

Abstract
The utilization of sequential palladium-catalyzed α-arylation and cyclization reactions provides a general approach to an array of isoquinolines and their corresponding N-oxides. This methodology allows the convergent combination of readily available precursors in a regioselective manner and in excellent overall yields. This powerful route to polysubstituted isoquinolines, which is not limited to electron rich moieties, also allows rapid access to analogues of biologically active compounds.
Keywords: alpha arylation, heterocycle, isoquinoline-N-oxide, one-pot
The isoquinoline motif and its derivatives form the cores of numerous natural products (1, 2), are the central components of a number of pharmaceutical agents (3–5), and can provide the scaffold for chiral ligands (6, 7) and valuable organic materials (8). However, traditional isoquinoline syntheses such as the Bischler-Napieralski (9, 10), Pictet-Spengler (11, 12), and Pomeranz-Fritsch reactions (13–15) all centre around the lynchpin of electrophilic aromatic substitution and are thus often limited to electron-rich carbocycles (Scheme 1A). It is also imperative to develop routes to highly-substituted isoquinolines to fully explore this chemical space, for example, for potential pharmaceutical targets. While recent synthetic efforts have greatly expanded the diversity of isoquinoline motifs available (16–29), new routes to isoquinolines are still highly desirable, particularly ones with the ability to directly access the isoquinoline moiety in a range of oxidation levels and which do not require highly-specialized starting materials.
Scheme 1.

Retrosynthetic Strategy.
Originally reported by Palucki and Buchwald (30), Hamann and Hartwig (31), and Miura and coworkers (32), the palladium-catalyzed α-arylation of enolates has recently emerged as a powerful new reaction in synthetic organic chemistry (33–35). Key to its success has been the design of appropriate ligands for palladium that have precisely engineered steric and electronic properties to enable the various steps in the catalytic cycle to proceed with maximum efficiency (36–39). The reaction’s utility has also been greatly enhanced by the development of robust preformed palladium catalysts. Properties such as air stability and very high reactivity in a range of systems have greatly expanded the potential uses of these catalysts (40–43). However, the α-arylation reaction has seen limited use in the de novo construction of aromatic compounds (44–51) and is still greatly underused in this regard, especially because the bond forming abilities provided by this powerful reaction can greatly simplify the design of routes to heavily-substituted aromatic compounds. Hence, we decided to explore the scope of this new catalytic method in the synthesis of important heteroaromatic ring systems, beginning our investigations with the isoquinoline nucleus.
Results
In our disconnection approach we envisaged that key pseudo-1,5-dicarbonyl intermediates 3 could be accessed via the palladium-catalyzed α-arylation of ketones 1 with aryl halides 2 possessing a protected aldehyde or ketone in the ortho-position (52). Subsequent treatment of these intermediates 3 with an acidic ammonium source would lead to concomitant acetal deprotection and aromatization to the corresponding isoquinolines 4. The attractiveness of this route stemmed from both its potential for regioselective installation of substituents at all positions on the isoquinoline nucleus and the fact that the coupling precursors were either commercially available or could be synthesized in a short sequence (Scheme 1B).
Initial optimization of the α-arylation conditions for the reaction of ketone 1a with bromide 2a (two commercially available substrates) showed that a catalyst loading of 2.0 mol% (DtBPF)PdCl2 or PdCl2(Amphos)2 was sufficient to obtain high yields of the arylated product 3a (Entries 3, 10, Table 1). Yields could be further increased when 5.0 mol% of catalyst and 200 mol% of ketone were used. Furthermore, the reaction was shown to proceed well with a decreased catalyst loading of 0.5 mol% (Entries 1, 2). As expected, iodide 2b showed comparable reactivity to bromide 2a under these conditions. Chloride 2c could also be employed as the aryl partner in the coupling reaction (Entry 9); this was a significant development, as the requisite aryl chlorides are both considerably cheaper than the corresponding aryl bromides and are commercially available in a greater variety of substitution patterns. As the aryl bromides presented the best compromise of reactivity and availability, these were used for further exemplification of the methodology.
