

Abstract
Photoreduction of [P2W18O62]6-, [S2Mo18O62]4-, and [S2W18O62]4- polyoxometalate anions (POMs) and oxidation of water occurs when water–ionic liquid and water–diethylether interfaces are irradiated with white light (275–750 nm) or sunlight. The ionic liquids (ILs) employed were aprotic ([Bmim]X; Bmim = (1-butyl-3-methylimidazolium,X = BF4,PF6) and protic (DEAS = diethanolamine hydrogen sulphate; DEAP = diethanolamine hydrogen phosphate). Photochemical formation of reduced POMs at both thermodynamically stable and unstable water–IL interfaces led to their initial diffusion into the aqueous phase and subsequent extraction into the IL phase. The mass transport was monitored visually by color change and by steady-state voltammetry at microelectrodes placed near the interface and in the bulk solution phases. However, no diffusion into the organic phase was observed when [P2W18O62]6- was photo-reduced at the water–diethylether interface. In all cases, water acted as the electron donor to give the overall process: 4POM + 2H2O + hν → 4POM- + 4H+ + O2. However, more highly reduced POM species are likely to be generated as intermediates. The rate of diffusion of photo-generated POM- was dependent on the initial concentration of oxidized POM and the viscosity of the IL (or mixed phase system produced in cases in which the interface is thermodynamically unstable). In the water-DEAS system, the evolution of dioxygen was monitored in situ in the aqueous phase by using a Clark-type oxygen sensor. Differences in the structures of bulk and interfacial water are implicated in the activation of water. An analogous series of reactions occurred upon irradiation of solid POM salts in the presence of water vapor.
Keywords: electrochemistry, water oxidation
It has been known for some time that photochemical reduction of polyoxometalate anions (POMs) in molecular solvents may occur in the presence of an efficient electron donor such as 2-propanol or benzyl alcohol (1–6). In recent studies, we have found that photoreduction of tetracyanoquinodimethane (TCNQ) to TCNQ- and [P2W18O62]6- to [P2W18O62]7- or more extensively reduced POMs occurs in “wet” ionic liquids (ILs) where water acts as an electron donor and is photo-oxidized to dioxygen (7, 8). Intriguingly, these processes do not occur in wet molecular organic solvents or in neat water itself. The net reactions that describe the photo-irradiation of [P2W18O62]6- in wet 1-butyl-3-methylimidazolium tetrafluoroborate [Bmim][BF4] (Fig. S1) are summarized in Eqs. 1 and 2:
| [1] |
| [2] |
However, while [P2W18O62]7- is the detected product it is probable that more extensively reduced POMs are generated (8) as intermediates in the overall water oxidation reaction [Eq. 3]:
| [3] |
The modified structure of water present as a solute in ILs relative to that found in bulk water is postulated to facilitate H2O acting as an electron donor (7–10). However, while multistep two- and four-electron-proton-coupled reaction schemes have been postulated, full mechanistic details have yet to be established (8).
To date, photochemical reactions have rarely been carried out in ILs even though these low temperature molten salts that consist entirely of ionic species have emerged as an important class of solvent (11–18). Nevertheless, it is known that IL media can provide a favorable solvent polarity effect for photo-induced electron transfer (19). Both protic and aprotic ILs are often sparingly soluble or immiscible in water, so certain mixtures of ILs and water give rise to two phases. Furthermore, on examining ILs as a medium for photooxidation of water (7, 8), it was noted that even if an IL is fully miscible with water, the kinetics of its dissolution are often sufficiently slow that two phases may persist for long periods of time. Consequently, studies on the photochemistry at both thermodynamically stable and unstable water–IL interfaces should be possible in a regime in which significant structural kinetic and thermodynamic differences occur relative to bulk solvents. Consequently, photooxidation of water at interfaces may be even more favorable than in bulk wet ILs.
