms and APA = 84.9 mV that matches well with the recorded trace of LQT1 cells with APD90 = 530.6 ± 18.8 ms and APA = 82.8 ± 2.2 mV (Fig. 1). The agreement between experimental data and simulated data supports the applicability of the modified model for quantitative analyses.
Table 1.
Comparison of simulated AP parameters with experimental values
| APA, mV | APD50, ms | APD90, ms | dV/dtMax, V/s | MDP, mV | APD90/APD50 | |
| Simulated control | 82.5 | 339.7 | 388.4 | 8.4 | −68.5 | 1.14 |
| Recorded control (n = 14) | 79.6 ± 1.6 | 330.7 ± 13.3 | 377.3 ± 14.0 | 16.3 ± 1.2 | −64.5 ± 1.4 | 1.14 ± 0.01 |
| Simulated LQT1 | 84.9 | 444.6 | 523.6 | 8.3 | −67.9 | 1.18 |
| Recorded LQT1 (n = 26) | 81.1 ± 1.3 | 463.5 ± 16.9 | 530.6 ± 18.8 | 13.4 ± 0.9 | −63.4 ± 1.0 | 1.15 ± 0.01 |
| Simulated LQT1 + compound | 84.0 | 323.0 | 360.4 | 18.1 | −79.3 | 1.12 |
| Recorded LQT1 + compound (n = 9) | 82.3 ± 2.2 | 340.8 ± 12.6 | 379.7 ± 13.3 | 12.0 ± 1.0 | −66.2 ± 1.5 | 1.12 ± 0.01 |
Key parameters are summarized of a simulated or recorded action potential from a healthy control and a patient with LQT1. For the simulation study, the values were obtained in the steady state. For the experimental study, the values were based on the real recording from small beating clusters of myocytes.
Therefore, we examined the robustness of the modified model by testing how reduction of INaL or increase in IKr would affect action potential in control or LQT1 cardiomyocytes. In adult human cardiomyocytes INaL is ∼0.055 nS/pF (11). Fig. 2A shows that when INaL is enhanced approximately sixfold (0.33 nS/pF) as seen in F1473C mutation in the SCN5A gene that leads to LQT3 (12), the control displays clear prolongation of APD. Specific sodium channel block therefore reduces this component and brings the duration back to 400 ms, similar to control values (Fig. 2A). However, in the LQT1 model, where IKs is reduced to 30%, the reduction of the normal INaL level was less effective on APD (Fig. 2B), consistent with an earlier report (13). Similarly, the INaL reduction had no significant effect on APD of the control either. This model-based prediction is in agreement with that of the experimental data, where specific sodium channel blockers are more effective to correct the defects in patients with LQT3 (14). In addition to INaL, IKr is another component critical for determining APD. Therefore, we examined whether an increase in repolarizing IKr could normalize the prolonged APD in LQT1 cardiomyocytes. The hERG potassium channel is the primary molecular determinant responsible for the IKr. A biophysical property of hERG absent in KCNQ1 is the rapid inactivation during depolarization. Thus, changing in channel inactivation by shifting its voltage sensitivity (V1/2) to a more positive value would slow the inactivation and increase channel availability during the repolarizing phase of the AP. To examine this, we progressively shifted the V1/2 from −87 mV to −70 mV, −40 mV, and −10 mV. The model predicts progressive shortening of APD in control cells (Fig. 2C). Importantly, in LQT1 cells the rightward shift of inactivation V1/2 could also progressively shorten the APD into the normal range (Fig. 2D). Another way to increase IKr density during the AP is to slow down the deactivation rate of hERG current. Thus, we simulated the effect of decreasing IKr decay rate on APD by increasing the time constant of the activation (deactivation) gate, τxr1. This change is not sufficient to shorten prolonged APDs in LQT1 cardiomyocytes. Instead, the model predicts a flattering of phase 4 diastolic depolarization and a reduction of firing frequency (Fig. 2E). This prediction is consistent with an earlier study demonstrating that IKr decay rate is one of the key determinants of the speed of pace-making depolarization (15). The simulation, despite the fact that some values used were based on empirical estimation or on earlier reports, is consistent with the experimental evidence that a sodium channel blocker is more effective in patients with LQT3, indicative of gene-specific rescue. In contrast, it argues an increase of IKr would be able to correct the APD in LQT1 cardiomyocytes. Importantly, the model specifies that a rightward shift of IKr inactivation V1/2 and not a slower deactivation rate is effective in restoring the prolonged APD in LQT1 patients to a normal range.