Table 1.
Optimization of the arylation conditions
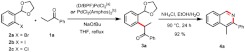 | |||||
| Entry | X | Cat. loading [mol%] | Ketone [mol%] | Time [h] | Yield [%] |
| 1 | Br | 0.5[a] | 120 | 18 | 71 |
| 2 | Br | 0.5[a] | 200 | 18 | 74 |
| 3 | Br | 2.0[a] | 120 | 18 | 82 |
| 4 | Br | 2.0[a] | 200 | 18 | 83 |
| 5 | Br | 5.0[a] | 200 | 18 | 89 |
| 6 | I | 2.0[a] | 120 | 18 | 79 |
| 7 | Cl | 5.0[a] | 200 | 18 | 30 |
| 8 | Cl | 5.0[b] | 200 | 18 | 45 |
| 9 | Cl | 5.0[b] × 2 | 200 | 96 | 74 |
| 10 |
Br |
2.0[b] |
120 |
18 |
82 |
For the final aromatization step, solutions of a variety of ammonium salts in EtOH/H2O were screened. The solutions needed to be of a certain acidity (approximately pH 5) to effect acetal hydrolysis at reasonable rates. It was found that heating intermediate 3a in a solution of ammonium chloride (1 M in 3∶1 EtOH/H2O) mediated deprotection and cyclization in excellent yield.
To examine the scope of this reaction, the substituents that could be tolerated during the arylation step and would be positioned on the heterocyclic ring of the resulting isoquinoline were investigated (Scheme 2). A range of ketones were arylated in good to excellent yields (68–92%) and a wide array of substituents was tolerated at the position to become C(3) on the isoquinoline; intermediates 3a,b,c,f,i with aryl, 3j with heteroaryl, and 3d,e,g,h with alkyl substitution. The regioselectivity of the arylation reaction enabled the regio-defined construction of the intermediate 3h where there was a choice of enolizable protons on the ketone. It was also pleasing to note that intermediate 3e, bearing a bulky quaternary substituent, could be synthesized. Even greater flexibility was achievable at the position to become C(4) on the isoquinoline and arylation reactions to produce intermediates 3a,d,f,g with alkyl, 3i with aryl, and 3c with heteroatom substitution proceeded efficiently. In the bromide coupling partner, both protected aldehydes and ketones were tolerated in the arylation reaction, enabling the construction of intermediates 3a–e where R1 = H and 3f–j where R1 = Me.
Scheme 2.

Exploring the scope of substituents R1, R3, and R4.
The subsequent deprotection and cyclization to produce isoquinolines 4a–e where R1 = Me also proceeded in excellent yields (79–99%). For intermediates 3f–j where R1 = Me, we noted that while deprotection was facile under the ammonium chloride conditions, the ensuing cyclization was sluggish. It was found, however, that following complete acetal hydrolysis, subsequent basification (to approximately pH 9) with ammonium bicarbonate solution (2 M in H2O) promoted cyclization of these substrates. Under these conditions, cyclization proceeded in good to excellent yields (81–93%) to produce isoquinolines 4f–j.
Our investigation of the scope of substitution possible on the carbocyclic ring involved varying the substituents on the benzene ring of the aryl bromide partner (when the required aryl bromide acetal is not itself a commercially available compound, it can generally be prepared in one step and at > 90% yield) (Scheme 3). The synthesis of isoquinolines with a wide variety of substitution was possible, covering all four positions, and including 4k,l,m,o,r with heteroatom, 4n with alkyl, 4p with aryl, and 4q,s with functionalized carbon substituents. The arylations proceeded in good to excellent yields (69–96%), as did the cyclizations (73–97%). Of particular note, isoquinolines 4p,r bearing a group at the C(5) position could be synthesized from sterically-hindered bromides. The use of milder bases in the arylation step allowed sensitive functionality such as a nitro group and a methyl ester to be carried through the synthesis (isoquinolines 4k,q). This approach could be used to synthesize isoquinolines 4k,l,m,q,s with electron poor and isoquinolines 4n,o,p,r with electron rich carbocyclic rings, confirming the wide applicability of this synthetic strategy—flexibility that is not offered by many conventional approaches. The benzene ring could also be replaced with a heteroarene to synthesize thieno-pyridine 4t, illustrating our ability to construct multiple heteroatom-containing ring systems, which are attractive pharmaceutical targets.