Solubility issues play an important role in determining which photoreactions may be studied at water–IL interfaces. Sodium and potassium salts of polyoxometalate anions are often very soluble in aqueous media. In contrast, tetraalkylammonium salts such as [Bu4N]6[P2W18O62] are sparingly soluble in water (or even insoluble) but are reasonably soluble in many ILs. However, dissolution of these salts in highly viscous aprotic ILs is often kinetically slow even though mM concentrations or above are thermodynamically accessible (20–22). Typically, reduced polyoxometalate anions are substantially more soluble in IL media. For example, one-electron reduced [P2W18O62]7- is highly soluble in many aprotic and protic ILs even in the presence of K+ counter ions (23, 24). It is apparent that a water–IL interface with a POM present in either phase can provide an environment where a variety of mass transport outcomes are possible.
ILs may act as both the solvent and electrolyte, so voltammetric detection of electroactive POM species is possible by careful placement of electrodes in the bulk solution and near the interface. A similar situation applies in an aqueous medium or organic solvent phases, provided an electrolyte is added. In principle, voltammetric monitoring of a two-phase system will allow the level of reactivity to be probed in both phases. An organic–aqueous interface provides another scenario to study photochemistry in which the structure of interfacial water is again expected to differ from that in bulk water. In this study we show that photooxidation of water in the presence of photoactive [P2W18O62]6-, [S2Mo18O62]4-, or [S2W18O62]4- anions occurs preferentially at water–IL and water–diethylether interfaces. We emphasize the use of electroanalytical techniques of transient and steady-state voltammetry to establish the POM redox level [Eq. 2] and to detect dioxygen and proton concentrations (Eq. 3; the Clark and pH electrodes).
Results and Discussion
As noted above, K6[P2W18O62] is highly soluble in water but has very limited solubility in the aprotic and protic ionic liquids used in this study. However, the solubility of the reduced species formed in photo-irradiation experiments (8) is significantly higher in the ILs. This is illustrated by cyclic voltammograms obtained when K6[P2W18O62] was adhered to a GC electrode and placed in contact with the IL DEAS (Fig. S1). Increased currents were detected upon repetitive cycling of the potential (Fig. S2), consistent with dissolution of the more soluble reduced salt of [P2W18O62]7- formed at the electrode surface. The mechanism appears to be analogous to that proposed for a POM-modified electrode in contact with [Bmim][PF6] (20) and is described by Eqs. 4 and 5:
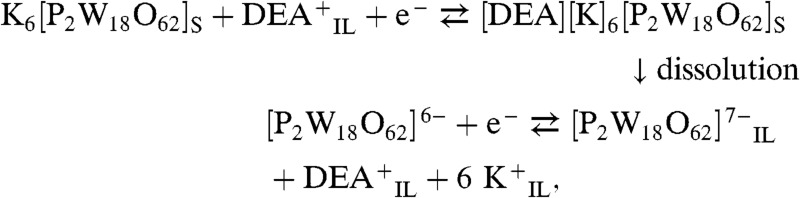 |
[4] |
| [5] |
Scanning to more negative potentials generates 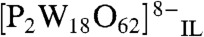 and more highly reduced forms [Eq. 5]. Formation of soluble reduced anions was confirmed visually by the appearance of a blue color in the IL near to the electrode surface.
and more highly reduced forms [Eq. 5]. Formation of soluble reduced anions was confirmed visually by the appearance of a blue color in the IL near to the electrode surface.
In order to ascertain if photoreduction of [P2W18O62]6- could occur at the thermodynamically unstable water–DEAS interface, photochemical experiments were initially undertaken on solutions contained in a soda glass vial with visual monitoring of solution color as a function of time (Fig. 1). In a previous study, it was shown that photoreduction of [P2W18O62]6- does not occur in either bulk water or bulk DEAS (8). In the present two-phase configuration, the upper phase was water, in which K6[P2W18O62] is highly soluble, and the lower phase was DEAS, in which the salt is essentially insoluble. Even though DEAS is highly miscible in water, the dissolution process (in the absence of convection) is slow, allowing a two-phase system to exist for more than 10 h. However, after sufficient time has elapsed, the upper phase can be described as water-rich and the lower one as DEAS-rich.
