

Abstract
Here we report on the effect of the mass transfer rate (kt) on the oxygen reduction reaction (ORR) catalyzed by Pt dendrimer-encapsulated nanoparticles (DENs) comprised of 147 and 55 atoms (Pt147 and Pt55). The experiments were carried out using a dual-electrode microelectrochemical device, which enables the study of the ORR under high kt conditions with simultaneous detection of H2O2. At low kt (0.02 to 0.12 cm s-1) the effective number of electrons involved in ORR, neff, is 3.7 for Pt147 and 3.4 for Pt55. As kt is increased, the mass-transfer-limited current for the ORR becomes significantly lower than the value predicted by the Levich equation for a 4-electron process regardless of catalyst size. However, the percentage of H2O2 detected remains constant, such that neff barely changes over the entire kt range explored (0.02 cm s-1). This suggests that mass transfer does not affect neff, which has implications for the mechanism of the ORR on Pt nanoparticles. Interestingly, there is a significant difference in neff for the two sizes of Pt DENs (neff = 3.7 and 3.5 for Pt147 and Pt55, respectively) that cannot be assigned to mass transfer effects and that we therefore attribute to a particle size effect.
Keywords: electrocatalysis, platinum nanoparticle
Here we report on the effect of mass transfer rate (kt) on the oxygen reduction reaction (ORR) catalyzed by Pt dendrimer-encapsulated nanoparticles (DENs) comprised of 147 and 55 atoms (Pt147 and Pt55, respectively). The experiments were carried out in the dual-electrode microelectrochemical device shown in Fig. 1 (1). This configuration enables the study of the ORR under high kt conditions with simultaneous detection of H2O2. This is important because the methods currently used to study ORR at high kt, such as ultramicroelectrodes (UMEs) (2), nanoelectrodes (3), and microjet electrodes (4) do not incorporate H2O2 detection. Accordingly, ORR data is generally interpreted based on steady-state, mass-transfer-limited currents alone. Using the microelectrochemical device, two principal effects are evident as kt is increased. First, the ORR current becomes significantly smaller than predicted by the Levich equation (5). Second, the effective number of electrons involved in the ORR, neff, remains constant over the entire kt range, for both Pt147 and Pt55. We draw three conclusions from these observations: (i) The decrease in ORR current at high kt is caused by kinetic effects; (ii) increasing kt does not affect the mechanism of the ORR catalyzed by Pt DENs; and (iii) there is a difference in neff between Pt147 and Pt55 that cannot be attributed to mass-transfer effects.
Fig. 1.

(A) Schematic representation of the hybrid PDMS/quartz microelectrochemical device used for ORR studies. Solution is introduced to the microchannel using a syringe pump connected to the reservoir on the left. A combined reference/counter electrode is placed in the other reservoir so that it is in close proximity to PPC microband electrodes. Note that this drawing is not to scale. In the actual device, the length of the microband electrodes is typically 40 μm with a gap of 15 μm between them. (B) Optical micrograph of the PPC microbands and the microchannel after platinization of the collector electrode and electrochemical deposition of Pt DENs onto the generator electrode. The outlet reservoir is visible to the right of the electrodes.
The electrochemical ORR has been the subject of extensive research and has been reviewed on several occasions (6–10). The generally accepted mechanism for the ORR at a metallic surface is a multielectron reaction with intermediates as shown in Scheme 1 (2, 7, 9, 11). Specifically, at the electrode surface, O2 can be reduced directly to H2O or H2O2 in 4- or 2-electron processes, respectively. Subsequently, electrogenerated H2O2 can disproportionate into H2O and O2, or by addition of two electrons, form H2O. According to Scheme 1, the yield of H2O2, quantified by neff, is determined by the relative rates of steps c, d, and e, as well as the rate of mass transfer of H2O2 away from the electrode surface (2).
Scheme 1.

The development of O2/H2 polymer electrolyte membrane fuel cells (PE-MFCs) as sustainable energy sources (12, 13) has led to increased interest in developing efficient, non-Pt catalysts for the ORR (14–18). In spite of this, Pt is still considered to be the most effective catalyst for the ORR, even though it is not optimal for this application due to its high overpotential and its low Earth abundance. In the absence of viable alternatives, interest has been focused on reducing the amount of Pt required for fuel cell applications. There are two ways to do this: dilute it with a second metal or decrease the size or total number of Pt monometallic catalyst particles. However, a decrease in Pt surface coverage (or, correspondingly, an increase in kt) has been associated with slower ORR kinetics (19, 20) and an increase in the amount of H2O2 produced (21–23). The presence of H2O2 is a major concern for fuel cell applications due to its corrosive nature and because it lowers the maximum attainable voltage (24, 25).
