

Abstract
The photochemical reactivity of osmocene in a biphasic water-organic solvent system has been investigated to probe its water splitting properties. The photoreduction of aqueous protons to hydrogen under anaerobic conditions induced by osmocene dissolved in 1,2-dichloroethane and the subsequent water splitting by the osmocenium metal-metal dimer formed during H2 production were studied by electrochemical methods, UV-visible spectrometry, gas chromatography, and nuclear magnetic resonance spectroscopy. Density functional theory computations were used to validate the reaction pathways.
Solar water splitting is undoubtedly a key challenge (1–3), and in the quest for solar fuels, hydrogen is definitely a major target with many industrial applications ranging from transportation to carbon dioxide mitigation (4).
Interestingly, some transition metal complexes like metallocenes have been reported to reduce protons to hydrogen. In 1988, cobaltocene was found to produce hydrogen in strong acidic solutions. The mechanism was investigated by pulse radiolysis, and it was found that the reaction kinetics was first order with respect to cobaltocene and the protons pointing to a protonation of the metal as the primary step (5). In 2009, Kunkely and Vogler reported that osmocene dissolved in strong acidic solutions could photogenerate hydrogen and that [Cp2OsIV(OH-)]+ could photogenerate oxygen (6). Proton reduction was shown to proceed also by the formation of a hydride followed under UV light by the formation of H2 and the dimer [Cp2OsIII-OsIIICp2]2+.
One of the major difficulties in studying water splitting by metallocenes is their very poor solubility in aqueous or even acidic solutions. When investigating the reduction of protons in organic phases where metallocenes are soluble, the other major drawback is the low solubility and dissociation of acids in organic solvents. One way to circumvent these issues is to carry out these reactions in biphasic systems.
In 2008, we studied the reduction of aqueous protons by decamethylferrocene (DMFc) in 1,2-dichloroethane (1,2-DCE), and, here again the data suggested that the first step is the protonation of the metal (7). In particular, we have shown that hydrogen bubbles can form at the interface and that hydrogen production is associated to the oxidation of the electron donor DMFc. More generally, we have shown that voltammetry at the interface between two immiscible electrolyte solutions (ITIES) is a very useful tool to study proton coupled electron transfer reactions involving aqueous protons and organic electron donors (8–10). This methodology has been also applied to molecular electrocatalysis where an amphiphilic catalyst is used to complex oxygen to facilitate its reduction as recently reviewed (10–13).
In this study, the photochemical reactivity of osmocene with water in biphasic systems has been investigated by electrochemical methods on solid electrodes and soft liquid-liquid interfaces to unravel the multistep reaction mechanisms. Density functional theory (DFT) computations were performed to shed light on the experimental findings and provide support for the reaction mechanisms. We demonstrate that, under anaerobic conditions, osmocene dissolved in 1,2-DCE can induce the photoreduction of aqueous protons to hydrogen as verified by gas chromatography; whereas, under aerobic conditions it can reduce oxygen to hydrogen peroxide as corroborated by UV-visible (UV-vis) spectroscopy. More importantly, we have investigated the water splitting properties of the osmocenium metal-metal dimer [Cp2OsIII-OsIIICp2]2+ and studied the oxidation of water.
Redox Voltammetry
Voltammetry at a glassy carbon electrode was used to investigate the redox properties of osmocene (Cp2Os) in 1,2-DCE under anaerobic conditions (Fig. 1A, solid line). When cycling to anodic potentials, one can observe the oxidation wave of Cp2Os, and on the reverse scan, the reduction of osmocenium [Cp2OsIII]+ followed at more negative potentials by another peak for the reduction of the dimer [Cp2OsIII-OsIIICp2]2+ similar to what has been described earlier by Geiger et al. (14) for ruthenocene. A very small peak at even more negative potentials is likely to be associated to the reduction of the ring dimer 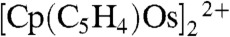 , again, by analogy to what has been observed for ruthenocene (14).
, again, by analogy to what has been observed for ruthenocene (14).
Fig. 1.

Electrochemical behavior of osmocene. (A) Cyclic voltammetry of 2 mM Cp2Os (solid line) and [Cp2Os]2TB2 (dotted line) in dry 1,2-DCE. Scan rate: 100 mV/s. Note the current (dotted line) was enlarged five times. (B) Cyclic voltammetry of 2 mM Cp2Os in dry 1,2-DCE at 5 mV/s (black), 40 mV/s (green), 400 mV/s (purple), 2,000 mV/s (red), and 5,000 mV/s (blue). (Inset) Zoom of the CV at 5 mV/s. (C) Cyclic voltammetry of 2 mM Cp2Os in dry DCM (black) and wet DCM containing 1% (vol/vol) H2O (red). Scan rate: 100 mV/s. (D) Cyclic voltammograms (50 mV·s-1) for Scheme 1 in the absence (x = 0, y = 1) and presence [x = 5, y = 1 (red), 0.1 (green), 0.01 (blue)] of osmocene in 1,2-DCE. (Inset) Half-wave potential dependence on pH. (A–C) Data were obtained on glassy carbon electrode under anaerobic conditions; supporting electrolyte: 25 mM BATB.