Scheme 3.

Exploring the scope of substituents R5, R6, R7, and R8.
The miscibility of THF with EtOH and H2O prompted investigation into a one-pot procedure for an arylation/cyclization sequence. After the arylation was complete, the reaction mixture was acidified to pH 5 with aqueous 1 M HCl and then the cyclization was carried out by the addition of the nitrogen source as previously described. The sequential one-pot protocol worked well for a number of substrates without significantly affecting the yield. This greatly improved the practicability of this synthetic procedure, and isoquinolines 4a and 4b, for example, could now be synthesized in one step from commercial materials (Scheme 4).
Scheme 4.

A One-pot protocol.
Whilst the majority of other isoquinoline syntheses can only give access to the isoquinoline manifold in one oxidation level, a strength of this method is that by variation of one of the reaction components, more oxidized isoquinoline scaffolds are easily accessible. The direct synthesis of isoquinoline N-oxides 5 was achievable by replacing the ammonium chloride in the cyclization step with the hydrochloride salt of hydroxylamine (Scheme 5). Conversion of the arylated intermediates 3 to the N-oxides 5 occurred more rapidly (2 h) than to the corresponding isoquinolines 4 and in excellent yields (86–99%). As a number of subsequent functionalization reactions on isoquinolines are performed via their N-oxides (53–56), we felt that this powerful and direct approach to their synthesis would prove extremely useful. In particular, it gives the potential to subsequently install a variety of substituents at C(1) without the need for long syntheses of complicated aryl halide precursors. Also, alternative procedures involving direct oxidations of isoquinolines to their N-oxides can have limited functional group tolerance and use a stoichiometric equivalent of an oxidizing agent in the process.
Scheme 5.

Isoquinoline N-oxides.
This de novo isoquinoline synthesis has the ability to construct complex isoquinoline scaffolds in a rapid and modular fashion. The simplicity and mild conditions of the one-pot procedure make it ideal for the manipulation of existing natural product frameworks to create libraries of potentially biologically active targets for medicinal screening. The addition of a basic isoquinoline nitrogen imparts a site on a molecule for varying lipophilicity, an essential consideration in lead optimisation (57). As many potent pharmaceuticals are derivatives of naturally-occurring steroidal hormones, we chose to exemplify this concept on derivatives of the key female sex hormone estrone and the testosterone metabolite androsterone. Application of the one-pot procedure enabled the synthesis of isoquinolines 8 and 9 in 63 and 59% yields, respectively, using commercially available bromide 2a (Scheme 6).
Scheme 6.

Derivatizing estrone and androsterone.
Conclusions
In summary, the utilization of sequential palladium-catalyzed α-arylation and cyclization reactions provides a general approach to an array of substituted isoquinolines and their corresponding N-oxides. The methodology presented here allows the convergent combination of readily-available precursors in a regioselective manner and in excellent overall yields (often > 70% from commercially available precursors). This powerful route to polysubstituted isoquinolines, which works equally well for both electron poor and electron rich moieties, also allows rapid access to analogues of biologically active compounds.
Methods
Full experimental details and compound characterization data are included in the SI Appendix.
General Procedure for the Palladium-Catalyzed α-Arylation Reaction.
A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. The palladium catalyst (2.0 or 5.0 mol%) and base (250 mol%) were added to the tube. The aryl halide (100 mol%) was dissolved in dry THF (5 mL mmol-1 substrate) and the resulting solution was added via syringe to the tube. The ketone (120 or 200 mol%) was then added via syringe to the tube. The rubber septum was replaced with a screw cap and the tube was heated at 70 °C for 18 h. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography furnished the requisite intermediate.