Fig. 2.

Simulation study of different compound effects on iPSC-derived cardiomyocytes from a healthy control and a patient with LQT1. (A) Simulated AP from a healthy control without compound treatment (INaL = 0.055 nS/pF) and a presumed patient with LQT3 without compound treatment (INaL = 0.33 nS/pF) or with INaL blocker treatment (INaL = 0.070 nS/pF). (B) Simulated action potential from a healthy control without compound treatment (INaL = 0.055 nS/pF, scale factor of GKs = 1), a patient with LQT1 without compound treatment (INaL = 0.055 nS/pF, scale factor of GKs = 0.3), a healthy control with INaL blocker treatment (INaL = 0.014 nS/pF, scale factor of GKs = 1), or a patient with LQT1 with INaL blocker treatment (INaL = 0.014 nS/pF, GKs =0.3). (C) Simulated APs from a healthy control (scale factor of GKs = 1, xr2 V1/2 = −87 mV) and a healthy control with a progressive shift in xr2 V1/2 = −70 mV, −40 mV, −10 mV, respectively, to show different doses of compound effect on inactivation V1/2. (D) Simulated APs from a patient with LQT1 without compound treatment (scale factor of GKs = 0.3, xr2 V1/2 = −87 mV) and a patient with LQT1 with progressive shift in xr2 V1/2 = −70 mV, −40 mV, or −10 mV, respectively. (E) Simulated AP from a patient with LQT1 without compound treatment (scale factor of txr1 = 1) and a patient with LQT1 with progressive decrease in IKr decay. Scale factor of txr1 = 2 or 3, respectively, to mimic different doses of compound effect on deactivation.
Identification of a Specific Gating Modulator.
Using automated electrophysiology, we have conducted a large compound library screen to identify and characterize small molecule modulators for hERG potassium channels (16). Although a vast majority of modulators were inhibitors, a small number of activators have been identified in the primary screen. On the basis of the model, we examined and selected hits that have biophysical characteristics consistent with affecting voltage sensitivity of inactivation. One scaffold with unique core structure was confirmed with a resynthesized compound. We then purchased or synthesized various derivatives to examine both potency and efficacy, using automated electrophysiology. ML-T531 (or CFK332-B) is one potent activator with highest efficacy (EC50 = 3.13 ± 0.47 μM (Table 2) and was selected for further investigation.
Table 2.
Structure–activity relationship (SAR) of ML-T531
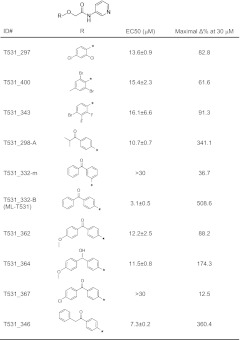 |
To experimentally characterize the specificity of ML-T531 on IKr and IKs, we performed manual voltage clamp recording of hERG currents, using a standard protocol with a Chinese hamster ovary (CHO) cell line stably expressing hERG channels. For comparison, recordings were made from a CHO cell line coexpressing cDNAs of KCNQ1 and KCNE1, the molecular determinants for IKs (17, 18). Fig. 3A shows that in the absence of compound, hERG currents display a current–voltage relationship in agreement with the literature. In the presence of ML-T531, a dramatic increase in steady-state current was observed. In contrast, under the same conditions, KCNQ1/E1 coexpression exhibited no potentiation, but a slight current reduction was observed (Fig. S1). In addition, ML-T531 has no noticeable effects on other cardiac channels including Cav1.2, Kir2.1, Nav1.5, and Kv4.3. These channels are the molecular determinants for ICa, IK1, INa, and Ito (Fig. S2).
Fig. 3.

ML-T531 effects on expressed hERG and KCNQ1/KCNE1 channels. (A) ML-T531, when tested at 10 μM, significantly potentiates hERG steady-state currents and tail currents in CHO cells. The holding potential was −80 mV. The steady-state currents were examined from −80 mV to +80 mV in 20-mV increments, whereas tail currents were elicited at −50 mV. (B) Steady-state current density of hERG either in the absence or in the presence of 10 μM ML-T531, plotted against the voltage. (C) ML-T531, when tested at 10 μM, has no significant effect on hERG activation V1/2. Tail currents were elicited at −50 mV and the G-V curve was fitted by the Boltzmann equation. (D) ML-T531, when tested at 10 μM, slows hERG deactivation rates. Tail current deactivation was examined from −130 mV to −80 mV and was fitted by a standard double-exponential equation. (E) Inactivation of hERG in the presence and the absence of 10 μM ML-T531 was measured at different voltages using the indicated voltage protocol.