The anodic peak and the first cathodic peak intensity vary linearly with the square root of the scan rate indicating a linear semi-infinite diffusion controlled process; whereas, the second cathodic peak at about 0.6 V shows some deviation from a linear dependence on the square root of scan rate. The three peaks can be explained by an ECdim mechanism, i.e., an electron transfer (1) followed by dimerization of the oxidized product [Cp2OsIII]+ to form the metal-metal dimer [Cp2OsIII-OsIIICp2]2+ (2) that can be reduced at more negative potentials than [Cp2OsIII]+ (3).
The dotted line in Fig. 1A shows the voltammetric response for the reduction of [Cp2OsIII-OsIIICp2]2+ prepared as a salt, [Cp2OsIII-OsIIICp2]TB2 using tetrakis(pentafluorophenyl)borate (TB-) as counter ion. Starting from a potential of 1.6 V, we can observe upon scanning to negative potentials first a small peak corresponding to the reduction of [Cp2OsIII]+ formed by disproportionation of the dimer (4) and then to the reduction of the dimer itself. At more negative potentials, the reduction of the ring dimer 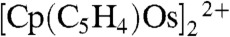 is also observed clearly showing that [Cp2OsIII-OsIIICp2]2+ can disproportionate and evolve slowly into
is also observed clearly showing that [Cp2OsIII-OsIIICp2]2+ can disproportionate and evolve slowly into 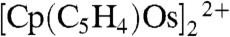 (5).
(5).
The dimerization mechanism given by [1–3] can be corroborated by scan rate dependence measurements (Fig. 1B). When varying from slow (40 mV/s) to high scan rates (5 V/s), the ratio between the forward and backward peak currents (ipa/ipc) tended to decrease to one indicating the presence of a dimerization step visible at slow scan rates. This mechanism can be further verified by the concentration dependence (Fig. S1). Indeed, as the concentration of Cp2Os decreases, the size of the second cathodic peak corresponding to the reduction of the dimer [Cp2OsIII-OsIIICp2]2+ (3) decreases as seen on Fig. S1 similar to previous observations on ruthenocene (14).
The slow disproportionation of the dimer [Cp2OsIII-OsIIICp2]2+ has been reported (15). This disproportionation can clearly be seen at low scan rates, e.g., 5 mV/s (Fig. 1B, Inset) where a steady-state current is clearly observed. Osmium (IV), as written in [4], has never been observed and would require too much energy to form; however, [4] illustrates that a disproportionation step must take place. Indeed, [Cp2OsIV]2+ can be readily reduced to osmium (III) on the electrode surface, considering the respective redox potentials (16). A recent chronopotentiometric study by the Compton’s group on ruthenocene clearly shows that the anion can play a role in stabilizing the oxidized species, namely RuIII and RuIV (17).
Digisim simulations were carried out to verify further the mechanism by [1–4] that qualitatively reproduced the experimental data thereby corroborating this reaction scheme (Fig. S1). In summary, we have
| [1] |
| [2] |
| [3] |
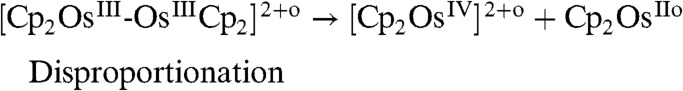 |
[4] |
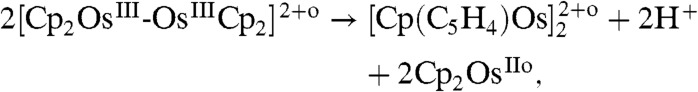 |
[5] |
where the superscript (o) indicates the organic phase.
When repeating this study in dichloromethane (DCM, Fig. 1C, black), the cathodic peak for the reduction of the dimer [Cp2OsIII-OsIIICp2]2+ appears only at higher concentrations corroborating previous observations (14). This difference between the two solvents may reflect differences in the rate of dimerization, the process being slower in DCM. In wet DCM, Fig. 1C (red line), the peak for the reduction of [Cp2OsIII]+ decreases indicating that oxidized osmocene reacts with water.
Proton nuclear magnetic resonance (1H NMR) was used to explore further the reactivity of osmocenium species with H2O (vide infra).
Ion Transfer Voltammetry
Cyclic voltammetry experiments were conducted to investigate the acid-base properties of osmocene at the water/1,2-DCE interface. Fig. 1D illustrates the cyclic voltammograms obtained in the absence (black) and presence (red, green, and blue) of osmocene in the organic phase under aerobic conditions (similar results were obtained under anaerobic conditions). In the blank cell (Fig. 1D, black), the potential window is limited at positive potentials by the transfer of H+ from water to 1,2-DCE; whereas, it is limited by the transfer of Cl- at negative potentials. After addition of osmocene to 1,2-DCE, a large current signal at positive potentials and a return peak on the backward scan were observed (Fig. 1D, red). The ratio of the area integrated for the forward and backward scans was found to be close to unity indicating a chemically reversible process. This current corresponds to an assisted proton transfer by osmocene acting as a base.