A number of studies have been reported that focus on the role of mass transfer in ORR efficiency. The analytical tools used for this purpose include the rotating disk and rotating ring-disk electrodes (RDE and RRDE) (21, 26–28), UMEs (2, 29), nanoelectrodes (3), microjet electrodes (4), and microfluidic thin-layer cells (22, 23). In most of these studies, kt is increased by decreasing the surface coverage of the catalyst (21–23, 27, 28). For example, Behm and coworkers studied the ORR using two-dimensional arrays of 100-nm Pt islands immobilized on glassy carbon (GC) electrodes (22, 23). The key finding was that the amount of electrogenerated H2O2 increased with decreasing catalyst coverage. A “desorption + readsorption + reaction” mechanism was proposed to explain this effect (26, 30). Under this assumption, more H2O2 is obtained at high kt (low catalyst loading or high flow rates), due to lower probability for H2O2 readsorption and further reaction on different Pt sites (Scheme 1) (26, 30). Kucernak and coworkers studied the effect of high kt conditions (up to 2 cm s-1) on the ORR catalyzed by single, submicron Pt particles affixed to the ends of nanoelectrodes (3). It was found that the steady-state mass-transfer-limited current for the ORR decreased with increasing kt. This was attributed to a shift in neff from 4 at kt = 0.01 cm s-1 to 3.5 at kt = 2 cm s-1 (3). However, because a collector electrode was not present, it was not possible to correlate these results to the presence of H2O2.
Here we study the effect of kt on the ORR using Pt DENs having diameters of 1–2 nm and low (< 0.3 nm) size polydispersity. In the microelectrochemical device shown in Fig. 1, Pt147 or Pt55 DENs are electrochemically immobilized onto a pyrolyzed photoresist carbon (PPC) generator electrode (1). This results in robust attachment of approximately 1 monolayer of Pt DENs onto the PPC surface (1). Importantly, a collector electrode is present downstream for amperometric detection of H2O2. By controlling the flow rate through the microchannel, it is possible to vary kt between 0.02 and 0.12 cm s-1. Indeed, such high mass transfer conditions are not achievable using the RRDE or UMEs with diameters larger than 2 μm. At low kt (0.02 cm s-1), which is similar to that achieved at the upper end of kt values accessible with the RRDE, neff is 3.7 for Pt147 and 3.4 for Pt55, in agreement with our previous findings (31). As kt increases, the mass-transfer-limited current for the ORR becomes significantly lower than the value predicted by the Levich equation for a 4-electron process for both Pt147 and Pt55. However, the percentage of H2O2 detected remains constant, such that neff does not change over the entire kt range employed. Interestingly, there is a significant difference in neff for the two sizes of Pt DENs (average neff = 3.7 and 3.5 for Pt147 and Pt55, respectively) that cannot be assigned to mass transfer effects.
The results reported here are significant for several reasons. First, we show that simultaneous measurement of the H2O2 current is vital for correct interpretation of ORR data. Second, while others have previously shown that the ORR catalyzed by large Pt nanoparticles is affected by changes in kt (22, 23) we show here that this does not apply for smaller Pt materials. Finally, we show that the size of very small Pt nanoparticles (diameters < 2 nm) has an effect on the mechanism of the ORR, and hence its product distribution. These observations are relevant to the development of efficient Pt electrocatalysts for the ORR.
Results and Discussion
Evaluation of ORR Activity for Pt147 and Pt55 at Low Mass Transfer Rates.
The ORR activity of Pt147 and Pt55 DENs was evaluated using the poly(dimethylsiloxane) (PDMS)/quartz hybrid microelectrochemical device shown in Fig. 1 and discussed in detail elsewhere (1). Following platinization of the collector (Fig. 1B) and immobilization of Pt DENs on the generator (1), the ORR measurements were carried out in air-saturated 0.10 M HClO4 electrolyte solution. To evaluate the ORR kinetics at low kt, solution was flowed through the device at 0.05 μL min-1 (0.04 cm s-1), corresponding to a kt value of 0.02 cm s-1. This is on the same order as that achieved using an RRDE.
We previously used the RDE to evaluate the ORR characteristics of Pt DENs having different sizes (31). The results showed that the steady-state limiting current for Pt147 was higher than for Pt55, an effect that was tentatively assigned to H2O2 becoming a significant product of the ORR for the smaller catalytic particles (31). We pointed out that if this were correct, it was a rare case of a very slight change in particle size resulting in a shift in product distribution. Fig. 2 shows generation-collection linear scan voltammograms (LSVs) for the ORR obtained using the microelectrochemical device for Pt147 (blue lines) and Pt55 (red lines) DENs at kt = 0.02 cm s-1. The potential of the generator electrode was swept from +0.80 to -0.10 V (vs. Ag/AgCl) at 50 mV s-1. The potential of the collector electrode was held at +1.10 V, where we observed reliable and stable mass-transfer-limited currents for H2O2 oxidation (1). The onset of catalytic current is at ∼+0.60 V, and a steady-state, mass-transfer-limited current is apparent over the range 0.00 to -0.10 V. There is a slight decrease in the ORR current at potentials more negative than 0 V due to hydrogen adsorption onto the catalyst (32). In addition, the adsorption of hydrogen is accompanied by an increase in hydrogen peroxide formation (manifested as an increase in the collector electrode current), an effect previously observed by others (32). To minimize artifacts arising from hydrogen adsorption, generator-collector current pairs were measured at the potential where the ORR current reached its maximum value.