General Procedure for Isoquinoline Formation Where R1 = H.
A solution of NH4Cl (1000 mol%, 1.0 M in 3∶1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 24 h. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NaHCO3 (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography furnished the requisite isoquinoline.
General Procedure for Isoquinoline Formation Where R1 = Me.
A solution of NH4Cl (1000 mol%, 1.0 M in 3∶1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 18 h. A solution of NH4HCO3 (2.0 M in H2O) was then added until the pH of the reaction mixture had been adjusted to approximately pH 9. The tube was resealed and heated for a further 24 h at 90 °C. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography furnished the requisite isoquinoline.
Supplementary Material
ACKNOWLEDGMENTS.
We would like to thank Johnson Matthey for their donation of palladium catalysts and St. John’s College, Oxford University for supporting this project.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206532109/-/DCSupplemental.
References
- 1.Bentley KW. β-Phenylethylamines and the isoquinoline alkaloids. Nat Prod Rep. 2006;23:444–463. doi: 10.1039/b509523a. [DOI] [PubMed] [Google Scholar]
- 2.Bentley KW. The Isoquinoline Alkaloids. Amsterdam: Harwood Academic Publishers; 1998. [Google Scholar]
- 3.Pike VW, et al. Radioligands for PET studies of central benzodiazepine receptors and PK (peripheral benzodiazepine) binding sites-current status. Nucl Med Biol. 1993;20:503–525. doi: 10.1016/0969-8051(93)90082-6. [DOI] [PubMed] [Google Scholar]
- 4.Weissman BA, Raveh L. Peripheral benzodiazepine receptors: On mice and human brain imaging. J Neurochem. 2003;84:432–437. doi: 10.1046/j.1471-4159.2003.01568.x. [DOI] [PubMed] [Google Scholar]
- 5.Rinehart KL. Antitumor compounds from tunicates. Med Res Rev. 2000;20:1–27. doi: 10.1002/(sici)1098-1128(200001)20:1<1::aid-med1>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.Lim CW, et al. Practical preparation and resolution of 1-(2′-diphenylphosphino-1′-naphthyl)isoquinoline: A useful ligand for catalytic asymmetric synthesis. Org Process Res Dev. 2003;7:379–384. [Google Scholar]
- 7.Alcock NW, Brown JM, Hulmes GI. Synthesis and resolution of 1-(2-diphenylphosphino-1-naphthyl)isoquinoline; A P-N chelating ligand for asymmetric catalysis. Tetrahedron Asymmetry. 1993;4:743–756. [Google Scholar]
- 8.Tsuboyama A, et al. Homoleptic cyclometalated iridium complexes with highly efficient red phosphorescence and application to organic light-emitting diode. J Am Chem Soc. 2003;125:12971–12979. doi: 10.1021/ja034732d. [DOI] [PubMed] [Google Scholar]
- 9.Bischler A, Napieralski B. A new method for the synthesis of isoquinolines. Chem Ber. 1893;26:1903–1908. [Google Scholar]
- 10.Whaley WM, Govindachari TR. In: Organic Reactions. Adams R, editor. Vol. 6. New York: Wiley; 1951. pp. 74–150. [Google Scholar]
- 11.Pictet A, Spengler T. Formation of isoquinoline derivatives by the action of methylal on phenylethylamine, phenylalanine and tyrosine. Chem Ber. 1911;44:2030–2036. [Google Scholar]
- 12.Whaley WM, Govindachari TR. In: Organic Reactions. Adams R, editor. Vol. 6. New York: Wiley; 1951. pp. 151–190. [Google Scholar]