By applying the end of potential window method proposed by Shao et al. (18), the half-wave potential for this assisted proton transfer was estimated. When plotted as a function of pH, a slope of 80 mV/pH (Fig. 1D, Inset) was obtained compared to the 60 mV/pH predicted by the Nernst equation for a single assisted proton transfer process across a liquid-liquid interface. Linear dependence of forward peak current on the square root of the scan rate and on the concentration of osmocene in 1,2-DCE indicates that the process is controlled by the diffusion of osmocene to the interface. From the data in Fig. 1D, we can deduce the acid dissociation constant in the organic phase. Indeed, the variation of the half-wave potential for an assisted ion transfer reaction is given by
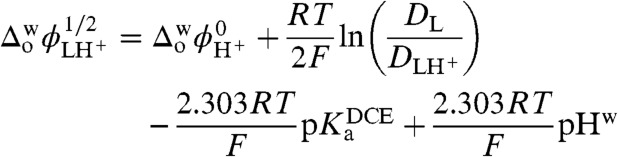 |
where  is the experimentally observed half-wave transfer potential of the facilitated proton transfer and
is the experimentally observed half-wave transfer potential of the facilitated proton transfer and 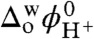 is the formal proton transfer potential equal to 0.55 V. DL and
is the formal proton transfer potential equal to 0.55 V. DL and  {L = Cp2Os, LH+ = [Cp2OsIV(H-)]+} represent the diffusion coefficients of the ligand and its protonated form. For simplicity, it can be assumed that
{L = Cp2Os, LH+ = [Cp2OsIV(H-)]+} represent the diffusion coefficients of the ligand and its protonated form. For simplicity, it can be assumed that 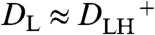 , that then yields
, that then yields 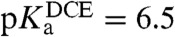 .
.
These results show that the forward current wave results from an assisted proton transfer by osmocene in the organic phase where the protonation of osmocene forms the corresponding hydride complex (19) as confirmed by NMR studies (vide infra). The return peak in the back scan can be ascribed to the interfacial deprotonation of osmocene hydride. Contrary to DMFc, where the hydride can further react to form hydrogen and decamethylferrocenium in the dark, here, the osmocene hydride is stable and can dissociate upon reversing the potential sweep.
Two-Phase Reactions
The reactivity of osmocene hydride [Cp2OsIV(H-)]+ was studied by biphasic reactions where the protonation of osmocene was carried out by contacting an aqueous acidic solution (HCl or H2SO4) containing lithium tetrakis-pentafluoro-phenylborate-diethyletharate (LiTB-DEE) with a solution of osmocene in 1,2-DCE containing, eventually, a supporting electrolyte bis(triphenylphosphoranylidene)-ammonium tetrakis-(pentafluorophenyl)borate (BATB). The role of the TB- anion in the aqueous phase is to act as a phase transfer catalyst of protons to the organic phase, DEE being a lipophilic base. This transfer is quantitative as reported earlier (20). For example, mixing an acidic solution containing 5 mM LiTB-DEE with an organic phase of the same volume results in the presence of about 5 mM hydrogen tetrakis-(pentafluorophenyl)borate diethyl etherate (HDEETB) in 1,2-DCE, labeled thereafter HTB for simplicity. Under anaerobic conditions, such a biphasic reaction with proton transfer leads to the formation of [Cp2OsIV(H-)]+ as indicated from ion transfer voltammetry and 1H NMR (vide infra).
Following the work of Kunkely and Vogler (6) showing that osmocene in strong acidic aqueous solution can generate hydrogen under UV irradiation with ϕ = 10-3 at λirr = 254 nm, we have studied its photolysis in the biphasic systems. A 1,2-DCE solution containing 5 mM osmocene and 5 mM BATB was placed in contact with an acidic aqueous solution (0.1 M HCl and 5 mM LiTB-DEE) within a Schlenk line using argon as inert gas. Again, TB- is used to transfer HTB to the 1,2-DCE phase. When the mixture was allowed to react overnight under dark conditions (12 h), no hydrogen was found in the headspace as revealed by gas chromatography analysis; however, when the two-phase reaction was carried out for 4 h under UV light, hydrogen was detected as shown in Fig. 2. The traces of oxygen and nitrogen found stem from an air leakage as evidenced by the ratio between the nitrogen and oxygen peaks.
Fig. 2.

Gas chromatogram of the headspace of a two-phase reaction under UV irradiation in anaerobic conditions.
1H NMR Analysis
Proton NMR was first used to verify the formation of the hydride complex [Cp2OsIV(H-)]+. Standard spectrum of the supporting electrolyte BATB shows peaks at 7.63 ppm and 7.47 ppm with small amount of water at 1.57 ppm. Cp2Os in the presence of BATB in deuterated chloroform (CDCl3) gives a singlet at 4.75 ppm (Fig. S2A) and, upon contacting with an aqueous phase of 0.05 M H2SO4 and 10 mM LiTB-DEE so as to transfer HTB to the organic phase, two additional peaks appeared at 5.44 ppm and -14.27 ppm standing for the ring protons and hydride in protonated osmocene, respectively (Fig. S2C). In addition, the peaks at 3.49 ppm and 1.23 ppm were from LiTB-DEE and showed no influence in the experiments (Fig. S2B). Using the supporting electrolyte BATB as internal calibration standard, and assuming that HTB concentration in the organic phase is about 10 mM, the concentration of [Cp2OsIV(H-)]+ is found to be 1.53 mM after 10 min. These NMR results indicate that osmocene is in equilibrium with its hydride form.
