

Abstract
Avibactam is a β-lactamase inhibitor that is in clinical development, combined with β-lactam partners, for the treatment of bacterial infections comprising Gram-negative organisms. Avibactam is a structural class of inhibitor that does not contain a β-lactam core but maintains the capacity to covalently acylate its β-lactamase targets. Using the TEM-1 enzyme, we characterized avibactam inhibition by measuring the on-rate for acylation and the off-rate for deacylation. The deacylation off-rate was 0.045 min−1, which allowed investigation of the deacylation route from TEM-1. Using NMR and MS, we showed that deacylation proceeds through regeneration of intact avibactam and not hydrolysis. Other than TEM-1, four additional clinically relevant β-lactamases were shown to release intact avibactam after being acylated. We showed that avibactam is a covalent, slowly reversible inhibitor, which is a unique mechanism of inhibition among β-lactamase inhibitors.
Keywords: antibacterial, drug discovery, enzymology
There is an urgent need for new antibacterial agents that are active against drug-resistant bacteria. In particular, some Gram-negative pathogens have accumulated enough resistance mechanisms to render them virtually untreatable by modern antibacterial chemotherapy (1, 2). A mainstay for treatment of Gram-negative infections is the β-lactam classes of drugs. The most common form of resistance to β-lactam antibiotics is the expression of various β-lactamase enzymes capable of hydrolyzing the β-lactam ring of β-lactam drugs, rendering them ineffective. As new β-lactams have been introduced into clinical use, a changing landscape of β-lactamases has been selected and disseminated. Presently, over 1,000 β-lactamases have been documented comprising several structural classes and a wide range of substrate promiscuities and catalytic efficiencies (3, 4).
In efforts to restore the efficacy of β-lactam antibiotics, β-lactamases have also been targeted with a variety of inhibitors (5, 6). The three inhibitors approved for clinical use are clavulanic acid, tazobactam, and sulbactam, all of which contain a β-lactam core. A challenge for the development of broad-spectrum β-lactamase inhibitors is the mechanistic diversity in β-lactamase enzymes, with the largest distinction being between the enzyme classes that use a serine residue as the nucleophilic species and the metallo-β-lactamases, which directly activate water for hydrolysis (7). A shared mechanistic feature of the marketed β-lactam–based inhibitors is their reaction with the serine enzymes to form a covalent acyl-enzyme intermediate. On ring opening, the acyl-enzyme intermediate can undergo additional rearrangements or be released through hydrolysis to regenerate the active β-lactamase enzyme (8). Originally designed to combat class A serine β-lactamase enzymes such as TEM-1, the clinical use of β-lactam–based inhibitors has been diminished by the emergence of enzymes against which they are ineffective. Despite intense investigation by pharmaceutical companies, no new β-lactamase inhibitor has reached the market in over 19 years.
Avibactam (Fig. 1) is a member of a class of inhibitors called the diazabicyclooctanes (DBOs) (9), and it is currently entering phases II and III clinical trials combined with ceftaroline and ceftazidime for the treatment of serious infections caused by Gram-negative organisms (http://clinicaltrials.gov). A design strategy for β-lactamase inhibitors has been to focus on scaffolds that maintain the capacity to rapidly acylate a wide range of β-lactamases while minimizing the liability of hydrolysis. Avibactam has been described as possessing these desirable properties (9), but the molecular understanding of how it inhibits its targets is not well-understood.
Fig. 1.

Structures of β-lactamase inhibitors used in this study.
In this work, the detailed enzymatic mechanism of inhibition for avibactam against the β-lactamase TEM-1 is described. Investigation of onset of and recovery from TEM-1 inhibition, coupled with biophysical measurements of the enzyme–inhibitor complex, leads us to propose a model for covalent β-lactamase inhibition that is reversible but not susceptible to hydrolysis. Profiling of additional clinically important class A and C β-lactamases suggests that this highly unusual reversible acylation and deacylation mechanism is a general mechanism of inhibition for avibactam.
Results
Onset of Acylation.