- 13.Pomeranz C. A new isoquinoline synthesis. Monatsh Chem. 1893;14:116–119. [Google Scholar]
- 14.Fritsch P. Syntheses in the isocoumarin and isoquinoline series. Chem Ber. 1893;26:419–422. [Google Scholar]
- 15.Gensler WJ. In: Organic Reactions. Adams R, editor. Vol. 6. New York: Wiley; 1951. pp. 191–206. [Google Scholar]
- 16.Jayakumar J, Parthasarathy K, Cheng C. One-pot synthesis of isoquinolinium salts by rhodium-catalyzed C-H bond activation: Application to the total synthesis of oxychelerythrine. Angew Chem Int Ed Engl. 2012;51:197–200. doi: 10.1002/anie.201105755. [DOI] [PubMed] [Google Scholar]
- 17.Too PC, Chua SH, Wong SH, Chiba S. Synthesis of azaheterocycles from aryl ketone O-acetyl oximes and internal alkynes by Cu-Rh bimetallic relay catalysts. J Org Chem. 2011;76:6159–6168. doi: 10.1021/jo200897q. [DOI] [PubMed] [Google Scholar]
- 18.Si C, Myers AG. A versatile synthesis of substituted isoquinolines. Angew Chem Int Ed Eng. 2011;50:10409–10413. doi: 10.1002/anie.201104769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaumontet M, Piccardi R, Baudoin O. Synthesis of 3,4-dihydroisoquinolines by a C(sp3)-H activation/electrocyclization strategy: Total synthesis of coralydine. Angew Chem Int Ed Eng. 2009;48:179–182. doi: 10.1002/anie.200804444. [DOI] [PubMed] [Google Scholar]
- 20.Chiba S, Xu Y, Wang Y. A Pd(II)-catalyzed ring-expansion reaction of cyclic 2-azidoalcohol derivatives: Synthesis of azaheterocycles. J Am Chem Soc. 2009;131:12886–12887. doi: 10.1021/ja9049564. [DOI] [PubMed] [Google Scholar]
- 21.Guimond N, Fagnou K. Isoquinoline synthesis via rhodium-catalyzed oxidative cross-coupling/cyclization of aryl aldimines and alkynes. J Am Chem Soc. 2009;131:12050–12051. doi: 10.1021/ja904380q. [DOI] [PubMed] [Google Scholar]
- 22.Sha F, Huang X. A multicomponent reaction of arynes, isocyanides, and terminal alkynes: Highly chemo- and regioselective synthesis of polysubstituted pyridines and isoquinolines. Angew Chem Int Ed Eng. 2009;48:3458–3461. doi: 10.1002/anie.200900212. [DOI] [PubMed] [Google Scholar]
- 23.Gilmore CD, Allan KM, Stoltz BM. Orthogonal synthesis of indolines and isoquinolines via aryne annulation. J Am Chem Soc. 2008;130:1558–1559. doi: 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Lu B, Jiang Y, Zhang Y, Ma D. Assembly of isoquinolines via CuI-catalyzed coupling of β-Keto esters and 2-halobenzylamines. Org Lett. 2008;10:2761–2763. doi: 10.1021/ol800900a. [DOI] [PubMed] [Google Scholar]
- 25.Fischer D, et al. Iodine-mediated electrophilic cyclization of 2-alkynyl-1-methylene azide aromatics leading to highly substituted isoquinolines and its application to the synthesis of norchelerythrine. J Am Chem Soc. 2008;130:15720–15725. doi: 10.1021/ja805326f. [DOI] [PubMed] [Google Scholar]
- 26.Huo Z, Tomeba H, Yamamoto Y. Iodine-mediated electrophilic cyclization of 2-alkynylbenzaldoximes leading to the formation of iodoisoquinoline N-oxides. Tetrahedron Lett. 2008;49:5531–5533. [Google Scholar]
- 27.Fischer D, Tomeba H, Pahadi NK, Patil NT, Yamamoto Y. Synthesis of 1,3,4-trisubstituted isoquinolines by iodine-mediated electrophilic cyclization of 2-alkynyl benzyl azides. Angew Chem Int Ed Eng. 2007;46:4764–4766. doi: 10.1002/anie.200701392. [DOI] [PubMed] [Google Scholar]