Upon further irradiation with UV light for 1 h, the signal for neutral osmocene disappeared and that for the hydride increased. This implied that all the neutral osmocene converted to osmium hydride. Besides, a new singlet peak at 5.75 ppm appeared that can be attributed to the osmocenium dimer [Cp2OsIII-OsIIICp2]2+ (Fig. 3A) that was observed at 5.88 ppm in deuterated nitromethane (CD3NO2) by Taube et al. (15). No other signals emerged from side reactions of osmocene under irradiation showing the stability of the cyclopentadienyl rings. This further indicated that the hydrogen detected by GC in the irradiated biphasic reactions was generated by osmium hydride and not by a loss of protons from the rings.
Fig. 3.

Optimized dimer structures obtained at the B3LYP-dDsC/cc-pVDZ level using the PCM model (ε = 10.36). (A) Metal-metal dimer [Cp2OsIII-OsIIICp2]2+, (B) water-sandwich dimer [Cp2OsIII-(H2O)-OsIIICp2]2+, and (C) ring dimer [(Cp(C5H4)Os)2]2+.
The overall reaction in the present biphasic systems under UV irradiation reads, as follows:
| [6] |
Reactivity of the metal-metal dimer [Cp2OsIII-OsIIICp2]2+ with water is an important step in water splitting. As a result, [Cp2OsIII-OsIIICp2]2+ was synthesized as reported (15) using tetrakis-(pentafluorophenyl)borate as counter ions instead of 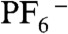 . A dark green complex was obtained, and the dimer structure was confirmed by 1H NMR in CD3NO2 (Fig. S3, freshly prepared) with a signal at 5.88 ppm, consistent to a precedent report (15). As soon as the dimer was dissolved in CD3NO2, it started to convert to another species (δ = 5.60 ppm) at a moderate speed even in dark and under anaerobic conditions. Upon addition of H2O (c.a. 0.4 μL) to the NMR tube under N2 protection, the newly appeared signal (δ = 5.60 ppm) broadened instantly suggesting the inclusion of water (Fig. S4). The intensity of the metal-metal dimer decreased; whereas, the signal at 5.60 ppm increased in an almost 1∶1 ratio strongly indicating that the metal-metal dimer may react with H2O to form a new species. In parallel, the ring dimer
. A dark green complex was obtained, and the dimer structure was confirmed by 1H NMR in CD3NO2 (Fig. S3, freshly prepared) with a signal at 5.88 ppm, consistent to a precedent report (15). As soon as the dimer was dissolved in CD3NO2, it started to convert to another species (δ = 5.60 ppm) at a moderate speed even in dark and under anaerobic conditions. Upon addition of H2O (c.a. 0.4 μL) to the NMR tube under N2 protection, the newly appeared signal (δ = 5.60 ppm) broadened instantly suggesting the inclusion of water (Fig. S4). The intensity of the metal-metal dimer decreased; whereas, the signal at 5.60 ppm increased in an almost 1∶1 ratio strongly indicating that the metal-metal dimer may react with H2O to form a new species. In parallel, the ring dimer 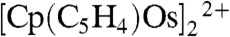 illustrated in Fig. 3C was then observed with signals at 6.19 ppm (2 H, triplet), 5.27 ppm (2 H, triplet), and 5.91 ppm (5 H, singlet) (15).
illustrated in Fig. 3C was then observed with signals at 6.19 ppm (2 H, triplet), 5.27 ppm (2 H, triplet), and 5.91 ppm (5 H, singlet) (15).
The inclusion of water results in the formation of a water sandwich dimer as illustrated in Fig. 3B (vide infra) and as given by [7],
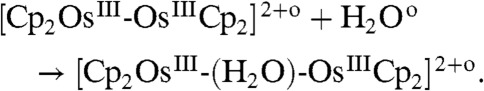 |
[7] |
Investigating the photolysis of this water-sandwich dimer is a key step toward elucidating water splitting as seen for other bimetallic dimers containing a metallic water bond, e.g., the well-known blue dimer (21). Photolysis products after irradiation (Xenon lamp, 150 W) were monitored by 1H NMR. Fig. S5 showed the intensity of the signal at 5.60 ppm for the water-sandwich dimer declining and new signals appearing with time.
Analysis of these data shows that the photolysis of the water-sandwich dimer followed [8] and [9] rather than [10–12] as demonstrated below by computations to form a hydroxo complex, i.e., [Cp2OsIV(OH-)]+ (δ = 6.14 ppm) and a hydride complex. The hydroxo species reacts further to a peroxo complex, i.e., Cp2OsIV-(O2) (δ = 6.02 and 5.56 ppm). These systems are light sensitive and could undergo further photolysis (Fig. S6),
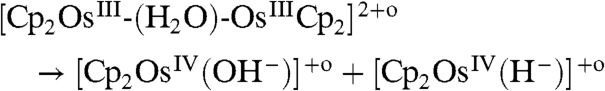 |
[8] |
| [9] |
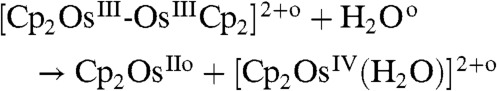 |
[10] |
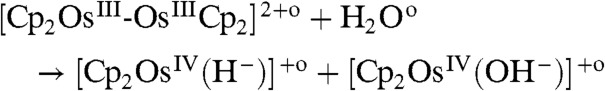 |
[11] |
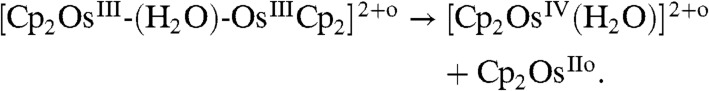 |
[12] |
To confirm the formation of [Cp2OsIV(OH-)]+, this complex was synthesized as reported earlier (22), and 1H NMR shows a signal at 6.11 ppm. The signals at 6.02 ppm and 5.56 ppm result from the photolysis product of [Cp2OsIV(OH-)]+ and can be attributed to Cp2OsIV-(O2) according to [9]. Peroxo Osmium (IV) complexes like [OsCl(O2)(dcpe)2] as well as many stable transition metals peroxo complexes are known (23).