Fig. 2.

Generation (upper curves) and collection (lower curves) LSVs obtained using Pt147 (blue lines) and Pt55 (red lines) DENs in air-saturated, aqueous 0.10 M HClO4. The potential of the generator electrode was scanned from +0.80 to -0.10 V (vs. Ag/AgCl) while holding the potential of the platinized collector at +1.10 V. The flow rate was 0.05 μL min-1 corresponding to kt = 0.02 cm s-1, and η was 40.0%. The scan rate was 50 mV s-1.
Importantly, the ORR limiting current for Pt147 DENs (55 nA) is noticeably higher than for Pt55 DENs (37 nA), although the LSVs have the same general shape. The downstream collector electrode makes it possible to determine the amount of H2O2 produced during ORR, at the same time the mass-transfer-limited currents are measured at the generator. The LSVs in Fig. 2 show that the amount of H2O2 oxidized at the collector electrode is higher for Pt55 DENs. As we hypothesized in our earlier publication, this indicates that the peroxide pathways play a more important role in the ORR catalyzed by smaller nanoparticles (31).
The value of neff was determined using the ratio of the steady-state mass-transfer-limited current for the ORR (igen) and the oxidation current for H2O2 (icol); that is, neff = 4 - [(2icol)/(ηigen)], where η represents the collection efficiency of the device (details on how η is calculated are provided in Materials and Methods) (33). In Fig. 2, icol has been background subtracted against the current value obtained by holding the collector electrode at a potential (1.10 V), where the ORR does not proceed. The measured η at 0.05 μL min-1 was 40.0%. Note that the collector current does not exhibit a clear, steady-state plateau (vide supra), and, as such, icol was measured at the potential where igen attained its maximum value (0 V). The calculated values of neff are 3.7 ± 0.1 and 3.4 ± 0.1 (average of three independent measurements), for Pt147 and Pt55, respectively. Using these values for neff, igen can be estimated using the Levich equation (Eq. 1) (5), as follows:
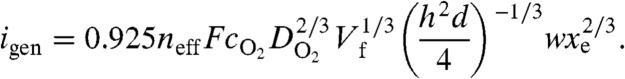 |
[1] |
Here, F is the Faraday constant, cO2 and DO2 are the bulk concentration and diffusion coefficient, respectively, of O2 [cO2 = 0.20 mM, (34) DO2 = 1.67 × 10-5 cm2 s-1] (35), Vf is the volume flow rate (0.05 μL min-1), xe and w are the length (40 μm) and the width (100 μm) of the electrode, respectively, d is the width of the microchannel (100 μm), and h is the height of the microchannel (20 μm). Using the previously determined neff values, igen is calculated to be 55 nA for Pt147 and 50 nA for Pt55. For Pt147, the calculated value is in excellent agreement with the measured igen of 55 nA. However, for Pt55, the calculated value is substantially higher than the experimentally determined igen (37 nA). Although we do not understand this result, such inconsistencies are common in the ORR RRDE literature (36, 37). It is possible that the relatively high scan rate employed (50 mV s-1) also contributes to this effect.
ORR Measurements at High Mass Transfer Rates.
To learn more about the effect of the mass transfer rate of O2 on neff, we acquired ORR generation collection LSVs while flowing air-saturated 0.10 M HClO4 through the microchannel at flow rates ranging from 0.05 to 10 μL min-1 (0.04 to 8.3 cm s-1). This corresponds to kt values from 0.02 to 0.12 cm s-1. This range is considerably higher than that accessible with the RRDE. Indeed, to achieve the upper limit of kt = 0.12 cm s-1, a RRDE would have to be rotated at 150,000 rpm, which is well above the typical maximum rotation rate of 7,000 rpm. Fig. 3A shows a plot of igen and simultaneously measured icol values obtained for Pt147 (blue dots) and Pt55 (red dots) as a function of flow. Values for igen and icol were obtained from both low-to-high and high-to-low flow rates, and were found to be independent of this parameter. This confirms that the Pt coverage is constant throughout the experiments (that is, that the Pt DENs remain attached to the electrode surface). In Fig. 3A, icol was normalized with respect to η for each flow rate. Recall that igen - icol current pairs were measured at the potential where igen reached its maximum value and icol values were background subtracted (vide supra). Fig. 3A shows that, for both Pt147 and Pt55 DENs, as kt increases, igen increases and begins to level off at flow rates higher than 2 μL min-1. The same pattern is observed for icol. Furthermore, across the entire flow-rate range, the ORR limiting current for Pt147 remains higher than that for Pt55.
Fig. 3.