The onset of inhibition of TEM-1 β-lactamase by avibactam was investigated using conventional and stopped-flow spectroscopy. Without stopped-flow spectroscopy, the addition of enzyme resulted in a time lag of ∼3 s in our apparatus and allowed investigation of avibactam concentrations up to 2.5 μM. Under these conditions, the observed pseudo first-order rate constant for formation of inhibited enzyme, kobs, was not saturable with respect to the concentration of avibactam. Stopped-flow equipment allowed interrogation of shorter time regimens from 250 ms to 7 s and higher avibactam concentrations from 8 to 50 μM. The kobs values from both studies overlaid with each other and remained linear at the highest avibactam concentration (Fig. 2). Although this linear relationship is required of a one-step inhibition mechanism, it is also consistent with a two-step binding and acylation process, where the initial binding constant is very weak (10). Covalent β-lactamase inhibitors follow a two-step binding and acylation reaction, and based on this precedent, avibactam inhibition was modeled to a two-step mechanism as described in Materials and Methods. Fitting the plot of kobs vs. inhibitor concentration to an equation discussed in Materials and Methods yielded a slope of 1.6 × 105 M−1s−1 (±0.1 × 105 M−1s−1 at 95% confidence interval). For a two-step mechanism with a weak affinity for the initial encounter complex, the slope value determined in this manner is the second-order rate constant for β-lactamase acylation. The contributing terms for the noncovalent Ki and the ring-opening chemical step k2 cannot be determined precisely (11).
Fig. 2.

kobs vs. I plot for avibactam onset of inhibition vs. TEM-1. The ranges of avibactam concentrations were 60 nM to 2.5 μM in a stirred cuvette and 0.8 to 50 μM by stopped flow. Data were fit as described in Materials and Methods. Error bars shown are ±SEM from the fit to kobs from the onset of inhibition time courses.
Off-Rate of Deacylation.
The experiments investigating the onset of avibactam inhibition revealed that the off-rate for return of enzyme activity was much slower than the time regimen for onset of acylation. Therefore, a separate experiment was performed to measure the deacylation off-rate. The most common routes of deacylation of β-lactamase inhibitors are through hydrolysis, chemical rearrangement, or a combination of these paths (8). Off-rates for deacylation from TEM-1 were measured using a jump dilution method (12) comparing avibactam with clavulanic acid and tazobactam. Avibactam displayed a slow return of activity with an off-rate of 0.045 ± 0.022 min−1, which converts to a residence time half-life (t1/2) of 16 ± 8 min (Fig. 3). In contrast, TEM-1 with clavulanic acid displayed a partial return of activity, which is attributed to rearrangement from the acylated enzyme form to additional irreversible acyl-enzyme species (13). TEM-1 inhibition by tazobactam follows a branched deacylation pathway that favors hydrolysis over rearrangement (14), which in the off-rate assay, manifested as a rapid return to nearly full activity.
Fig. 3.

Recovery of activity time courses for β-lactamase inhibitors. Avibactam (AVI), tazobactam (TAZ) and clavulanic acid (CLA) were incubated with TEM-1 to allow acylation and then diluted to follow enzyme reactivation. Results shown are the averages of three measurements. Data for the avibactam time course were fit to Eq. 1 as described in Materials and Methods, yielding a koff of 0.045 ± 0.022 min−1 (mean ± 2 SD).
The measured off-rate for avibactam suggested that slow deacylation through hydrolysis or reversibility was occurring, and it is in contrast to previously reported extremely long t1/2 values of >1 or >7 d for avibactam inhibition of TEM-1 (15, 16). We investigated the methods used in these earlier studies and concluded that these data resulted from measuring initial rates of enzyme activity within seconds after dilution of a 1 μM acyl-enzyme (EI*) complex. Under these conditions, we were able to reproduce those earlier results (Fig. S1). When only the initial rates are measured, the percent activity observed is very small, because it is a measure of the amount of free enzyme present in the 1 μM EI* sample. If the EI* acyl-enzyme is stable to hydrolysis, then sampling it over the course of days will not change the amount of free TEM-1 or initial percent enzyme activity observed. By following the return of activity in a continuous manner for 1 h at a much lower EI* concentration (25 pM as in Fig. 2), the deacylation from TEM-1 can be measured.
Lack of Hydrolysis or Rearrangement.
The recovery of enzyme activity on dilution of the acylated enzyme led to experiments designed to identify the deacylation routes involved. Avibactam has been assumed to covalently acylate its β-lactamase target based on acyl-enzyme adducts seen in electrospray ionization MS (16). To eliminate the possibility of a tight noncovalent interaction, the acylated TEM-1 was subjected to strong denaturing conditions followed by MS. The retention of the acyl-enzyme adduct (Fig. S2) under these conditions supports the covalent nature of this linkage. The observation that enzyme inhibition was maintained for >24 h at 1 μM acyl-enzyme (Fig. S1) suggested that the carbamoyl acyl-enzyme intermediate is stable to hydrolysis. This suggestion was confirmed by 1H NMR spectroscopy. As opposed to tazobactam, which was hydrolyzed by TEM-1 (Fig. 4A), avibactam remained intact after 24 h of incubation with a high concentration of TEM-1 (Fig. 4B), and therefore, it is not turned over as a substrate of the enzyme. In addition, this sample was analyzed for small-molecule species by MS in negative and positive ion modes, and only the mass of avibactam was detected. We estimate that our quantitative method could have detected >5% loss of avibactam, which sets a limit for a first-order hydrolytic rate constant at <0.002 h−1. This finding translates to a half-life of >14 d for the hydrolytic stability of the acyl-enzyme under these conditions.