- 28.Xiang Z, et al. Concise synthesis of isoquinoline via the Ugi and Heck reactions. Org Lett. 2004;6:3155–3158. doi: 10.1021/ol048791n. [DOI] [PubMed] [Google Scholar]
- 29.Roesch KR, Zhang H, Larock RC. Synthesis of isoquinolines and pyridines by the palladium-catalyzed iminoannulation of internal alkynes. J Org Chem. 2001;66:8042–8051. doi: 10.1021/jo0105540. [DOI] [PubMed] [Google Scholar]
- 30.Palucki M, Buchwald SL. Palladium-catalyzed α-arylation of ketones. J Am Chem Soc. 1997;119:11108–11109. [Google Scholar]
- 31.Hamann BC, Hartwig JF. Palladium-catalyzed direct α-arylation of ketones: Rate acceleration by sterically hindered chelating ligands and reductive elimination from a transition metal enolate complex. J Am Chem Soc. 1997;119:12382–12383. [Google Scholar]
- 32.Satoh T, Kawamura Y, Miura M, Nomura M. Palladium-catalyzed regioselective mono- and diarylation reactions of 2-phenylphenols and naphthols with aryl halides. Angew Chem Int Ed Eng. 1997;36:1740–1742. [Google Scholar]
- 33.Bellina F, Rossi R. Transition metal-catalyzed direct arylation of substrates with activated sp3-hybridized C-H bonds and some of their synthetic equivalents with aryl halides and pseudohalides. Chem Rev. 2010;110:1082–1146. doi: 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]
- 34.Johansson CCC, Colacot TJ. Metal-catalyzed α-arylation of carbonyl and related molecules: Novel trends in C–C bond formation by C–H bond functionalization. Angew Chem Int Ed Eng. 2010;49:676–707. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]
- 35.Novák P, Martin R. Pd-catalyzed α-arylation of carbonyl and related compounds: Recent developments and perspectives. Curr Org Chem. 2011;15:3233–3262. [Google Scholar]
- 36.Cullen WR, Kim TJ, Einstein FWB, Jones T. Structure of the hydrogenation catalyst [(PP)Rh(NBD)]ClO4, PP = (η5-(Me3C)2PC5H4)2Fe, and some comparative rate studies. Organometallics. 1983;2:714–719. [Google Scholar]
- 37.Butler IR, Cullen WR, Kim TJ, Rettig SJ, Trotter J. 1,1′-Bis(alkylarylphosphino)ferrocenes: Synthesis, metal complex formation, and crystal structure of three metal complexes of Fe(η5-C5H4PPh2)2. Organometallics. 1985;4:972–980. [Google Scholar]
- 38.Hamann BC, Hartwig JF. Sterically hindered chelating alkyl phosphines provide large rate accelerations in palladium-catalyzed amination of aryl iodides, bromides, and chlorides, and the first amination of aryl tosylates. J Am Chem Soc. 1998;120:7369–7370. [Google Scholar]
- 39.Kawatsura M, Hartwig JF. Simple, highly active palladium catalysts for ketone and malonate arylation: Dissecting the importance of chelation and steric hindrance. J Am Chem Soc. 1999;121:1473–1478. [Google Scholar]
- 40.Colacot TJ, Shea HA. Cp2Fe(PR2)2PdCl2(R = i- Pr ,t-Bu) Complexes as air-stable catalysts for challenging Suzuki coupling reactions. Org Lett. 2004;6:3731–3734. doi: 10.1021/ol048598t. [DOI] [PubMed] [Google Scholar]
- 41.Guram AS, et al. New air-stable catalysts for general and efficient Suzuki-Miyaura cross-coupling reactions of heteroaryl chlorides. Org Lett. 2006;8:1787–1789. doi: 10.1021/ol060268g. [DOI] [PubMed] [Google Scholar]