(A) Plot of igen (ORR mass-transfer-limited current) and simultaneously acquired icol (H2O2 oxidation current) for Pt147 (blue circles) and Pt55 (red circles) measured while flowing air-saturated, aqueous 0.10 M HClO4 through the microchannel at flow rates ranging from 0.05 to 10 μL min-1 (kt from 0.02 to 0.12 cm s-1). Note that icol was normalized with respect to η (η values for 0.05, 0.075, 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 μL min-1 flow rates were 40.0, 37.2, 36.3, 34.7, 34.2, 34.0, 33.5, 33.3 and 32.7%, respectively). (B) Plot of igen from A vs. 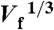 and kt, for Pt147 (blue circles) and Pt55 (red circles). The dashed black lines were calculated using the Levich equation (Eq. 1) for a 4-electron (upper line) and a 2-electron (lower line) process.
and kt, for Pt147 (blue circles) and Pt55 (red circles). The dashed black lines were calculated using the Levich equation (Eq. 1) for a 4-electron (upper line) and a 2-electron (lower line) process.
It is interesting to compare the experimental values of igen with the theoretical values predicted by the Levich equation (Eq. 1) for 4- and 2-electron ORR processes. In Fig. 3B, the igen values from Fig. 3A are plotted vs. 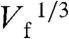 and kt. The current for 4- and 2-electron ORR processes are plotted on the same graph (dashed lines). For Pt147, Fig. 3B indicates that as kt increases, igen appears to drift from the 4-electron to the 2-electron ORR pathway (Scheme 1). Interestingly, at Vf higher than 1 μL min-1, the measured igen for Pt55 is smaller than the value predicted by Eq. 1 for a 2-electron process, an observation that will be discussed later. Importantly, for both Pt147 and Pt55 DENs, the plot of igen vs.
and kt. The current for 4- and 2-electron ORR processes are plotted on the same graph (dashed lines). For Pt147, Fig. 3B indicates that as kt increases, igen appears to drift from the 4-electron to the 2-electron ORR pathway (Scheme 1). Interestingly, at Vf higher than 1 μL min-1, the measured igen for Pt55 is smaller than the value predicted by Eq. 1 for a 2-electron process, an observation that will be discussed later. Importantly, for both Pt147 and Pt55 DENs, the plot of igen vs. 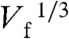 is not linear. A similar trend in the ORR limiting current was observed during Pt-catalyzed ORR studies using UMEs (2) and nanoelectrodes (3), and for Cu using hydrodynamic electrodes (4). These results were interpreted in terms of O2 reduction not following a simple 4- or 2-electron transfer process, but instead undergoing a transition from 4 electrons at the lowest kt values towards 2 electrons as the mass transfer rate increased. It is important to note that in these earlier reports a collector electrode was not used, and therefore the amount of H2O2 generator could not be measured directly and then correlated to the ORR current. Because we (1) and others (36, 37) have noted that the mass-transfer-limited current for the ORR is often unreliable for the determination of neff, an accurate calculation requires the ratio of icol to igen. Here, the concomitant measurement of H2O2 oxidation current allows the calculation of neff at each kt value. Fig. 4 shows a plot of neff vs. Vf and kt, calculated for data presented in Fig. 3A. At all but the lowest flow rates, the value of neff (that is, the percentage of H2O2 collected during ORR) is nearly constant. Consequently, although the magnitude of igen suggests a shift in the mechanism of ORR with the change in mass transfer rate, neff remains constant over the entire kt range. The average neff is 3.7 and 3.5 for Pt147 and Pt55, respectively, similar to the values calculated at kt = 0.02 cm s-1 (3.7 for Pt147 and 3.4 for Pt55). We attribute the shift in igen observed here, and previously by others (2–4) to kinetic effects: As kt increases, some O2 molecules flow past the generator electrode faster than they can be reduced. This results in a decrease in igen from its theoretical mass-transfer-limited value. However, the mechanism of ORR remains unaffected. Although we do not currently have a definitive explanation for the difference in neff between Pt DENs of different sizes, it is possible that the decrease in particle size leads to a different O2 binding mechanism onto the Pt surface, which ultimately affects the ORR pathway. For small Pt NPs, molecular O2 can bind to corner and edge sites in the so-called single-bond binding geometry, in which one oxygen atom is against the metal surface and the second is exposed to the solvent. Such single-bond binding can lead to the formation of OOH groups, and ultimately, H2O2. Experimental and theoretical work is currently underway to fully understand this effect.