Fig. 1.

Photo-irradiation of a thermodynamically unstable two-phase water–DEAS system with a white light (275–750 nm) source located at a distance of 3 cm from the side of the soda glass vial (see Fig. 2). The initial aqueous phase contained K6[P2W18O62] (2.0 mM) and KCl (0.1 M). Irradiation time: (A) none; (B) 5; (C) 10; (D) 20; (E) 60 min.
To achieve the thermodynamically unstable two-phase condition, an aqueous solution containing [P2W18O62]6- was added slowly and dropwise onto the top of the DEAS phase in the soda glass vial using a Pasteur pipette whose tip rested against the edge of the vial, as shown in Fig. 2A. This approach minimized intermixing of the two solvents by convection: Miscibility was achieved only after standing for more than 10 h (predominantly by diffusion, a very slow process). Water has the lower viscosity (approximately 1 vs. > 4,356 mPa s) and density (1.00 vs. 1.21 g cm-3) (25–27). Thus, careful dropwise addition of the aqueous solution onto the IL surface was preferred to the alternative of addition of the IL onto water. The addition of water (5 mL) to DEAS (2 mL), in the manner described, leads to the formation of two phases, with the bottom one being the denser IL phase (Fig. 1A). Prior to irradiation with white light, cylic voltammetric monitoring of the [P2W18O62]6-/7- process (Fig. 3) confirmed that oxidized [P2W18O62]6- remained present in the aqueous phase and that its concentration in the DEAS phase was negligible (below the detection limit of 10 μM). Note that electrolyte KCl (0.1 M) was also present in the aqueous phase in experiments involving voltammetric monitoring of the aqueous phase. A single phase was formed rapidly when the two-phase system was shaken vigorously. It follows that partial mixing of the two neat liquids must occur in the interfacial region (Fig. 1) in a time-dependent manner.
Fig. 2.

Schematic representation of the cell arrangement for photochemical experiments at a water–IL interface. (A) Dropwise addition of the aqueous phase onto the IL phase; (B) in situ voltammetric monitoring.
Fig. 3.

Cyclic voltammograms (ν = 0.15 V s-1) obtained at a GC electrode before irradiation with white light in (A) the aqueous phase (K6[P2W18O62], 0.3 mM; KCl, 0.1 M) and (B) the DEAS phase of the thermodynamically unstable two-phase system.
In photochemical experiments, the initial water–DEAS two-phase system was irradiated from the side with white light (λ = 275–750 nm). The light source was located at a distance of 3 cm from the vial in a manner that ensured that both phases were exposed to the same light intensity (Fig. 2A). No change in temperature (± 0.1 °C) was detected in either phase during the course of this experiment. Very similar results were obtained using UV light (275–320 nm) because the [P2W18O62]6- anion only absorbs weakly in the visible wavelength region.
Initially, both phases were colorless. Five to 10 min of irradiation with white light generated blue coloration solely in the water–DEAS interfacial region (Fig. 1 B and C). The blue color is characteristic of reduced [P2W18O62]n- (n > 6). After 10 min, the color diffused slowly into the bulk water phase but not into the DEAS phase (Fig. 1C). Previously, photoreduction of [P2W18O62]6- was shown to occur in aqueous media containing DEAS concentrations≥0.08 M (8). It is likely that these conditions are present in the interfacial region in the present experiment. The time required for blue material to be detected in the aqueous phase decreased with increasing [P2W18O62]6- concentration (0.1 to 3 mM), consistent with the extent of photoreduction being dependent on the polyoxometalate anion concentration. After 20 min of irradiation, some blue material was seen to be transported into the DEAS phase (Fig. 1D). Continued photoreduction led to further diffusion of color into the IL phase until, after about 1 h, this phase was uniformly blue (Fig. 1E).