- 42.Grasa GA, Colacot TJ. α-Arylation of ketones using highly active, air-stable (DtBPF)PdX2 (X = Cl, Br) catalysts. Org Lett. 2007;9:5489–5492. doi: 10.1021/ol702430a. [DOI] [PubMed] [Google Scholar]
- 43.Grasa GA, Colacot TJ. A highly practical and general route for α-arylations of ketones using Bis-phosphinoferrocene-based palladium catalysts. Org Process Res Dev. 2008;12:522–529. [Google Scholar]
- 44.Barluenga J, Jiménez-Aquino A, Aznar F, Valdés C. Modular synthesis of indoles from imines and o-Dihaloarenes or o-Chlorosulfonates by a Pd-catalyzed cascade process. J Am Chem Soc. 2009;131:4031–4041. doi: 10.1021/ja808652a. [DOI] [PubMed] [Google Scholar]
- 45.Eidamshaus C, Burch JD. One-pot synthesis of benzofurans via palladium-catalyzed enolate arylation with o-bromophenols. Org Lett. 2008;10:4211–4214. doi: 10.1021/ol801510n. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Wang Y, Sun Z, Ma D. Elaboration of 2-(Trifluoromethyl)indoles via a cascade coupling/condensation/deacylation Process. Org Lett. 2008;10:625–628. doi: 10.1021/ol7029382. [DOI] [PubMed] [Google Scholar]
- 47.Barluenga J, Jiménez-Aquino A, Valdés C, Aznar F. The azaallylic anion as a synthon for Pd-catalyzed synthesis of heterocycles: Domino two-and three-component synthesis of indoles. Angew Chem Int Ed Eng. 2007;46:1529–1532. doi: 10.1002/anie.200604407. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y, Xie X, Ma D. Facile access to polysubstituted indoles via a cascade Cu-catalyzed arylation-condensation process. J Org Chem. 2007;72:9329–9334. doi: 10.1021/jo702059q. [DOI] [PubMed] [Google Scholar]
- 49.Tanimori S, Ura H, Kirihata M. Copper-catalyzed synthesis of 2,3-disubstituted indoles. Eur J Org Chem. 2007:3977–3980. [Google Scholar]
- 50.Churruca F, SanMartin R, Tellitu I, Domínguez E. A new, expeditious entry to the benzophenanthrofuran framework by a Pd-catalyzed C- and O-arylation/PIFA-mediated oxidative coupling sequence. Eur J Org Chem. 2005;12:2481–2490. [Google Scholar]
- 51.Terao Y, Satoh T, Miura M, Nomura M. Palladium-catalyzed cross-coupling of benzyl ketones and α,β-unsaturated carbonyl and phenolic compounds with O-dibromobenzenes to produce cyclic products. Bull Chem Soc Jpn. 1999;72:2345–2350. [Google Scholar]
- 52.Fox JM, Huang X, Chieffi A, Buchwald SL. Highly active and selective catalysts for the formation of α-aryl ketones. J Am Chem Soc. 2000;122:1360–1370. [Google Scholar]
- 53.Campeau L, et al. Palladium-catalyzed direct arylation of azine and azole N-oxides: Reaction development, scope and applications in synthesis. J Am Chem Soc. 2009;131:3291–3306. doi: 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]
- 54.Kanyiva KS, Nakao Y, Hiyama T. Nickel-catalyzed addition of pyridine-N-oxides across alkynes. Angew Chem Int Ed Eng. 2007;46:8872–8874. doi: 10.1002/anie.200703758. [DOI] [PubMed] [Google Scholar]
- 55.Ren H, Luo Y, Ye S, Wu J. Silver triflate catalyzed reaction of 2-alkynylbenzaldoxime with aryne. Org Lett. 2011;13:2552–2555. doi: 10.1021/ol200629y. [DOI] [PubMed] [Google Scholar]
- 56.Londregan AT, Jennings S, Wei L. Mild addition of nucleophiles to pyridine-N-oxides. Org Lett. 2011;13:1840–1843. doi: 10.1021/ol200352g. [DOI] [PubMed] [Google Scholar]
- 57.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.