is not linear. A similar trend in the ORR limiting current was observed during Pt-catalyzed ORR studies using UMEs (2) and nanoelectrodes (3), and for Cu using hydrodynamic electrodes (4). These results were interpreted in terms of O2 reduction not following a simple 4- or 2-electron transfer process, but instead undergoing a transition from 4 electrons at the lowest kt values towards 2 electrons as the mass transfer rate increased. It is important to note that in these earlier reports a collector electrode was not used, and therefore the amount of H2O2 generator could not be measured directly and then correlated to the ORR current. Because we (1) and others (36, 37) have noted that the mass-transfer-limited current for the ORR is often unreliable for the determination of neff, an accurate calculation requires the ratio of icol to igen. Here, the concomitant measurement of H2O2 oxidation current allows the calculation of neff at each kt value. Fig. 4 shows a plot of neff vs. Vf and kt, calculated for data presented in Fig. 3A. At all but the lowest flow rates, the value of neff (that is, the percentage of H2O2 collected during ORR) is nearly constant. Consequently, although the magnitude of igen suggests a shift in the mechanism of ORR with the change in mass transfer rate, neff remains constant over the entire kt range. The average neff is 3.7 and 3.5 for Pt147 and Pt55, respectively, similar to the values calculated at kt = 0.02 cm s-1 (3.7 for Pt147 and 3.4 for Pt55). We attribute the shift in igen observed here, and previously by others (2–4) to kinetic effects: As kt increases, some O2 molecules flow past the generator electrode faster than they can be reduced. This results in a decrease in igen from its theoretical mass-transfer-limited value. However, the mechanism of ORR remains unaffected. Although we do not currently have a definitive explanation for the difference in neff between Pt DENs of different sizes, it is possible that the decrease in particle size leads to a different O2 binding mechanism onto the Pt surface, which ultimately affects the ORR pathway. For small Pt NPs, molecular O2 can bind to corner and edge sites in the so-called single-bond binding geometry, in which one oxygen atom is against the metal surface and the second is exposed to the solvent. Such single-bond binding can lead to the formation of OOH groups, and ultimately, H2O2. Experimental and theoretical work is currently underway to fully understand this effect.
Fig. 4.

The value of neff calculated from the experimental results in Fig. 3A for Pt147 (blue circles) and Pt55 (red circles) vs. 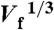 and kt. The black lines represent the average values of neff across the entire mass transfer range shown.
and kt. The black lines represent the average values of neff across the entire mass transfer range shown.
Conclusions
Using a dual-electrode microelectrochemical device we investigated the effect of mass transfer on the ORR catalyzed by Pt147 and Pt55 DENs. At low values of kt (0.02 cm s-1), neff was 3.7 for Pt147 and 3.4 for Pt55, in agreement with previous findings obtained using a RDE (31). As kt was increased from 0.02 to 0.12 cm s-1, the measured ORR mass-transfer-limited current for both Pt147 and Pt55 was smaller than the value predicted by the Levich equation, similar to results obtained by others (2–4). Previously, this was interpreted as a transition of the ORR from four electrons at the lowest kt values towards two electrons as the mass transfer rate increased. Here, we observed that the percentage of H2O2 produced was unaffected by kt, and neff was nearly constant across the entire kt range. Hence, the concomitant measurement of H2O2 allows us to conclude that the decrease in the expected mass-transfer-limited current for ORR at high kt is in fact due to kinetic effects, rather than a change in the ORR mechanism.
Importantly, we also observed an unambiguous nanoparticle size effect on the mechanism of ORR, with an average neff higher for Pt147 (3.7) than for Pt55 (3.5). It is certainly surprising that a change in particle size of just 100 atoms is able to shift the reaction mechanism. Accordingly, we are investigating this effect in more detail, as it may have an important role to play in ORR electrocatalysis specifically and electrocatalysis generally. The results of these studies will be reported in due course.
Materials and Methods
Chemicals.
PDMS channels were prepared using a Sylgard 184 elastomer kit obtained from K. R. Anderson, Inc. Quartz microscope slides (25 mm × 75 mm, 1 mm thick) were purchased from Technical Glass Products. Positive-tone photoresist (AZ 1518) and developer (AZ 400K) were purchased from Capitol Scientific, Inc. Sixth-generation poly(amidoamine) dendrimers terminated with hydroxyl groups (G6-OH) were purchased as a 5% (wt/wt) solution in methanol (Dendritech, Inc.). Prior to use, the methanol was removed under vacuum and the dendrimer was reconstituted in water at a concentration of 100 μM. K2PtCl4 (99.99%), NaBH4 (99.99%), and LiClO4 (99.99%) were purchased from Sigma-Aldrich, Inc.; HClO4 (Ultrex II) was from J. T. Baker and ferrocenemethanol (FcMeOH, 97%) was from Acros Organics. These reagents were used without further purification. Deionized water having a resistivity of 18.2 MΩ cm was used for all experiments (Milli-Q gradient system, Millipore).
Preparation of Microelectrochemical Devices.
The procedure for fabricating PPC microband electrodes is described in detail elsewhere (1). Briefly, PPC microbands, 40 μm wide and separated by a 15 μm gap, were fabricated by slow pyrolysis of patterned AZ 1518 photoresist at 1,000 °C under a forming gas of 5% H2 and 95% N2 (1). The thickness of the resulting PPC features, measured using profilometry, was 300 nm. PDMS microfluidic channels, 6 mm long, 100 μm wide and 20 μm high, were prepared using a previously described replica micromolding method (1, 38). Two reservoirs (1.0 mm diameter) were punched at the ends of each channel to accommodate introduction of solution. The PDMS layer and quartz substrate were exposed to an air plasma (60 W, model PDC-32G, Harrick Scientific) for 45 s, joined together under an optical microscope, and then placed over a hot plate at 80 °C for 5 min. A syringe pump (Pump 11, Pico Plus Elite, Harvard Apparatus), connected to the inlet (left side) reservoir using Teflon tubing, was used to push solution through the microelectrochemical device.