To further characterize ML-T531 effects on hERG channels, steady-state current density was plotted against voltages from −80 mV to +40 mV (Fig. 3B). The enhancement of current was much more pronounced at positive compared with negative voltages. To examine whether ML-T531 affects the conductance–voltage (G-V) relationship, the relative conductance at −50 mV was plotted against step voltages (Fig. 3C). ML-T531 at 10 μM caused no detectable change in the activation V1/2. Further characterization of ML-T531 effects on inactivation and deactivation was performed by measuring the time constants in the presence or absence of 10 μM ML-T531. Fig. 3D shows that ML-T531 slowed down the deactivation rate at all voltages tested, indicating its capability to increase occupancy of the channel open states (Fig. S3). Furthermore, it caused a rightward shift of inactivation V1/2 by 28 ± 1.0 mV (n = 8) (Fig. 3E and Fig. S4). On the basis of the results obtained from heterologously expressed channels, we simulated the effects in LQT1 cells (Table 1).
Action Potential of iPSC-Derived Control and LQT1 Cardiomyocytes.
To examine the effects of ML-T531 on native currents, we performed single-cell voltage clamp experiments measuring native IKr and IKs currents from healthy human iPSC-derived cardiomyocytes. Using a published protocol for human iPSC-derived cardiomyocytes (19), the combined potassium currents mainly contributed by IKs and IKr were isolated (SI Experimental Procedures and Fig. 4). In the presence of 10 μM ML-T531, the current density consistently increased at all voltages tested for both steady-state and tail currents (Fig. 4 A and B). Moreover, under these conditions ML-T531 enhanced voltage-dependent potassium current amplitudes in the presence of an Iks-specific inhibitor chromanol 293B. The activation kinetics and rectification properties of the enhanced current are consistent with those of IKr (Fig. S5). These results combined with specificity tests (Fig. S2) argue for IKr augmentation being the primary effect of ML-T531 in native human cardiomyocytes. To examine whether the LQT1 phenotype could be rescued by the compensatory effect of increasing IKr, we recorded the spontaneous action potentials of human cardiomyocytes derived from both control and LQT1 iPSCs (8). The experiments were carried out using spontaneously beating cardiomyocytes with ventricular-like action potentials. These cells have APA greater than 70 mV, maximum diastolic potential (MDP) more negative than −50 mV, and APD90/APD50 smaller than 1.25. Consistent with earlier reports (8), Fig. 5B shows that LQT1 patient-derived cardiomyocytes displayed significantly longer APD values (APD90 = 530.6 ± 18.8 ms, n = 26) compared with those derived from the healthy control individual (APD90 = 377.3 ± 14.0 ms, n = 14). Next, we investigated the effect of 10 μM ML-T531 on spontaneous APs of LQT1 cardiomyocytes. ML-T531 significantly shortened the APD of the diseased cardiomyocytes and normalized the APD values comparable to those recorded from the healthy control (Fig. 5 A and C and Table 1). In addition, ML-T531 caused a hyperpolarizing shift in the MDP values (Fig. 5C and Table 1). The above two major effects are consistent with our modeling results for a compound that causes a rightward shift in inactivation V1/2 and thereby enhances potassium currents during repolarization. Indeed, when the compound effects on recombinant hERG were incorporated into the model, the predicted AP parameters during compound treatment correlated well with those obtained experimentally (Table 1). Furthermore, ML-T531 shortened the APD90 of LQT1 cardiomyocytes in a dose-dependent manner (Fig. 5D). When fitted by the Hill equation, the dose–response curve gives rise to an EC50 value of 5.71 ± 1.42 μM (Fig. 5E), which is comparable to the EC50 (3.13 ± 0.47 μM) obtained in a CHO recombinant system. The similarity of EC50 values measured in LQT1 human cardiomyocytes to that in hERG-CHO cells supports that IKr or hERG is the major target of this compound in native cells. Taken together, our results demonstrate the possibility of normalizing the LQT1-related disease phenotype by targeting the voltage dependence (V1/2) of inactivation of hERG channels.
Fig. 4.