Fig. 4.

Hydrolysis of β-lactamase inhibitors. (A) 1H Carr–Purcell–Meiboom–Gill NMR spectra of 40 μM tazobactam alone (Upper) and 40 μM tazobactam + 4 μM TEM-1 sampled after 5 min at 37 °C (Lower). (B) 1H Carr–Purcell–Meiboom–Gill NMR spectra of 40 μM avibactam alone (overlay 1) and 40 μM avibactam + 4 μM TEM-1 sampled from 5 min to 24 h at 37 °C (overlays 2–5). Experiments were performed as described in SI Materials and Methods. Signals originating from the TEM-1 enzyme are labeled with asterisks.
Other classes of β-lactamase inhibitors undergo chemical rearrangements from their initial acyl-enzyme species, and such rearrangements are observable at high ratios of inhibitor to enzyme (17, 18). To investigate this possibility, a high concentration of avibactam was incubated with TEM-1, and the off-rate was measured (Fig. S3A). Compared with clavulanic acid, which follows a branched deacylation pathway, the off-rate and level of activity return were unaffected by a high concentration of avibactam. In addition, analysis of TEM-1 by MS at a high concentration of avibactam did not reveal additional acyl-enzyme adducts (Fig. S3B), which further supports the lack of chemical rearrangements or a branched pathway for avibactam deacylation.
Equilibration of Acyl-Enzyme with Free Enzyme and Free Inhibitor.
Because we did not observe irreversible deacylation pathways through hydrolysis or chemical rearrangement, we explored the possibility of deacylation occurring through a reversible reaction to release intact avibactam. In a reversible mechanism, the position of the equilibrium between the acyl-enzyme, free enzyme, and free inhibitor will depend on the total concentrations of the enzyme and inhibitor relative to the bound complex Kd. Three independent methods were used to substantiate the reversible mechanism. A solution of acyl-enzyme (EI*) at 1 μM was prepared and diluted serially, and each acyl-enzyme concentration was allowed to equilibrate for 2 h. Assays were then performed to measure the proportions of acyl-enzyme, free enzyme, and free inhibitor present after equilibration. First, protein MS showed that the percentage of free enzyme increased as the extent of the dilution of the acyl-enzyme complex increased (Fig. 5A). Second, the percentage of enzyme activity, which is a measure of free enzyme, increased as the extent of dilution increased (Fig. 5B). Third, the amount of intact, free avibactam was quantified using small-molecule MS. The percentage of free avibactam in solution increased as the extent of dilution of the acyl-enzyme complex increased (Fig. 5B), consistent with deacylation reforming and releasing intact avibactam. The equilibrium titrations by the three methods overlaid with each other. Each measurement represents a measurement of Ki*, which had an average value of 2.1 ± 1.0 nM. An independent measurement of Kd was made using isothermal titration calorimetry in a competitive titration with a previously described reversible ligand (19) (Fig. S4). The isothermal titration calorimetry-measured Kd was 3.3 ± 0.4 nM. The agreement between Ki* and Kd supports the two-step mechanistic model, where Ki is large relative to Ki* and k−2 is much lower than k2.
Fig. 5.

Equilibration of avibactam-TEM-1 acyl-enzyme. (A) Mass spectra of acylated TEM-1 (EI*) after dilution to various concentrations and equilibration for 2 h at 37 °C. (B) Fit of measurement of equilibria between avibactam-TEM-1 acyl-enzyme complex and free avibactam + TEM-1 as a function of complex dilution. The percent avibactam bound was measured by TEM-1 protein MS (blue squares), avibactam MS (green triangles), and initial enzyme activity (red circles). For the avibactam MS titration, the observed % free avibactam was used to calculate the % bound by assuming a mass balance that fraction bound is equal to (1 − fraction unbound). Error bars shown are ±SEM from three measurements for each detection technique. For calculation of Ki*, data for the different detection methods were fit independently assuming the tight-binding condition (39). The value of Ki* determined by the three techniques is 2.1 ± 1.0 nM (mean ± 2 SD), and the solid line indicates the fit to this value.