Steady-state microelectrode voltammetric measurements on the [P2W18O62]6-/7- couple were undertaken in the aqueous phase after 20 min of irradiation by using the photoelectrochemical cell shown in Fig. 2B. Results are displayed in Fig. 4. Close to the interface, the anion was found to be at the one electron-reduced redox level [P2W18O62]7-. In the bulk phase, [P2W18O62]7- was still the major component but with a significant concentration of [P2W18O62]6- still present. The magnitude of the limiting current was higher in the bulk aqueous solution than in the region just above the interface (Fig. 4). This reflects the higher concentration of viscous DEAS in the interfacial region and correspondingly lower diffusion coefficient values. The diffusion coefficient for [P2W18O62]6- in water–DEAS mixed solvent media is a function of DEAS concentration (Table S1).
Fig. 4.

Steady-state voltammograms (GC microelectrode; d = 15 μm); ν = 0.002 V s-1) obtained in the aqueous phase (K6[P2W18O62], 0.3 mM; KCl, 0.1 M) after 20 min of irradiation of the water–DEAS two-phase system. Voltammograms obtained just above the interface (‐‐‐) and in the bulk region (---) of the aqueous solution phase.
The mass transport process is driven by the differential solubilities and distribution coefficients of the reactant and product as well as their diffusion coefficients in the water and IL phases. K6[P2W18O62] is highly soluble in water but not in the IL; the reduced material is more soluble in the IL than in water. The overall mass transport observations are rationalized by the fact that the diffusion coefficient of [P2W18O62]7- in the IL is two orders of magnitude smaller than in molecular solvents (i.e., the product anion diffused into the aqueous phase prior to its extraction into the IL phase). Eventual transfer of [P2W18O62]7- to the IL layer was confirmed by cyclic voltammetric experiments (Fig. 5).
Fig. 5.

Cyclic voltammograms obtained at a GC working electrode (ν = 0.15 V s-1) in photo-irradiation experiments of a water–DEAS two-phase systems initially containing K6[P2W18O62] (0.2 mM) in the aqueous phase. The electrode was located in the DEAS ionic liquid phase (see Fig. 2B). (A) Before irradiation with white light; (B) after 2 h irradiation and (C) after 4 h irradiation. The increase in faradic current is primarily due to the transfer of photo-generated [P2W18O62]7- into the IL phase.
The production of dioxygen via Eq. 2 in the upper aqueous phase in the water–DEAS system was monitored by a Clark-type electrode located 1 cm above the interface. The aqueous phase containing [P2W18O62]6- was initially degassed for 10 min with dinitrogen to minimize the dioxygen concentration. This solution was then added to dinitrogen-degassed DEAS using the procedure described in Fig. 2A. The vial was then sealed from the atmosphere (8). No significant change in [O2] from the background level of about 120 μM was apparent initially but a significant increase occurred after about 6 min of irradiation with white light (Fig. 6). The delay is attributed to the time required for diffusion of O2 to the Clark electrode. After prolonged photolysis, the oxygen concentration reached a maximum value and then decreased slowly (Fig. 6). [P2W18O62]7- reacts very slowly with O2 (28) to regenerate [P2W18O62]6-. Clearly, if this reaction were fast, [P2W18O62]7- would not be detected. More highly reduced anions react rapidly with O2 so [P2W18O62]- is the “sink” (28).
Fig. 6.

Concentration of dioxygen in the aqueous phase in a sealed glass vial. Dioxygen was detected by a Clark-type electrode as a function of irradiation time during photoilumination with white light of the two-phase water–DEAS system. K6[P2W18O62] (0.3 mM) and KCl (0.1 M) was present in the initial aqueous phase.
Two-phase irradiation experiments were also carried out under the conditions described above but with [Bmim][PF6] as the IL. Water is only sparingly soluble in this aprotic IL and the system now represents a thermodynamically stable two-phase scenario. However, photoreduction again occurred preferentially at the interface with initial diffusion of [P2W18O62]7- into the aqueous phase followed by subsequent extraction into the IL phase.