Preparation of Pt DENs.
Pt DENs comprised of 147 and 55 atoms were prepared according to our previously published procedure (31, 39). A 0.10 mM aqueous solution of G6-OH was mixed with sufficient aqueous 0.10 M K2PtCl4 such that the final Pt2+-to-dendrimer ratios were 147∶1 or 55∶1. After stirring for three days (40), a 10-fold molar excess of aqueous 1.00 M NaBH4 was added to reduce Pt2+. This solution was kept capped for 24 h to maximize reduction of Pt2+ (40). Note that we have previously shown that reduction of the Pt2+/G6-OH precursor by 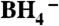 does not yield fully reduced Pt DENs (40, 41). However, electrochemical cycling does result in complete reduction (vide infra) (40, 41). The average diameters and size distributions of the Pt DENs were 1.7 nm and 1.4 nm for Pt147 and Pt55, respectively (31). After synthesis, the quality of the Pt DENs was verified by UV-vis absorption spectroscopy (31, 42). Before using for electrochemical experiments, Pt DENs were dialyzed for 24 h in 4.0 L of Millipore water (Milli-Q Gradient PF-06073) to remove salts and other impurities. After dialysis, but prior to immobilization onto PPC electrodes, the DEN solution was mixed with sufficient LiClO4 to yield a 0.10 M solution.
does not yield fully reduced Pt DENs (40, 41). However, electrochemical cycling does result in complete reduction (vide infra) (40, 41). The average diameters and size distributions of the Pt DENs were 1.7 nm and 1.4 nm for Pt147 and Pt55, respectively (31). After synthesis, the quality of the Pt DENs was verified by UV-vis absorption spectroscopy (31, 42). Before using for electrochemical experiments, Pt DENs were dialyzed for 24 h in 4.0 L of Millipore water (Milli-Q Gradient PF-06073) to remove salts and other impurities. After dialysis, but prior to immobilization onto PPC electrodes, the DEN solution was mixed with sufficient LiClO4 to yield a 0.10 M solution.
Electrochemistry.
The microelectrochemical device was configured in a three-electrode arrangement, comprising the two PPC microbands as working electrodes. A Ag/AgCl reference electrode, which also acted as a counter electrode was positioned in the outlet (right-hand side) reservoir (Scheme 1). All potentials are referenced to Ag/AgCl (3.4 M KCl, model 66-EE009 “no-leak” Ag/AgCl, Dionex). LSV with two working electrodes was carried out using a computer-based bipotentiostat (Model CHI760B potentiostat, CH Instruments). All electrochemical experiments were performed at 22 ± 1 °C.
Prior to the immobilization of Pt DENs on the generator, Pt was electrodeposited onto the PPC collector electrode to increase its sensitivity for amperometric H2O2 detection (Fig. 1B) (1). After platinization, LSVs were acquired at the PPC generator electrode in the ORR region of interest (+0.80 to -0.10 V vs. Ag/AgCl) using aqueous 0.10 M HClO4, to check that the generator electrode had not been contaminated with Pt during the platinization step. Pt DENs were then immobilized on the PPC generator using a previously reported procedure (1, 31, 39). Briefly, a 10.0 μM Pt DEN solution containing 0.10 M LiClO4 was flowed through the microchannel at 0.05 μL min-1 while the electrode potential was swept three times between +0.10 and +1.00 V (vs. Ag/AgCl) at 10 mV s-1. We previously showed that this results in robust attachment of approximately 1 monolayer of Pt DENs onto both GC and PPC surfaces (1, 31). Before acquiring ORR generation collection LSVs, the DEN-modified electrodes were scanned 20 times at 100 mV s-1 between +1.25 V and -0.20 V (vs. Ag/AgCl) in air-saturated 0.10 M HClO4 solution. As we have shown previously, these conditioning scans (43) are commonplace and required to achieve stable and reproducible LSVs (39).
Upon completion of the ORR measurements, η was determined for each microelectrochemical device using the following method. Generation collection LSVs were recorded while pumping an aqueous solution containing 0.10 mM FcMeOH and 0.10 M KNO3 through the microchannel at flow rates ranging from 0.05 to 10 μL min-1 (0.04 to 8.3 cm s-1). At each flow rate, the values of η were calculated as the ratio of the steady-state mass-transfer-limited currents at the collector and generator electrodes, for the electrolysis of FcMeOH. The calculated values of η ranged from 32.7–40.0%.
ACKNOWLEDGMENTS.