Regeneration of intact avibactam was also shown by an acyl-enzyme transfer experiment. TEM-1 was acylated, free avibactam was removed by centrifugal ultrafiltration, and then, a second β-lactamase, CTX-M-15, was added (Fig. 6A). The acylated protein MS peak for TEM-1 decreased with a concomitant appearance of acylated CTX-M-15, consistent with the hypothesis that intact avibactam was released from TEM-1 to allow acylation of CTX-M-15. The percentage of acylated TEM-1 and CTX-M-15 equilibrated to a greater percentage of acylated CTX-M-15 (Fig. 6B), consistent with the higher affinity of CTX-M-15 (5 nM IC50) for avibactam vs. TEM-1 (8 nM IC50) (16).
Fig. 6.

Acyl-enzyme exchange between TEM-1 and CTX-M-15. (A) Time course of acyl-enzyme exchange from acyl-TEM-1 to apo-CTX-M-15 detected by MS. The peaks corresponding to apo- and acyl-enzyme forms are labeled. (B) Plot of apo- and acyl-enzyme species of TEM-1 and CTX-M-15 over time. (C) Acyl-enzyme exchange to apo-TEM-1 from donor-acylated CTX-M-15, KPC-2, E. cloacae P99, and P. aeruginosa AmpC. Time courses are shown in Fig. S5 and the percentages of acyl-enzyme species at the final time point for each reaction are depicted.,
To assess the generality of the reversible mechanism, additional β-lactamases were tested in acyl-enzyme exchange experiments. TEM-1 was used as the apo acceptor enzyme from four donor acylated class A and C enzymes: CTX-M-15, KPC-2, Enterobacter cloacae P99, and Pseudomonas aeruginosa AmpC (Fig. 6C). In each case, the mass equivalent to avibactam was observed to migrate in a time-dependent manner from the acylated donor enzyme onto the acceptor TEM-1 (Fig. S5). The observation of acyl-enzyme transfer suggests that acylation of these other β-lactamases is reversible and that alternate deacylation routes through rearrangement or hydrolysis, if present, are kinetically less significant. Among these enzymes, as measured by IC50, avibactam is a more potent inhibitor of TEM-1 and CTX-M-15 than KPC-2, P99, or AmpC (16). Reflecting these potency differences, the competition reactions equilibrated to different proportions of acyl-TEM-1.
A proposed kinetic scheme can now be defined for avibactam inhibition of the TEM-1 class A β-lactamase (Fig. 7). In the absence of hydrolytic or chemical rearrangement pathways, avibactam can be modeled as a two-step, reversible covalent inhibitor. The upper bound of the acylation on-rate could not be determined and is limited by either a low-affinity noncovalent encounter complex (Ki) or the rate of serine nucleophilic attack to form the carbamoyl acyl-enzyme (k2). The off-rate from the acyl-enzyme, with a t1/2 value of 16 min, is the rate-limiting step for the regeneration of free TEM-1 enzyme.
Fig. 7.

Scheme for the inhibition of TEM-1 by avibactam.
Discussion
The non–β-lactam nature of the DBO scaffold prompted a detailed investigation into its mode of inhibition of the model β-lactamase, TEM-1. Previous work with avibactam and TEM-1 used a two-step model for inhibition, where the ring-opening acylation step was treated as irreversible (16, 20). Our observation of reversible acylation led us to use a two-step reversible model that is also used to describe slow-binding inhibitors (10). However, unlike slow-binding inhibitors, where the rate of formation of the initial enzyme inhibitor complex (E to EI) is faster than the rate of conversion to a more stable inhibited complex (EI to EI*), for avibactam inhibition of TEM-1, we could not separate the rate of formation of EI from the conversion to EI* because of the combined effect of a low-affinity encounter complex and a rapid acylation rate.
As an enzyme class, β-lactamases are highly catalytically efficient, such that on-rates for β-lactam substrates can approach the limit of diffusion control (21). The design of inhibitors based on β-lactam scaffolds has sought to drive inhibitor effectiveness by maintaining efficient enzyme acylation while slowing or eliminating hydrolysis. Avibactam is not a β-lactam, but the measurement that we made of the apparent second-order enzyme inactivation rate constant against TEM-1 of >1.6 × 105 M−1s−1 is comparable with the inactivation efficiency values reported for a variety of β-lactam–based compounds with magnitudes of 104 to 106 M−1s−1 (22–24). In this light, the DBO bridged bicyclic scaffold seems comparable with β-lactams in terms of acylation efficiency by the TEM-1 catalytic machinery.