In related experiments, two-phase systems were formed with acetonitrile (9% water) as the phase containing dissolved K6[P2W18O62], [Bu4N]4[S2W18O62] or [Bu4N]4[S2Mo18O62] and 0.1 M Bu4NPF6 as the supporting electrolyte. The IL phases tested were DEAS, DEAP, [Bmim][BF4], or [Bmim][PF6]. In all cases, photoreduction of the polyoxometalate anion occurred preferentially at the interface to produce [P2W18O62]7-, [S2Mo18O62]5-, or [S2W18O62]5- followed by diffusion into the water-acetonitrile phase (0.1 M Bu4NPF6, voltammetric detection). Again, at longer times, the reduced anions were transported into the IL phase. [S2Mo18O62]5- is further reduced to [S2Mo18O62]6- on longer light exposure times. Unlike the other POM anions, [S2Mo18O62]4- is photochemically reduced very slowly to [S2Mo18O62]5- in wet acetonitrile, confirming that the role of reversible potential also is significant (8, 28). In general terms the overall reaction is represented by Eq. 6:
| [6] |
The significance of the interfacial region was supported by data obtained in analogous experiments involving a thermodynamically stable (immiscible) aqueous-organic solvent two-phase system. A biphasic system consisting of an aqueous solution of K6[P2W18O62] (0.3 mM; 0.1 M KCl) in contact with diethyl ether phase was examined; on this occasion the diethyl ether phase is on top (ρ = 0.7134 vs. 1.0000 g cm-3 for water) (26). Irradiation with white light for 10 min gave the outcome shown in Fig. 7. Steady-state voltammograms were obtained with a GC microelectrode placed at three different locations (A–C) in the aqueous phase (Fig. 8). All voltammograms exhibit similar total limiting currents but reveal differences in the redox level in the three zones; the oxidation current is higher for the locations closer to the interface, indicating increased proportions of reduced anion [P2W18O62]7-. No extraction into the ether phase occurred in this system as the POM salts are insoluble in ether, irrespective of redox level.
Fig. 7.

Two phases water-diethylether system after irradiation with white light for 10 min. The ether phase is at the top with the aqueous phase at the bottom. K6[P2W18O62] (0.3 mM) and KCl (0.1 M) was present in the initial aqueous phase. Steady-state voltammograms were obtained at the indicated positions (A, B, C) in order to evaluate the redox level of the polyoxometalate after irradiation (see Fig. 8).
Fig. 8.

Steady-state voltammograms (GC microelectrode: d = 15 μm; ν = 0.002 V s-1) probing the redox level of [P2W18O62]6-/7- in the water-diethylether system after irradiation with white light for 10 min at three locations in the aqueous phase (see positions A–C in Fig. 7). K6[P2W18O62] (0.3 mM) and KCl (0.1 M) was present in the initial aqueous phase.
In addition, a decrease of pH from 6.35 to 3.90 occurred in region A during the experiment shown in Fig. 7. This is consistent with production of protons via Eq. 3. Dioxygen evolution was detected by a Clark-type electrode experiment. A control reaction involving irradiation of a suspension of K6[P2W18O62] in diethylether did not change color over 2 h. Photooxidation does not occur for the salt dissolved in neat water.
It is apparent that the interface between the liquid phases plays a key role in photoreduction of the POM anion. This was again apparent in the experiment of Fig. S3 where reduced anion can be detected at the water–diethylether interface after irradiation with sunlight for 3 min. Longer exposure times led to diffusion of reduced product into the aqueous phase. Equivalent changes occurred in the water–DEAS system upon exposure to sunlight; as with white light, diffusion into the aqueous phase occurred initially, then, after 4 h, the products extracted into the IL phase.
Photoreduction was also observed when solid polyoxometalate salts were in contact with water vapor. Soda glass vials containing powdered salts were used for these experiments. A micro droplet of water was adhered to the vial edge and immediately sealed from the atmosphere. It was placed on a hot plate so that evaporation of water occurred followed, upon cooling, by uniform condensation over the solid. Controls lacking the droplet and subjected to high vacuum were treated equivalently. The vials were exposed to sunlight (23–31 °C for 12 h per day) and the color changes monitored over a two-week period. Surface color changes to green/blue were seen after a few hours for (Bu4N)4[S2Mo18O62] and after a few days for K6[P2W18O62]. The controls exhibited no change in color. The observations are consistent with photoreduction of the salts and oxidation of water. Similar experiments under 2-propanol vapour produced color changes within 1 h.