DFT Computations
DFT computations were carried out to gain further insight into the dimerization of osmocenium as well as the mechanism of the water splitting.
According to reaction [2], the dimerization of [Cp2OsIII]+ into the metal-metal dimer (Fig. 3A) is exothermic in 1,2-DCE (-16 and -18 kcal/mol at B3LYP-dDsC and M06L, respectively, though highly endothermic in the gas phase). The dimer A has a direct metal-metal bond of 3.013 Å, in good agreement with the crystal structure (15) bond length (3.039 Å). In biphasic systems, the organic phase is saturated with water and in such conditions another type of dimer (water-sandwich dimer B in Fig. 3) can be considered in which a water molecule is incorporated in the cavity between two osmocenium ions according to reactions [7] or [13],
| [13] |
As evident from Table 1 (see also Table S1), the formation of water-sandwich dimer B from [Cp2OsIII]+ by [13] is favored (-13 kcal/mol) compared to the inclusion of water in the metal-metal dimer A (7) that is 3 (5) kcal/mol at the B3LYP-dDsC (M06L) level including continuum solvation. This suggests that it is much more likely to form directly the water-sandwich dimer B from osmocenium [Cp2OsIII]+ rather than by incorporating a water molecule into already formed metal-metal dimer A.
Table 1.
Relative ΔH298 energies (in kcal/mol) for different dimerization reactions computed at B3LYP-dDsC/cc-pVDZ and M06L/cc-pVDZ levels in 1,2-DCE
| Reaction no. | B3LYP-dDsC | M06L |
| 2 | −16 | −18 |
| 5 | 23 | 17 |
| 7 | 3 | 5 |
| 13 | −13 | −13 |
The metal-metal dimer A can undergo a reaction according to [5] yielding osmocene, protons, and a ring dimer C (Fig. 3) where two cyclopentadienyl rings are σ-bonded to the Os centers (15). This reaction is, however thermodynamically unfavorable by +23 kcal/mol (B3LYP-dDsC).
Thus, according to DFT, osmocenium ions dimerize upon oxidation of osmocene to two types of dimer, i.e., metal-metal A and water sandwich dimer B depending on the conditions.
In accord with the experimental findings, the protonation of osmocene to [Cp2OsIV(H-)]+ is an energetically favorable process [-22 (-20) kcal/mol at B3LYP-dDsC (M06L) in 1,2-DCE]. The computations, moreover, indicate that the protonation occurs at the metal center with the formation of a Os-H bond of 1.602 Å and not at the Cp rings. As seen above, the osmocene-hydride can react under UV illumination,
| [14] |
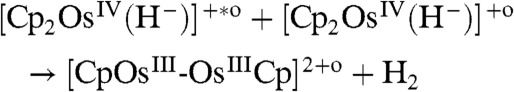 |
[15] |
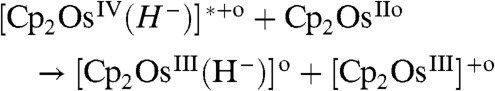 |
[16] |
| [17] |
The mechanism in [14–17] has been corroborated by TD-DFT excitation energies showing that [Cp2OsIV(H-)]+ is much easier to photoexcite than Cp2Os. The vertical excitation energy for [Cp2OsIV(H-)]+ corresponds to a wavelength of 266 nm (∼107 kcal/mol) and is associated with a HOMO to LUMO transition. The excited [Cp2OsIV(H-)]+∗ can react with a nonexcited partner to form the metal-metal dimer A along with H2 (15). Alternatively, the reaction can also occur with two excited protonated species. Another possible pathway is given in [16] in which the excited [Cp2OsIV(H-)]+∗ reacts with osmocene forming osmocenium ions that, as discussed above can dimerize according to [2]. The [Cp2OsIII(H-)] species can then be protonated to yield H2.
Formation of the metal-metal dimer A and the water-sandwich B is a key step for the water splitting reaction. Closer inspection of Table 2 indicates that the water-sandwich dimer could split water into [Cp2OsIV(OH-)]+ and [Cp2OsIV(H-)]+ in the dark according to [8]; whereas, reactions [10] and [12] are unfavorable by 16 and 13 kcal/mol, respectively (B3LYP-dDsC level in 1,2-DCE). In the next step, the hydroxo complex [Cp2OsIV(OH-)]+ can undergo a photolysis according to [9].