In contrast to β-lactam–based inhibitors, for which acylation is a one-way, kinetically irreversible reaction, the acylation reaction of avibactam with TEM-1 was shown to be slowly reversible, with a half-life of enzyme recovery of 16 min. Before the discovery of the DBO series, other acylating β-lactamase inhibitors had been shown to rapidly decompose off the acyl-enzyme species through routes of hydrolysis or chemical rearrangement (8, 25). Deacylation through ring closing to regenerate the intact inhibitor is not seen with β-lactam–based inhibitors, presumably because of the high ring strain of the four-membered lactam ring (26). In avibactam, the site of attack by the catalytic serine is the carbonyl of a five-membered cyclic urea, which may be less intrinsically strained than a β-lactam, thereby permitting ring closing and intact inhibitor regeneration for the DBO scaffold vs. hydrolytic release and inhibitor destruction for the β-lactam scaffold.
Across serine hydrolase enzymes, inhibitors that acylate through ester or carbamate linkages are kinetically irreversible as long as they are protected from hydrolytic turnover. Compared with the ester linkage, the carbamoyl acyl-enzyme linkage is generally more stable to enzyme-assisted hydrolysis (27, 28), and inhibitors that acylate through a carbamate have been reported with extremely long residence times, indicating stability to hydrolytic deacylation (27, 29). In this light, the hydrolytic stability of the avibactam acyl-enzyme is not without precedent. Deacylation through intramolecular ring closure, however, is an unusual finding, particularly as the exclusive deacylation route. Reversible acylation was shown with a monocyclic γ-lactam inhibitor of elastase, a serine hydrolase with mechanistic comparability to β-lactamases (30). Nevertheless, reversible acylation/deacylation through a carbamate linkage and a lack of hydrolysis seem to be unique features of avibactam among β-lactamase inhibitors. This mechanism also underscores the chemical use of avibactam compared with β-lactam–based inhibitors, because on release, the regenerated avibactam is intact and competent to reacylate its β-lactamase target and initiate another cycle of inhibition.
For inhibitors that display slow-off kinetics, often, the rate-limiting step involves a protein or ligand conformation change. Discerning the precise adjustments in a protein–ligand complex that govern a slow kinetic step requires a sophisticated understanding of ligand binding and protein dynamics (31, 32). With avibactam and its β-lactamase targets, the acyl-enzyme species may be able to resist hydrolysis because of its carbamate linkage, but reformation of the cyclic urea is a remarkable feat. Understanding how the deacylating ring-closing chemistry is promoted will require future work with both static X-ray structures and solution-based biophysical techniques.
Importantly, the reversible acylation behavior for TEM-1 and avibactam is not restricted to the TEM-1 model β-lactamase. Acyl-enzyme transfer was shown using additional, clinically problematic enzymes of classes A and C. The broad spectrum of inhibition of avibactam across classes A and C enzymes distinguishes it from other inhibitor classes, especially β-lactam–based series (6). Furthermore, the ability of avibactam to restore the minimum inhibitory concentration (MIC) activity of its cephalosporin partners may be enhanced by its slow-off kinetic profile. The factors that translate β-lactamase inhibition into MIC restoration are complex to dissect; nevertheless, inhibitors that span a wide range of off-rates have been described, from fast-off boronic acids to truly irreversible β-lactam–based compounds (5). Studies that carefully account for permeability differences across scaffolds could shed light on whether the enzyme off-rate magnitudes do result in microbiological differentiation.
In the context of slowly reversible inhibitors, the concept of the residence time of the inhibited target has been shown to be a key driver of pharmacokinetic-pharmacodynamic (PK-PD) relationships for enzyme targets in a wide variety of therapeutic areas (33, 34). Avibactam, combined with ceftaroline or ceftazidime, has shown efficacy in preclinical animal models and also in phase II clinical trials of patients with Gram-negative infections (35–38). The increased understanding of the mechanism of avibactam inhibition described in the present work may provide insight into the PK–PD relationship for β-lactam/avibactam combination therapies.