This work demonstrated that photoreduction of [P2W18O62]6-, [S2Mo18O62]4-, and [S2W18O62]4- anions occurred at the interfaces of two-phase systems under conditions in which water acts as the electron donor. It is proposed that photo-oxidation of water is facilitated at an interface where the structure of water is different from that in neat H2O or when water is dissolved in a molecular organic solvent. In particular, hydrogen bonding is expected to be weaker when water is present in the gas phase or at an interface. While the electrochemical methodology used in this study provides qualitative information on the overall reaction, quantitative photochemical data and the photophysics of the reactions are yet to be established. Thus, while Eq. 6 is written as an overall one-electron reduction step with formation of final product POM-, it is probable that multi-electron reduced anions are produced as intermediate photo products arising directly from oxidation of water. They would then be expected to react with oxidized anion, H+, or O2 to generate POM- as the detected final product (8).
Quantum yields, ground and excited state potentials, plus the activity of H+ at the interface are needed to quantify the photochemistry. The present study raises a series of important photochemical, photophysical, and interfacial questions that are in general beyond the scope of electrochemical methodology. It is relevant that studies in acid-base behavior and catalysis in emulsions have also revealed the role that differences in structure play in the chemistry of water at interfaces (29–32).
Materials and Methods
Reagents.
K6[α-P2W18O62]14H2O, [Bu4N]4[α-S2W18O62], and [Bu4N]4[α-S2Mo18O62] were synthesized according to literature procedures (28, 33–35). The aprotic ILs [Bmim][BF4] and [Bmim][PF6] (Merck, high purity grade, ≥99.0%) were used as received. The protic ionic liquids DEAS and DEAP were synthesized and purified according to published procedures (36). Structures of all ILs are provided in Fig. S1.
Electrochemistry.
In most cases, the POM salt was dissolved in the solvent of interest. Voltammetric experiments in such single-phase solutions were conducted at (20 ± 1) °C according to the procedure described before (8). For electrochemistry in two-phase environments, a specially designed electrochemical cell arrangement was developed for voltammetric monitoring of reactions taking place upon irradiation with light in a two-phase water/IL system (Fig. 2B). Detailed electrochemical precedures are present in SI Text.
Photochemistry.
Both phases and the interfacial regions of aqueous–IL or aqueous–diethylether systems were irradiated with white light (275–750 nm) using a Rofin Polilight PL6 Xe source placed 3 cm from the size wall of the soda glass vial (see Fig. 2A). The oxygen concentration and pH values in aqueous media were estimated with a Clark-type gas sensor and a pH Meter. Further details of the light source, Clark sensor, and pH Meter are provided in ref. 8.
Supplementary Material
ACKNOWLEDGMENTS.
The authors gratefully acknowledge financial support from the Australian Research Council.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203818109/-/DCSupplemental.