Table 2.
Relative ΔH298 energies (in kcal/mol) for reactions of water splitting computed at B3LYP-dDsC/cc-pVDZ and M06L/cc-pVDZ levels in 1,2-DCE solvent
| Reaction no. | B3LYP-dDsC | M06L |
| 8 | −4 | −2 |
| 10 | 16 | 14 |
| 11 | 1 | 4 |
| 12 | 13 | 9 |
The computed proton NMR chemical shifts of [Cp2OsIII-OsIIICp2]2+ and [Cp2OsIII-(H2O)-OsIIICp2]2+ dimers as well as the [Cp2OsIV(OH-)]+ product of the photoinduced water splitting give the same averaged 1H shift (5.4 ppm) for the protons in the cyclopentadienyl rings of the two dimers and 0.2 ppm (i.e., 5.6 ppm) larger shift for the Cp protons in [Cp2OsIV(OH-)]+ system. For Cp2OsIV-(O2) illustrated in Fig. 4, two averaged peaks (one for each nonequivalent Cp ring) are computed at 5.0 and 5.3 ppm shifted by 0.6 and 0.3 ppm with respect to the corresponding value in [Cp2OsIV(OH-)]+. These results are in line with the above analysis of the experimental NMR data.
Fig. 4.
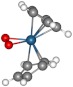
Optimized structure for Cp2OsIV-(O2), computational details as in Fig. 3.
Oxygen Detection in Biphasic Systems
Most attempts to detect oxygen from biphasic reactions were not successful using osmocene or dimer A as starting material in the organic phase. This outcome is mainly due to the fact that osmocene hydride reacts strongly with oxygen even in the dark to form hydrogen peroxide. This reaction has been studied and is reported in Figs. S7 and S8. When doing photolysis, H2O2 can further decompose back to water and O2.
Conclusion
All in all, the experiments unambiguously demonstrate that the irradiation of a biphasic system containing aqueous protons, a phase transfer catalyst, and osmocene in the organic phase results in the production of hydrogen and of osmocenium dimers A or B. The water-sandwich dimer can split water to form the complexes [Cp2OsIV(OH-)]+ and [Cp2OsIV(H-)]+ observed by 1H NMR and corroborated by DFT computations. Photolysis of [Cp2OsIV(OH-)]+ yields Cp2OsIV-(O2) that can further react to form O2. It is clear that the photoproduction of hydrogen remains a low yield reaction with a very low quantum efficiency (ϕ = 0.026% at λirr = 365 nm) and cannot be envisaged for a water splitting process; however, this study shows that bimetallic dimers can offer a route to split water and that mixed metal dimers, e.g., Cp2Os-RuCp2 could be potential candidates.
Experimental Section
Chemicals.
All chemicals were used as received without further purification. All the aqueous solutions were prepared with ultra pure water (18.2 MΩ·cm). Osmocene was supplied by ABCR. LiTB-DEE and DCM were provided by Sigma-Aldrich. DCM was stored over preactivated 3A molecular sieves. Lithium chloride anhydrous (LiCl, ≥99%), bis(triphenylphosphoranylidene)-ammonium chloride (BACl, ≥98%), and 1,2-dichloroethane (1,2-DCE, ≥99.8%) were obtained from Fluka. Hydrochloric acid (HCl, 32%) was ordered from Merck. BATB was prepared by metathesis of 1∶1 mixtures of BACl and LiTB, respectively, in a methanol/water mixture (2/1, vol/vol) followed by recrystallization from acetone. HTB was prepared by shaking 5 mM LiTB and 100 mM HCl in the water phase with pure 1,2-DCE for 1 h. HTB was recovered in the organic phase whereas the aqueous phase contained only LiCl and HCl. CDCl3 was from ARMAR Chemicals. Argon (48) was supplied by Carbagas. CD3NO2 was supplied by ABCR.
Electrochemical Measurements.
Electrochemical measurements at the water/1,2-DCE interface were conducted in a four-electrode configuration with a commercial potentiostat (PGSTAT 30, Metrohm). The electrochemical cell used was a three-compartment glass cell featuring a cylindrical vessel where the water/1,2-DCE interface with a geometric area of 1.53 cm2 was formed. Two platinum counter electrodes were positioned in the aqueous and organic phases, respectively, to supply the current flow. The external potential was applied between two silver/silver chloride (Ag/AgCl) reference electrodes connected to the aqueous and organic phases by means of a Luggin capillary. Electrolyte compositions are illustrated in Scheme 1. The applied potential difference was converted to the Galvani potential difference (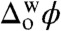 ) based on cyclic voltammetry measurements of the reversible half-wave potential of the tetraethylammonium cation (TEA+) transfer (0.019 V) (24). All the electrochemical measurements were performed at ambient temperature (23 ± 2 °C) with air-saturated solutions unless specified otherwise.
) based on cyclic voltammetry measurements of the reversible half-wave potential of the tetraethylammonium cation (TEA+) transfer (0.019 V) (24). All the electrochemical measurements were performed at ambient temperature (23 ± 2 °C) with air-saturated solutions unless specified otherwise.
Scheme 1.