Materials and Methods
Reagents were purchased from Sigma unless otherwise indicated. Nitrocefin was purchased from Acme Bioscience. All assays were performed at 37 °C in 100 mM phosphate buffer (50 mM monobasic + 50 mM dibasic sodium phosphate) adjusted to pH 7.0 and, for the enzyme assays, supplemented with 0.1 mg/mL BSA. Avibactam was prepared by Novexel SA (16). TEM-1 was prepared as described previously (16), with replacement of SP Sepharose (GE Healthcare) for the zinc chelation step. CTX-M-15, KPC-2, E. cloacae P99, and P. aeruginosa AmpC β-lactamases were prepared by Novexel SA; enzyme activity was assessed by measuring nitrocefin hydrolysis rates, and it was found to be equivalent to the values reported by Novexel SA.
Acylation Kinetic Measurements.
Using a Cary 400 Bio UV visual spectrophotometer (Varian) outfitted with a temperature controller, reactions were initiated in stirred 1-cm quartz cuvettetes by adding 20 μL 2.5 nM TEM-1 and 980 μL 204 μM nitrocefin solution in the presence or absence of avibactam. Enzyme activity was monitored using a continuous measurement of 460 nm absorbance in 0.1-s intervals between measurements. For data analysis, the offset between reaction initiation and the first absorbance read was 3 s. Experiments at higher avibactam concentrations were performed on a Bio-Logic SFM-4 Stopped-Flow/Quench-Flow instrument using a cuvettete with a 2-mm path length. A three-syringe method was used to give a constant final concentration of 2 nM TEM-1 and 200 μM nitrocefin. The total flow rate was adjusted to 3 mL/s. Absorbance was recorded continuously at 490 nm in 0.002-s intervals. For data analysis, the offset between reaction initiation and the first absorbance read was 250 ms.
Data for TEM-1 and avibactam were fit to the two-step, reversible inhibition model  . Time courses were fit to Eq. 1 (10) to obtain k, also known as the pseudo first-order rate constant, kobs (Eq. 1):
. Time courses were fit to Eq. 1 (10) to obtain k, also known as the pseudo first-order rate constant, kobs (Eq. 1):
In the on-rate experiment, V0 represents uninhibited enzyme velocity, and it was measured in a reaction with TEM-1 and no avibactam. VS represents the fully inhibited enzyme velocity, and it was estimated using a reaction with no TEM-1. Eq. 2 was used to derive the apparent second-order rate constant for enzyme inactivation (Eq. 2):
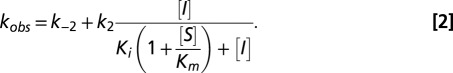 |
In the case where Ki is large relative to [I], then Eq. 2 simplifies to (Eq. 3)
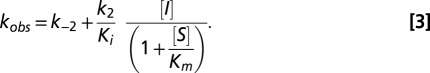 |
The reported value for the kobs vs. [I] slope (the 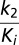 second-order rate constant) includes an adjustment for the (1 + S/Km) term to account for the nitrocefin substrate concentration (200 μM relative to the nitrocefin Km of 76 μM). The reported 95% confidence interval was calculated using MATLAB software (Mathworks). In the plot of kobs vs. [I], k−2 was not precisely measurable, and it was determined independently with a separate off-rate experiment.
second-order rate constant) includes an adjustment for the (1 + S/Km) term to account for the nitrocefin substrate concentration (200 μM relative to the nitrocefin Km of 76 μM). The reported 95% confidence interval was calculated using MATLAB software (Mathworks). In the plot of kobs vs. [I], k−2 was not precisely measurable, and it was determined independently with a separate off-rate experiment.
Deacylation koff Measurement.
Enzyme (1 μM) was incubated with inhibitor (5 μM avibactam, 20 μM tazobactam, or 100 μM clavulanic acid) for 5 min at 37 °C and diluted 4,000-fold in the assay buffer with or without inhibitor. The free enzyme control was diluted in the absence of the inhibitor. The background absorbance control omitted the enzyme. After dilution, TEM-1 activity was assayed in a 96-well microtiter plate by adding 20 μL to 180 μL 400 μM nitrocefin for a final TEM-1 concentration of 25 pM. Absorbance at 490 nm was monitored continuously in a Spectramax plate reader (Molecular Devices). Data were fit to Eq. 1 to obtain koff. In the off-rate experiment, V0 represents fully inhibited enzyme velocity, and it was estimated using a reaction with no TEM-1. VS represents the uninhibited enzyme velocity, and it was measured in a reaction with TEM-1 and no avibactam. The koff value is reported as ±2 SD from three separate determinations.
Preparation of TEM-1 Acyl-Enzyme.