References
- 1.Argitis P, Papaconstantinou E. Photocatalytic multielectron photoreduction of 18-tungstodiphosphate in the presence of organic compounds—production of hydrogen. J Photochem. 1985;30:445–451. [Google Scholar]
- 2.Ward MD, Brazdil JF, Grasselli RK. Photocatalytic alcohol dehydrogenation using ammonium heptamolybdate. J Phys Chem. 1984;88:4210–4213. [Google Scholar]
- 3.Hill CL, Bouchard DA. Catalytic photochemical dehydrogenation of organic substrates by polyoxometalates. J Am Chem Soc. 1985;107:5148–5157. [Google Scholar]
- 4.Papaconstantinou E, Dimotikali D, Politou A. Photochemistry of heteropoly electrolytes. The 18-molybdodiphosphate. Inorg Chim Acta. 1980;43:155–158. [Google Scholar]
- 5.Yamase T, Takabayashi N, Kaji M. Solution photochemistry of tetrakis(tetrabutylammonium) decatungstate(VI) and catalytic hydrogen evolution from alcohols. J Chem Soc, Dalton Trans. 1984:793–799. [Google Scholar]
- 6.Papaconstantinou E. Photochemistry of polyoxometallates of molybdenum and tungsten and/or vanadium. Chem Soc Rev. 1989;18:1–31. [Google Scholar]
- 7.Zhao C, Bond AM. Photoinduced oxidation of water to oxygen in the ionic liquid BMIMBF4 as the counter reaction in the fabrication of exceptionally long semiconducting silver-Tetracyanoquinodimethane nanowires. J Am Chem Soc. 2009;131:4279–4287. doi: 10.1021/ja806893t. [DOI] [PubMed] [Google Scholar]
- 8.Bernardini G, Zhao C, Wedd AG, Bond AM. Ionic liquid-enhanced photooxidation of water using the polyoxometalate anion [P2W18O62]6- as the sensitizer. Inorg Chem. 2011;50:5899–5909. doi: 10.1021/ic1016627. [DOI] [PubMed] [Google Scholar]
- 9.Menjoge A, Dixon J, Brennecke JF, Maginn EJ, Vasenkov S. Influence of water on diffusion in imidazolium-based ionic liquids: A pulsed field gradient NMR study. J Phys Chem B. 2009;113:6353–6359. doi: 10.1021/jp900902n. [DOI] [PubMed] [Google Scholar]
- 10.Raju SG, Balasubramanian S. Aqueous solution of [bmim][PF6]: Ion and solvent effects on structure and dynamics. J Phys Chem B. 2009;113:4799–4806. doi: 10.1021/jp8111777. [DOI] [PubMed] [Google Scholar]
- 11.Stark A, MacLean BL, Singer RD. 1-Ethyl-3-methylimidazolium halogenoaluminate ionic liquids as solvents for Friedel-Crafts acylation reactions of ferrocene. J Chem Soc, Dalton Trans. 1999:63–66. [Google Scholar]
- 12.Earle MJ, Seddon KR, Adams CJ, Roberts G. Friedel-Crafts reactions in room temperature ionic liquids. Chem Commun. 1998:2097–2098. [Google Scholar]
- 13.Fischer T, Sethi A, Welton T, Woolf J. Diels-Alder reactions in room-temperature ionic liquids. Tetrahedron Lett. 1999;40:793–796. [Google Scholar]
- 14.Earle MJ, McCormac PB, Seddon KR. Regioselective alkylation in ionic liquids. Chem Commun. 1998:2245–2246. [Google Scholar]
- 15.Monteiro AL, Zinn FK, de Souza RF, Dupont J. Asymmetric hydrogenation of 2-arylacrylic acids catalyzed by immobilized Ru-BINAP complex in 1-n-butyl-3-methylimidazolium tetrafluoroborate molten salt. Tetrahedron: Asymmetry. 1997;8:177–179. [Google Scholar]
- 16.Simon LC, Dupont J, de Souza RF. Two-phase n-butenes dimerization by nickel complexes in molten salt media. Appl Catal A: Gen. 1998;175:215–220. [Google Scholar]
- 17.Dullius JEL, et al. Selective catalytic hydrodimerization of 1,3-butadiene by palladium compounds dissolved in ionic liquids. Organometallics. 1998;17:815–819. [Google Scholar]
- 18.Chauvin Y, Olivier H, Wyrvalski CN, Simon LC, de Souza RF. Oligomerization of n-Butenes catalyzed by nickel complexes dissolved in organochloroaluminate ionic liquids. J Catal. 1997;165:275–278. [Google Scholar]