We thank Prof. Julie MacPherson (Warwick University) for enlightening discussions. We gratefully acknowledge financial support from the US Department of Energy, Office of Basic Energy Sciences (Grant No. DE-FG02-09ER16090). Sustained support from the Robert A. Welch Foundation (Grant F-0032) is also acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Dumitrescu I, Yancey DF, Crooks RM. Dual-electrode microfluidic cell for characterizing electrocatalysts. Lab Chip. 2012;12:986–993. doi: 10.1039/c2lc21181e. [DOI] [PubMed] [Google Scholar]
- 2.Pletcher D, Sotiropoulos SA. Study of cathodic oxygen reduction at platinum using microelectrodes. J Electroanal Chem. 1993;356:109–119. [Google Scholar]
- 3.Chen S, Kucernak A. Electrocatalysis under conditions of high mass transport rate: Oxygen reduction on single submicrometer-sized Pt particles supported on carbon. J Phys Chem B. 2004;108:3262–3276. [Google Scholar]
- 4.Colley AL, Macpherson JV, Unwin PR. Effect of high rates of mass transport on oxygen reduction at copper electrodes: Implications for aluminium corrosion. Electrochem Commun. 2008;10:1334–1336. [Google Scholar]
- 5.Levich VG. Physicochemical Hydrodynamics. 2nd Ed. Englewood Cliffs, NJ: Prentice Hall; 1962. [Google Scholar]
- 6.Yeager E. Electrocatalysts for O2 reduction. Electrochim Acta. 1984;29:1527–1537. [Google Scholar]
- 7.Yeager E. Dioxygen electrocatalysis: Mechanisms in relation to catalyst structure. J Mol Catal. 1986;38:5–25. [Google Scholar]
- 8.Kinoshita K. Electrochemical Oxygen Technology. New York: Wiley; 1992. [Google Scholar]
- 9.William M, Jai P. Fundamental Understanding of Electrode Processes in Memory of Professor Ernest B. Yeager. Pennington, NJ: The Electrochemical Society; 2003. [Google Scholar]
- 10.Savéant JM. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem Rev. 2008;108:2348–2378. doi: 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- 11.Wroblowa HS, Yen Chi P, Razumney G. Electroreduction of oxygen: A new mechanistic criterion. J Electroanal Chem Interfac Electrochem. 1976;69:195–201. [Google Scholar]
- 12.Costamagna P, Srinivasan S. Quantum jumps in the PEMFC science and technology from the 1960s to the year 2000: Part I. Fundamental scientific aspects. J Power Sources. 2001;102:242–252. [Google Scholar]
- 13.O’Hayre R, Suk-Won C, Colella W, Prinz FB. Fuel Cell Fundamentals. Hoboken, NJ: Wiley; 2006. [Google Scholar]
- 14.Marković NM, Schmidt TJ, Stamenković V, Ross PN. Oxygen reduction reaction on Pt and Pt bimetallic surfaces: A selective review. Fuel Cells. 2001;1:105–116. [Google Scholar]
- 15.Spendelow JS, Wieckowski A. Electrocatalysis of oxygen reduction and small alcohol oxidation in alkaline media. Phys Chem Chem Phys. 2007;9:2654–2675. doi: 10.1039/b703315j. [DOI] [PubMed] [Google Scholar]
- 16.Manthiram A, Vadivel Murugan A, Sarkar A, Muraliganth T. Nanostructured electrode materials for electrochemical energy storage and conversion. Energy Environ Sci. 2008;1:621–638. [Google Scholar]
- 17.Peng Z, Yang H. Designer platinum nanoparticles: Control of shape, composition in alloy, nanostructure and electrocatalytic property. Nano Today. 2009;4:143–164. [Google Scholar]
- 18.Gewirth AA, Thorum MS. Electroreduction of dioxygen for fuel-cell applications: Materials and challenges. Inorg Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]
- 19.Gasteiger HA, Panels JE, Yan SG. Dependence of PEM fuel cell performance on catalyst loading. J Power Sources. 2004;127:162–171. [Google Scholar]
- 20.Neyerlin KC, Gu W, Jorne J, Gasteiger HA. Study of the exchange current density for the hydrogen oxidation and evolution reactions. J Electrochem Soc. 2007;154:B631–B635. [Google Scholar]
- 21.Inaba M, Yamada H, Tokunaga J, Tasaka A. Effect of agglomeration of Pt/C catalyst on hydrogen peroxide formation. Electrochem Solid-State Lett. 2004;7:A474–A476. [Google Scholar]
- 22.Schneider A, et al. Transport effects in the oxygen reduction reaction on nanostructured, planar glassy carbon supported Pt/GC model electrodes. Phys Chem Chem Phys. 2008;10:1931–1943. doi: 10.1039/b719775f. [DOI] [PubMed] [Google Scholar]
- 23.Seidel YE, et al. Mesoscopic mass transport effects in electrocatalytic processes. Faraday Discuss. 2008;140:167–184. doi: 10.1039/b806437g. [DOI] [PubMed] [Google Scholar]