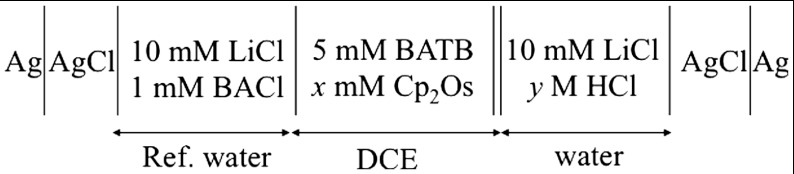
Composition of electrochemical cell used for ion transfer voltammetry.
Classic voltammetry at a glassy carbon electrode (2 mm Ø) was carried out in 1,2-DCE with 25 mM BATB as supporting electrolyte in a conventional three-electrode cell in which a silver wire was used as a quasi reference electrode and a platinum wire as the counter electrode.
Shake Flask Experiments.
Biphasic shake flask experiments were performed by mixing 2 mL of 1,2-DCE solution containing 5 mM BATB and 5 mM osmocene with 2 mL of an aqueous solution of 5 mM LiTB and 100 mM HCl. Magnetic stirring was used to emulsify the two-phases throughout the experiment. The head space was analyzed by gas chromatography using a Perkin-Elmer GC (Clarus 400) equipped with packed 5 Å molecular sieves and 80/100 mesh using a TCD detector and argon as the carrier gas.
NMR Measurements.
NMR experiments were carried out with a Bruker Biospin Avance-400. Two same vials containing 10 mM osmocene and 10 mM BATB in CDCl3 and 10 mM LiTB at pH = 1 (adjusted by H2SO4) in water were stirred under anaerobic conditions for 10 min. One organic phase was analyzed by NMR, the second being further irradiated by UV light (Philips, TLDK 30W/05) for 1 h prior to NMR analysis. CD3NO2 was used for the NMR measurements of the synthesized dimer. Photolysis of dimer A was carried out in CD3NO2.
Computational Details.
Geometries were fully optimized using a locally modified version of Gamess US (2010.1) (25, 26) and the PCM model (ε = 10.36 for 1,2-DCE) (27) at the B3LYP (28) -dDsC (29, 30) /cc-pVDZ (31, 32) level. For Os the LANL2DZ effective core potential of Hay and Wadt was used. The density-dependent dispersion correction (i.e., dDsC) accounts for the effect of long-range weak interactions that are, by construction, absent in standard density functionals and that are crucial for the correct description of the dimer complexes studied herein (33). Alternatively, computations were also performed at the M06L (34) /cc-pVDZ level using the Gaussian 09 program (35) and the same implicit solvent model. The low-lying excited states of the species of interest were computed at the B3LYP level using time-dependent density functional theory (TD-DFT) (36, 37). Unrestricted Kohn–Sham formalism was used to explore the spin multiplicity of the ground state of the investigated species. The nature of all stationary points was verified by analytic computation of vibrational frequencies that were also used for the computation of zero-point vibrational energies and molecular partition functions for use in computing 298 K thermal contributions to enthalpy employing the usual rigid-rotator harmonic oscillator approximation (38). The proton NMR chemical shielding computations were performed in nitromethane using the Gauge-Invariant Atomic Orbitals (GIAO) approximations (39) at the B3LYP/IGLO-III level for C, H, O and all-electron WTBS (40) basis set for Os.
Supplementary Material
Acknowledgments.
T.K.T. acknowledges the financial support by Marie–Heim–Voegtlin Grant PMPDP2_134151/1. C.C. thanks the Sandoz Family Foundation and the Swiss National Science Foundation Grant 200021-121577/1. L.E.P.A. is grateful to the Swiss National Science Foundation—National Center of Competence in Research program Molecular Ultrafast Science and Technology for financial support. The work in LSCI is supported by a starting grant from the European Research Council under the European Community’s Seventh Framework Programme (FP7 2007–2013)/European Research Council Grant agreement no. 257096.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203743109/-/DCSupplemental.