- 19.Kavarnos GJ, Turro NJ. Photosensitization by reversible electron transfer: Theories, experimental evidence, and examples. Chem Rev. 1986;86:401–449. [Google Scholar]
- 20.Zhang J, et al. Voltammetric Studies on the reduction of polyoxometalate anions in ionic liquids. Inorg Chem. 2005;44:5123–5132. doi: 10.1021/ic050032t. [DOI] [PubMed] [Google Scholar]
- 21.Bond AM. Broadening Electrochemical Horizons. Oxford, UK: Oxford Univ Press; 2002. [Google Scholar]
- 22.Scholz F, Meyer B. Electroanalytical Chemistry. New York: Marcel Dekker; 1998. [Google Scholar]
- 23.Zhang J, Bond AM. Practical considerations associated with voltammetric studies in room temperature ionic liquids. Analyst. 2005;130:1132–1147. doi: 10.1039/b504721h. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Bond AM, Richardt PJS, Wedd AG. Voltammetric reduction of α- and γ∗-[S2W18O62]4- and α-, β-, and γ-[SiW12O40]4-: Isomeric dependence of reversible potentials of polyoxometalate anions using data obtained by novel dissolution and conventional solution-phase processes. Inorg Chem. 2004;43:8263–8271. doi: 10.1021/ic049043x. [DOI] [PubMed] [Google Scholar]
- 25.Sawyer DT, Sobkowiak A, Roberts JL. Electrochemistry for Chemists. 2nd Ed. New York: Wiley; 1995. [Google Scholar]
- 26.Berstad DA, et al. Accurate measurements of the viscosity of water in the temperature range 19.5–25.5 °C. Physica A. 1988;151:246–280. [Google Scholar]
- 27.Lide DR. CRC Handbook of Chemistry and Physics. 70th Ed. Boca Raton, FL: CRC Press; 1990. [Google Scholar]
- 28.Bernardini G, Wedd AG, Bond AM. Reactivity of one-, two-, three- and four-electron reduced forms of α-[P2W18O62]6- generated by controlled potential electrolysis in water. Inorg Chim Acta. 2011;374:327–333. [Google Scholar]
- 29.Cooper JB, Way DM, Bond AM, Wedd AG. A green heteropoly blue: Isolation of a stable, odd oxidation level in a Dawson molybdate anion, [S2Mo18O62]5- Inorg Chem. 1993;32:2416–2420. [Google Scholar]
- 30.Narayan S, et al. “On Water”: Unique reactivity of organic compounds in aqueous suspension. Angew Chem Int Ed Engl. 2005;117:3219–3219. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- 31.Gray-Weale A, Beattie JK. An explanation for the charge on water’s surface. Phys Chem Chem Phys. 2009;11:10994–11005. doi: 10.1039/b901806a. [DOI] [PubMed] [Google Scholar]
- 32.Beattie JK, McErlean CSP, Phippen CBW. The mechanism of on-water catalysis. Chem Eur J. 2010;16:8972–8974. doi: 10.1002/chem.201001705. [DOI] [PubMed] [Google Scholar]
- 33.Mbomekalle I-M, Lu YW, Keita B, Nadjo L. Simple, high yield and reagent-saving synthesis of pure α-K6P2W18O62•14H2O. Inorg Chem Commun. 2004;7:86–90. [Google Scholar]
- 34.Graham CR, Finke RG. The classic Wells–Dawson polyoxometalate, K6[α-P2W18O62]•14H2O Answering an 88 year-old question: What is its preferred, optimum synthesis? Inorg Chem. 2008;47:3679–3686. doi: 10.1021/ic702295y. [DOI] [PubMed] [Google Scholar]
- 35.Richardt PJS, Gable RW, Bond AM, Wedd AG. Synthesis and redox characterization of the polyoxo anion, γ∗-[S2W18O62]4-: A unique fast oxidation pathway determines the characteristic reversible electrochemical behavior of polyoxometalate anions in acidic media. Inorg Chem. 2001;40:703–709. doi: 10.1021/ic000793q. [DOI] [PubMed] [Google Scholar]
- 36.Zhao C, et al. Electrochemistry of room temperature protic ionic liquids. J Phys Chem B. 2008;112:6923–6936. doi: 10.1021/jp711804j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.