- 24.Borup R, et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem Rev. 2007;107:3904–3951. doi: 10.1021/cr050182l. [DOI] [PubMed] [Google Scholar]
- 25.Sethuraman VA, Weidner JW, Haug AT, Pemberton M, Protsailo LV. Importance of catalyst stability vis-à-vis hydrogen peroxide formation rates in PEM fuel cell electrodes. Electrochim Acta. 2009;54:5571–5582. [Google Scholar]
- 26.Ke K, Hatanaka T, Morimoto Y. Reconsideration of the quantitative characterization of the reaction intermediate on electrocatalysts by a rotating ring-disk electrode: The intrinsic yield of H2O2 on Pt/C. Electrochim Acta. 2011;56:2098–2104. [Google Scholar]
- 27.Ruvinskiy P, Bonnefont A, Savinova E. Further insight into the oxygen reduction reaction on Pt nanoparticles supported on spatially structured catalytic layers. Electrocatal. 2011;2:123–133. [Google Scholar]
- 28.Ruvinskiy PS, Bonnefont A, Pham-Huu C, Savinova ER. Using ordered carbon nanomaterials for shedding light on the mechanism of the cathodic oxygen reduction reaction. Langmuir. 2011;27:9018–9027. doi: 10.1021/la2006343. [DOI] [PubMed] [Google Scholar]
- 29.Birkin PR, Elliott JM, Watson YE. Electrochemical reduction of oxygen on mesoporous platinum microelectrodes. Chem Commun. 2000:1693–1694. [Google Scholar]
- 30.Fuhrmann J, et al. The role of reactive reaction intermediates in two-step heterogeneous electrocatalytic reactions: A model study. Fuel Cells. 2011;11:501–510. [Google Scholar]
- 31.Ye H, Crooks JA, Crooks RM. Effect of particle size on the kinetics of the electrocatalytic oxygen reduction reaction catalyzed by Pt dendrimer-encapsulated nanoparticles. Langmuir. 2007;23:11901–11906. doi: 10.1021/la702297m. [DOI] [PubMed] [Google Scholar]
- 32.Stamenkovic VM, Markovic N, Ross PN., Jr Structure-relationships in electrocatalysis: Oxygen reduction and hydrogen oxidation reactions on Pt(111) and Pt(100) in solutions containing chloride ions. J Electroanal Chem. 2001;500:44–51. [Google Scholar]
- 33.Antoine O, Durand R. RRDE study of oxygen reduction on Pt nanoparticles inside Nafion: H2O2 production in PEMFC cathode conditions. J Appl Electrochem. 2000;30:839–844. [Google Scholar]
- 34.Truesdale GA, Downing AL, Lowden GF. The solubility of oxygen in pure water and sea-water. J Appl Chem. 1955;5:53–62. [Google Scholar]
- 35.Zagal J, Bindra P, Yeager E. A mechanistic study of O2 reduction on water soluble phthalocyanines adsorbed on graphite electrodes. J Electrochem Soc. 1980;127:1506–1517. [Google Scholar]
- 36.Luo J, Njoki PN, Lin Y, Wang L, Zhong CJ. Activity-composition correlation of AuPt alloy nanoparticle catalysts in electrocatalytic reduction of oxygen. Electrochem Commun. 2006;8:581–587. [Google Scholar]
- 37.Chen W, Ny D, Chen S. SnO2-Au hybrid nanoparticles as effective catalysts for oxygen electroreduction in alkaline media. J Power Sources. 2010;195:412–418. [Google Scholar]
- 38.Hlushkou D, Perdue RK, Dhopeshwarkar R, Crooks RM, Tallarek U. Electric field gradient focusing in microchannels with embedded bipolar electrode. Lab Chip. 2009;9:1903–1913. doi: 10.1039/b822404h. [DOI] [PubMed] [Google Scholar]
- 39.Ye H, Crooks RM. Electrocatalytic O2 reduction at glassy carbon electrodes modified with dendrimer-encapsulated Pt nanoparticles. J Am Chem Soc. 2005;127:4930–4934. doi: 10.1021/ja0435900. [DOI] [PubMed] [Google Scholar]
- 40.Knecht MR, et al. Synthesis and characterization of Pt dendrimer-encapsulated nanoparticles: Effect of the template on nanoparticle formation. Chem Mater. 2008;20:5218–5228. [Google Scholar]
- 41.Myers VS, Frenkel AI, Crooks RM. In situ structural characterization of platinum dendrimer-encapsulated oxygen reduction electrocatalysts. Langmuir. 2012;28:1596–1603. doi: 10.1021/la203756z. [DOI] [PubMed] [Google Scholar]
- 42.Ye H, Scott RWJ, Crooks RM. Synthesis, characterization, and surface immobilization of platinum and palladium nanoparticles encapsulated within amine-terminated poly(amidoamine) dendrimers. Langmuir. 2004;20:2915–2920. doi: 10.1021/la0361060. [DOI] [PubMed] [Google Scholar]
- 43.Garsany Y, Baturina OA, Swider-Lyons KE, Kocha SS. Experimental methods for quantifying the activity of platinum electrocatalysts for the oxygen reduction reaction. Anal Chem. 2010;82:6321–6328. doi: 10.1021/ac100306c. [DOI] [PubMed] [Google Scholar]