In a 200 μL reaction volume, 1 μM TEM-1 was incubated with and without 5 μM avibactam for 5 min at 37 °C and subjected to two ultrafiltration cartridge (UFC) steps to remove excess inhibitor (Ultrafree-0.5 with Biomax membrane, 5-kDa cutoff; Millipore). Centrifugation at 10,600 × g for 8 min was performed at 4 °C. After each ultrafiltration step, 20 μL retentate were diluted with 180 μL assay buffer to restore the original enzyme concentration. After two UFC treatments, the amount of free avibactam was quantified by liquid chromotography/MS/MS and found to be <5% of the original concentration. Loss of protein during UFC was assessed by measuring TEM-1 activity (on 4,000-fold dilution) in the acyl-enzyme sample compared with non-UFC–treated enzyme, and loss was found to be <5%.
Equilibration of Acyl-Enzyme with Free Enzyme.
Acyl-TEM-1 (1 μM), prepared as described above, was diluted threefold serially in eight steps (from 1 μM to 150 pM) and equilibrated for at least 2 h at 37 °C. Samples were analyzed by enzyme activity, protein MS, and small-molecule MS as described in SI Materials and Methods.
Acyl-Enzyme Exchange.
For the acyl-enzyme exchange experiment from acyl-TEM-1 to apo-CTX-M-15, acylated TEM-1 and apo-CTX-M-15 were combined to final concentrations of 1 μM each and incubated at 37 °C. Aliquots (30 μL) were collected at indicated time points and analyzed as described in SI Materials and Methods. Details for the acyl-enzyme exchange to apo-TEM-1 from acyl-CTX-M-15, acyl-KPC-2, acyl-P99, and acyl-AmpC are also described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the scientists at Novexel SA for previous work with avibactam and providing enzymes and avibactam. We also acknowledge Jason Thresher, Jim Whiteaker, and Stephania Livchak for purification of TEM-1. Zhiping You is acknowledged for statistical analysis.
Footnotes
Conflict of interest statement: All authors are present or past employees of AstraZeneca, as stated in the affiliations, and potentially own stock and/or hold stock options in the company.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205073109/-/DCSupplemental.
References
- 1.Arias CA, Murray BE. Antibiotic-resistant bugs in the 21st century—a clinical super-challenge. N Engl J Med. 2009;360:439–443. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 2.Nordmann P, Poirel L, Toleman MA, Walsh TR. Does broad-spectrum β-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by Gram-negative bacteria? J Antimicrob Chemother. 2011;66:689–692. doi: 10.1093/jac/dkq520. [DOI] [PubMed] [Google Scholar]
- 3.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bush K, Fisher JF. Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from gram-negative bacteria. Annu Rev Microbiol. 2011;65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 5.Bebrone C, et al. Current challenges in antimicrobial chemotherapy: Focus on ß-lactamase inhibition. Drugs. 2010;70:651–679. doi: 10.2165/11318430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 6.Biondi S, Long S, Panunzio M, Qin WL. Current trends in β-lactam based β-lactamases inhibitors. Curr Med Chem. 2011;18:4223–4236. doi: 10.2174/092986711797189655. [DOI] [PubMed] [Google Scholar]
- 7.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 8.Fisher JF, Mobashery S. Three decades of the class A β-lactamase acyl-enzyme. Curr Protein Pept Sci. 2009;10:401–407. doi: 10.2174/138920309789351967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr Opin Microbiol. 2011;14:550–555. doi: 10.1016/j.mib.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 10.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 11.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1985. pp. 140–141. [Google Scholar]
- 12.Copeland RA, Basavapathruni A, Moyer M, Scott MP. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal Biochem. 2011;416:206–210. doi: 10.1016/j.ab.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Brown RP, Aplin RT, Schofield CJ. Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: Observation of intermediates by electrospray ionization mass spectrometry. Biochemistry. 1996;35:12421–12432. doi: 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- 14.Bush K, Macalintal C, Rasmussen BA, Lee VJ, Yang Y. Kinetic interactions of tazobactam with β-lactamases from all major structural classes. Antimicrob Agents Chemother. 1993;37:851–858. doi: 10.1128/aac.37.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnefoy A, et al. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J Antimicrob Chemother. 2004;54:410–417. doi: 10.1093/jac/dkh358. [DOI] [PubMed] [Google Scholar]