ML-T531 potentiates native potassium currents in iPSC-derived cardiomyocytes. (A) A pair of representative traces from a single differentiated cardiomyocyte in the absence and the presence of 10 μM ML-T531 as indicated. The holding potential was −60 mV. Steady-state currents were examined from −10 mV to +50 mV in 20-mV increments, whereas tail currents were elicited at −30 mV. Except for IKr and IKs, other major ionic components including INa, IK1, Ito, and ICaL were pharmacologically suppressed under the recording conditions (Experimental Procedures). (B) Summary of the compound effects on steady-state current (Upper) and tail current (Lower) at the indicated voltages (n = 11). (*P < 0.05; **P < 0.001).
Fig. 5.

ML-T531 normalizes the disease phenotype of cardiomycytes derived from a patient with LQT1. (A) A train of action potentials recorded from cardiomycytes derived from a patient with LQT1. The time window of ML-T531 application is indicated. Representative action potentials from indicated areas were expanded to appreciate the difference in APD. (Lower) Parameters for individual action potentials are shown in heatmap format. Data are z-score normalized to the first 20 potentials in the train, with color intensity representing the number of SDs of change from this baseline. The plotted parameters are as indicated on the left. (B) Comparison of APD90 recorded from healthy control cells and cells from a patient with LQT1. (C) Overlay of representative action potential traces of cardiomyocytes from a patient with LQT1 at baseline (black), in the presence of 10 μM ML-T531(red), and after washout (blue). (D) ML-T531 effects on action potential durations (APD90) of cardiomyocytes derived from a patient with LQT1 at the indicated concentrations (n = 3 or more). (E) Dose–response curve of ML-T531 on shortening APD90 in LQT1 cardiomyocytes. The curve was fitted by a Hill equation with a Hill coefficient of 2.0.
Discussion
The modified ionic components in the current model relative to the ten Tusscher model are consistent with the notion that in vitro differentiated cardiomyocytes display electrophysiological characteristics that are closer to the fetal counterparts (10). These characteristics include a lower IK1 component, reduced value of dV/dt during the initial stage of action potential, and more depolarized MDPs. The revised model enables several valuable predictions testable by experiments using human cells. Almost all model parameters were based on published work with a few modifications (9). The conductance of the IK1 value was reduced to 3% of the normal value. This change is supported by the autonomous beating commonly seen in these cells and agrees with results of RT-PCR studies that suggest a noticeable reduction of normal levels of IK1 (20). The ICaL component was doubled in the simulations to account for the consistently longer APDs recorded from control cells compared with the original model. The revised values, even though some have qualitative support, were assigned artificially to achieve closer correlation between modeled and recorded action potential traces, either in the presence or in the absence of the compound treatments (Table S1). Future directions may include quantification of individual current components so that a more refined model may be established. Because the initial variables for this quantitative analysis were directly adapted from the original ten Tusscher model, clearly our simulated action potentials tend to gradually reach a steady state and gain some immature phenotypes, such as a depolarized MDP and a slower dV/dtmax (Fig. S6 A–F). Under steady state, while we note that LQT3 phenotype become less severe, which is likely due to reduced INaL availability under relatively deporlarized MDP, the conclusion remains essentially true; i.e., modulation of voltage dependence of inactivation is more effective than slowing of deactivation (Fig. S6G). To examine whether the general conclusion could be applicable to adult cardiomyocytes, we also tested the idea using original ten Tusscher values and obtained similar conclusion (Fig. S6H).
Cardiomyocytes have several potassium channels. These channels, although permeable to potassium, typically display distinct gating properties. Therefore, it is possible that compensatory rescue as described in this study requires specific gating modulation achievable by modifying specific biophysical properties of the targeted channel. Indeed, it was noted that 10 μM ML-T531 decreased AP firing frequency (Fig. 5A), which is consistent with our modeling results, because ML-T531 slows down the IKr deactivation (Fig. 3D). To examine this hypothesis more closely, we tested ginsenoside (Rg3), which potentiates hERG current by predominantly slowing the deactivation kinetics (Fig. S7 A and B) (21). Indeed, we found that when this compound is applied to the beating cardiomyocytes, the AP firing frequency was greatly reduced (Fig. S7 D and G). However, there was minimal change in shape or duration of action potentials (Fig. S7 C and F). This result provides the direct support for use of a specific mechanism of gating modulation to achieve compensatory rescue.
The two potassium currents, IKr and IKs, play a critical role in repolarizing the membrane to terminate an AP. Reports suggest a possible interaction between a mutant KCNQ1 channel and hERG, resulting in change of hERG trafficking to the membrane (22). However, in our LQT1 iPSC-derived cardiomyocyte model, expression levels of genes coding for IKr were similar to those of control cells (Fig. 1A) as well as IKr current density, as previously reported (11). Reduction of IKs current has been modeled both pharmacologically using a KCNQ1 inhibitor (23) and genetically with transgenic rabbits overexpressing loss-of-function pore mutants of KCNQ1 (4). Using these animal or pharmacological models, the attempts to rescue prolongation of APDs have been carried out using either an IKr activator (24) or an opener of ATP-sensitive potassium channels (25). Pharmacological interventions aiming at influencing the APD have been in use for many years, such as class III antiarrhythmics for APD prolongation. The notion of compensatory rescue of prolonged APDs has been previously tested by enhancing KATP current (26). Unlike the prolongation of the APD, the pharmacological shortening of a pathologically prolonged APD has been investigated only in very few studies using patient samples that are not readily available (27). Our results using patient iPSC-derived myocytes with disease phenotypes provide an important proof of principle of a therapeutic intervention to shorten APD.
The hERG activator ML-T531 has a distinct chemical structure compared with the previously described compounds that potentiate hERG currents (28–33). The reported activators may be classified according to the modes of action, including slowing of deactivation and modulation of voltage dependence of inactivation. Not all of them have effects on APD in tested animal models. ML-T531 has a pronounced effect on voltage sensitivity of hERG inactivation similar to that reported for ICA-105574. Interestingly, an earlier study has shown that ICA-105574 was capable of shortening APD in isolated cardiomyocytes from guinea pig (28). In contrast, RPR260243, which primarily acts in slowing hERG deactivation, has pronounced effects on T-wave amplitude in guinea pig hearts but almost no effects on QT duration even at 30 μM, the highest concentration tested (30). These experiments are in agreement with our results that reduction of voltage dependence for inactivation is more effective in modulating APD.
Whereas IKr may involve more than the hERG channel, it is thought that the hERG subunits are the primary determinants for the pharmacology of IKr (34). We noted that ML-T531 has nearly identical potency (EC50) measured by shortening of action potential in cardiomyocytes and by steady current increase in heterologously expressed hERG channels. This correlation argues for the compound specificity on the native hERG channel in human cardiac cells. Our experiments demonstrate an application of human iPSC technologies by testing compound effects on correcting an abnormal functional phenotype due to an inherited cardiac disorder. Future optimization via structure-activity studies may afford improved potency and specificity, thereby increasing its utility.
Experimental Procedures
The general method of modeling is similar to that used in the ten Tusscher model (9) and SI Experimental Procedures (35). Human iPS cell lines from the patient with long QT1 syndrome and the healthy control (8) were maintained in feeder-free mTeSR1 medium. For whole-cell patch-clamp recordings, beating clusters 2–3 mo old were microdissected and disaggregated as previously reported. Traditional whole-cell voltage-clamp recording was performed at room temperature. Additional details are described in SI Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Drs. Leslie Tung and David Milan and members of the M.L. laboratory for valuable discussions and comments on the manuscript, Shunyou Long and Kaiping Xu for technical assistance, and Alison Neal for editorial assistance. This work is supported by grants to M.L. from the National Institutes of Health (GM078579 and MH084691) and the Maryland Stem Cell Research Fund (2010-MSCRFE-0164-00) and to K-L.L. from the European Research Council (ERC 261053) and the German Research Foundation (Research Unit 923, Mo 2217/1-1 and La 1238 3-1/4-1).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205266109/-/DCSupplemental.