- 16.Stachyra T, et al. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-β-lactam β-lactamase inhibitor. Antimicrob Agents Chemother. 2010;54:5132–5138. doi: 10.1128/AAC.00568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, et al. Mechanism of inhibition of the class A β -lactamases PC1 and TEM-1 by tazobactam. Observation of reaction products by electrospray ionization mass spectrometry. J Biol Chem. 2000;275:26674–26682. doi: 10.1074/jbc.M002369200. [DOI] [PubMed] [Google Scholar]
- 18.Tabei K, et al. Mechanism of inactivation of β-lactamases by novel 6-methylidene penems elucidated using electrospray ionization mass spectrometry. J Med Chem. 2004;47:3674–3688. doi: 10.1021/jm049903j. [DOI] [PubMed] [Google Scholar]
- 19.Martin R, Gold M, Jones JB. Inhibition of the RTEM-1 β-lactamase by boronic acids. Bioorg Med Chem Lett. 1994;4:1229–1234. [Google Scholar]
- 20.De Meester F, et al. Automated analysis of enzyme inactivation phenomena. Application to β-lactamases and DD-peptidases. Biochem Pharmacol. 1987;36:2393–2403. doi: 10.1016/0006-2952(87)90609-5. [DOI] [PubMed] [Google Scholar]
- 21.Christensen H, Martin MT, Waley SG. β-lactamases as fully efficient enzymes. Determination of all the rate constants in the acyl-enzyme mechanism. Biochem J. 1990;266:853–861. [PMC free article] [PubMed] [Google Scholar]
- 22.De Meester F, et al. 6-β-Iodopenicillanate as a probe for the classification of β-lactamases. Biochem J. 1986;239:575–580. doi: 10.1042/bj2390575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bulychev A, et al. Potent mechanism-based inhibition of the TEM-1 β-lactamase by novel N-sulfonyloxy β-lactams. J Am Chem Soc. 1995;117:5938–5943. [Google Scholar]
- 24.Charnas RL, Knowles JR. Inhibition of the RTEM β-lactamase from Escherichia coli. Interaction of enzyme with derivatives of olivanic acid. Biochemistry. 1981;20:2732–2737. doi: 10.1021/bi00513a005. [DOI] [PubMed] [Google Scholar]
- 25.Matagne A, Ghuysen MF, Frère JM. Interactions between active-site-serine β-lactamases and mechanism-based inactivators: A kinetic study and an overview. Biochem J. 1993;295:705–711. doi: 10.1042/bj2950705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitarch J, Ruiz-Lopez MF, Silla E, Pascual-Ahuir J, Tunon I. Neutral and alkaline hydrolysis of model β-lactam antibiotics. An ab initio study of water catalysis. J Am Chem Soc. 1998;120:2146–2155. [Google Scholar]
- 27.Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 28.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn K, et al. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 30.Westwood NJ, Claridge TDW, Edwards PN, Schofield CJ. Reversible acylation of elastase by gamma-lactam analogs of β-lactam inhibitors. Bioorg Med Chem Lett. 1997;7:2973–2978. [Google Scholar]
- 31.Garvey EP. Structural mechanisms of slow-onset, two-step enzyme inhibition. Curr Chem Biol. 2010;4:64–73. [Google Scholar]
- 32.Fieulaine S, et al. Trapping conformational states along ligand-binding dynamics of peptide deformylase: The impact of induced fit on enzyme catalysis. PLoS Biol. 2011;9:e1001066. doi: 10.1371/journal.pbio.1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu H, Tonge PJ. Drug-target residence time: Critical information for lead optimization. Curr Opin Chem Biol. 2010;14:467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- 35.Endimiani A, et al. Evaluation of ceftazidime and NXL104 in two murine models of infection due to KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 2011;55:82–85. doi: 10.1128/AAC.01198-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiskirchen DE, Crandon JL, Furtado GH, Williams G, Nicolau DP. In vivo efficacy of a human-simulated regimen of ceftaroline combined with NXL104 against extended-spectrum-β-lactamase (ESBL)-producing and non-ESBL-producing Enterobacteriaceae. Antimicrob Agents Chemother. 2011;55:3220–3225. doi: 10.1128/AAC.00024-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lucasti C, Popescu I, Ramesh M, Lipka J, Sable C. Efficacy and safety of ceftazidime/NXL104 plus metronidazole vs. meropenem in the treatment of complicated intra-abdominal infections in hospitalised adults. Clin Microbiol Infect. 2011;17:S437. doi: 10.1093/jac/dks523. [DOI] [PubMed] [Google Scholar]
- 38.Vazquez J, Gonzalez Patzan LD, Lipka J, Sable C. Efficacy, safety and tolerability of ceftazidime/NXL104 vs. imipenem cilastatin in the treatment of complicated urinary tract infections in hospitalised adults. Clin Microbiol Infect. 2011;17:S438. [Google Scholar]
- 39.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. New York: Wiley; 2005. pp. 179–185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.