References
- 1.Sanguinetti MC. Long QT syndrome: Ionic basis and arrhythmia mechanism in long QT syndrome type 1. J Cardiovasc Electrophysiol. 2000;11:710–712. doi: 10.1111/j.1540-8167.2000.tb00035.x. [DOI] [PubMed] [Google Scholar]
- 2.Sanguinetti MC, Zou A. Molecular physiology of cardiac delayed rectifier K+ channels. Heart Vessels Suppl. 1997;12:170–172. [PubMed] [Google Scholar]
- 3.Hedley PL, et al. The genetic basis of long QT and short QT syndromes: A mutation update. Hum Mutat. 2009;30:1486–1511. doi: 10.1002/humu.21106. [DOI] [PubMed] [Google Scholar]
- 4.Brunner M, et al. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nerbonne JM. Studying cardiac arrhythmias in the mouse—a reasonable model for probing mechanisms? Trends Cardiovasc Med. 2004;14:83–93. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Salata JJ, et al. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol. 1998;54:220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- 7.Mruk K, Kobertz WR. Discovery of a novel activator of KCNQ1-KCNE1 K channel complexes. PLoS ONE. 2009;4 doi: 10.1371/journal.pone.0004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 9.ten Tusscher KH, Noble D, Noble PJ, Panfilov AV. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1573–H1589. doi: 10.1152/ajpheart.00794.2003. [DOI] [PubMed] [Google Scholar]
- 10.Rajala K, Pekkanen-Mattila M, Aalto-Setälä K. Cardiac differentiation of pluripotent stem cells. Stem Cells Int. 2011;2011:383–709. doi: 10.4061/2011/383709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardona K, et al. Effects of late sodium current enhancement during LQT-related arrhythmias. A simulation study. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:3237–3240. doi: 10.1109/IEMBS.2010.5627184. [DOI] [PubMed] [Google Scholar]
- 12.Bankston JR, et al. A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PLoS ONE. 2007;2 doi: 10.1371/journal.pone.0001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz PJ, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 15.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993;73:197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- 16.Du F, et al. hERGCentral: A large database to store, retrieve, and analyze compound-human Ether-à-go-go related gene channel interactions to facilitate cardiotoxicity assessment in drug development. Assay Drug Dev Technol. 2011;9:580–588. doi: 10.1089/adt.2011.0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barhanin J, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 18.Sanguinetti MC, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 19.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 20.Satin J, et al. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004;559:479–496. doi: 10.1113/jphysiol.2004.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SH, et al. Ginsenoside Rg(3) decelerates hERG K(+) channel deactivation through Ser631 residue interaction. Eur J Pharmacol. 2011;663:59–67. doi: 10.1016/j.ejphar.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Biliczki P, et al. Trafficking-deficient long QT syndrome mutation KCNQ1-T587M confers severe clinical phenotype by impairment of KCNH2 membrane localization: Evidence for clinically significant IKr-IKs alpha-subunit interaction. Heart Rhythm. 2009;6(12):1792–1801. doi: 10.1016/j.hrthm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu W, Antzelevitch C. Effects of a K(+) channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation. 2000;102:706–712. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- 24.Bentzen BH, et al. Pharmacological activation of Kv11.1 in transgenic long QT-1 rabbits. J Cardiovasc Pharmacol. 2011;57:223–230. doi: 10.1097/FJC.0b013e318203a44d. [DOI] [PubMed] [Google Scholar]
- 25.Biermann J, et al. Nicorandil normalizes prolonged repolarisation in the first transgenic rabbit model with Long-QT syndrome 1 both in vitro and in vivo. Eur J Pharmacol. 2011;650:309–316. doi: 10.1016/j.ejphar.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahangir A, Terzic A. K(ATP) channel therapeutics at the bedside. J Mol Cell Cardiol. 2005;39:99–112. doi: 10.1016/j.yjmcc.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu W, et al. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation. 1998;97:1581–1588. doi: 10.1161/01.cir.97.16.1581. [DOI] [PubMed] [Google Scholar]
- 28.Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-à-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574) Mol Pharmacol. 2010;77:58–68. doi: 10.1124/mol.109.059543. [DOI] [PubMed] [Google Scholar]
- 29.Hansen RS, et al. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) Mol Pharmacol. 2006;69:266–277. doi: 10.1124/mol.105.015859. [DOI] [PubMed] [Google Scholar]
- 30.Kang J, et al. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol Pharmacol. 2005;67:827–836. doi: 10.1124/mol.104.006577. [DOI] [PubMed] [Google Scholar]
- 31.Su Z, et al. Electrophysiologic characterization of a novel hERG channel activator. Biochem Pharmacol. 2009;77:1383–1390. doi: 10.1016/j.bcp.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Recanatini M, Roberti M, Tseng GN. Probing the binding sites and mechanisms of action of two human ether-a-go-go-related gene channel activators, 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) and 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD307243) Mol Pharmacol. 2008;73:1709–1721. doi: 10.1124/mol.108.045591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, et al. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68:876–884. doi: 10.1124/mol.105.014035. [DOI] [PubMed] [Google Scholar]
- 34.Weerapura M, Nattel S, Chartier D, Caballero R, Hébert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J Physiol. 2002;540:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benson AP, Al-Owais M, Holden AV. Quantitative prediction of the arrhythmogenic effects of de novo hERG mutations in computational models of human ventricular tissues. Eur Biophys J. 2011;40:627–639. doi: 10.1007/s00249-010-0663-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.