References
- 1.Pagliaro M, Konstandopoulos AG, Ciriminna R, Palmisano G. Solar hydrogen: Fuel of the near future. Energy Environ Sci. 2010;3:279–287. [Google Scholar]
- 2.Walter MG, et al. Solar water splitting cells. Chem Rev. 2010;110:6446–6473. doi: 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- 3.Bard AJ, Fox MA. Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen. Acc Chem Res. 1995;28:141–145. [Google Scholar]
- 4.Cook TR, et al. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem Rev. 2010;110:6474–6502. doi: 10.1021/cr100246c. [DOI] [PubMed] [Google Scholar]
- 5.Koelle U, Infelta PP, Grätzel M. Kinetics and mechanism of the reduction of protons to hydrogen by cobaltocene. Inorg Chem. 1988;27:879–883. [Google Scholar]
- 6.Kunkely H, Vogler A. Water splitting by light with osmocene as photocatalyst. Angew Chem Int Edit. 2009;48:1685–1687. doi: 10.1002/anie.200804712. [DOI] [PubMed] [Google Scholar]
- 7.Hatay I, et al. Hydrogen evolution at liquid-liquid interfaces. Angew Chem Int Edit. 2009;48:5139–5142. doi: 10.1002/anie.200901757. [DOI] [PubMed] [Google Scholar]
- 8.Hatay I, et al. Proton-coupled oxygen reduction at liquid-liquid interfaces catalyzed by cobalt porphine. J Am Chem Soc. 2009;131:13453–13459. doi: 10.1021/ja904569p. [DOI] [PubMed] [Google Scholar]
- 9.Partovi-Nia R, et al. Proton pump for O2 reduction catalyzed by 5, 10, 15, 20-tetraphenylporphyrinatocobalt(II) Chem Eur J. 2009;15:2335–2340. doi: 10.1002/chem.200801807. [DOI] [PubMed] [Google Scholar]
- 10.Su B, et al. Molecular electrocatalysis for oxygen reduction by cobalt porphyrins adsorbed at liquid/liquid interfaces. J Am Chem Soc. 2010;132:2655–2662. doi: 10.1021/ja908488s. [DOI] [PubMed] [Google Scholar]
- 11.Schaming D, et al. Artificial photosynthesis at soft interfaces. CHIMIA Int J Chem. 2011;65:356–359. doi: 10.2533/chimia.2011.356. [DOI] [PubMed] [Google Scholar]
- 12.Olaya AJ, et al. Four-electron oxygen reduction by tetrathiafulvalene. J Am Chem Soc. 2011;133:12115–12123. doi: 10.1021/ja203251u. [DOI] [PubMed] [Google Scholar]
- 13.Olaya AJ, et al. Self-assembled molecular rafts at liquid|liquid interfaces for four-electron oxygen reduction. J Am Chem Soc. 2012;134:498–506. doi: 10.1021/ja2087322. [DOI] [PubMed] [Google Scholar]
- 14.Swarts JC, Nafady A, Roudebush JH, Trupia S, Geiger WE. One-electron oxidation of ruthenocene: Reactions of the ruthenocenium ion in gentle electrolyte media. Inorg Chem. 2009;48:2156–2165. doi: 10.1021/ic802105b. [DOI] [PubMed] [Google Scholar]
- 15.Droege MW, Harman WD, Taube H. Higher oxidation state chemistry of osmocene: Dimeric nature of the osmocenium ion. Inorg Chem. 1987;26:1309–1315. [Google Scholar]
- 16.Kuwana T, Bublitz DE, Hoh G. Chronopotentiometric studies on the oxidation of ferrocene, ruthenocene, osmocene and some of their derivatives. J Am Chem Soc. 1960;82:5811–5817. [Google Scholar]
- 17.Rogers EI, Lawrence NS, Compton RG. The electrochemical oxidization of ruthenocene in various room temperature ionic liquids. J Electroanal Chem. 2011;657:144–149. [Google Scholar]
- 18.Shao Y, Stewart AA, Girault HH. Determination of the half-wave potential of the species limiting the potential window. Measurement of Gibbs transfer energies at the water/1,2-dichloroethane interface. J Chem Soc Faraday T. 1991;87:2593–2597. [Google Scholar]
- 19.Shubina ES, Krylov AN, Kreindlin AZ, Rybinskaya MI, Epstein LM. Hydrogen-bonded complexes involving the metal atom and protonation of metallocenes of the iron subgroup. J Organomet Chem. 1994;465:259–262. [Google Scholar]
- 20.Su B, et al. H2O2 generation by decamethylferrocene at a liquid|liquid interface. Angew Chem Int Edit. 2008;47:4675–4678. doi: 10.1002/anie.200801004. [DOI] [PubMed] [Google Scholar]
- 21.Concepcion JJ, Jurss JW, Templeton JL, Meyer TJ. Mediator-assisted water oxidation by the ruthenium blue dimer cis,cis-[(bpy)2(H2O)RuORu(OH2)(bpy)2]4+ Proc Natl Acad Sci USA. 2008;105:17632–17635. doi: 10.1073/pnas.0807153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer EO, Grubert H. Über Aromatenkomplexe von Metallen, XXIX. Di-cyclopentadienyl-osmium. Chem Ber. 1959;92:2302–2309. [Google Scholar]
- 23.Mezzetti A, Zangrando E, Del Zotto A, Rigo P. Dioxygen addition to five-coordinate osmium(II) complexes. X-Ray crystal structure of [OsH(η2-O2)(dcpe)2]bph4 [dcpe = 1,2-bis(dicyclohexylphosphino)ethane] J Chem Soc Chem Comm. 1994:1597–1598. [Google Scholar]
- 24.Wandlowski T, Mareček V, Samec Z. Galvani potential scales for water-nitrobenzene and water-1,2-dichloroethane interfaces. Electrochim Acta. 1990;35:1173–1175. [Google Scholar]
- 25.Schmidt MW, et al. General atomic and molecular electronic structure system. J Comput Chem. 1993;14:1347–1363. [Google Scholar]
- 26.Gordon MS, Schmidt MW. Advances in electronic structure theory: GAMESS a decade later. In: Dykstra CE, Frenking G, Kim KS, Scuseria GE, editors. Theory and Applications of Computational Chemistry, The First Forty Years. Amsterdam: Elsevier; 2005. pp. 1167–1189. [Google Scholar]
- 27.Miertuš S, Scrocco E, Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981;55:117–129. [Google Scholar]
- 28.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 29.Steinmann SN, Corminboeuf C. A system-dependent density-based dispersion correction. J Chem Theory Comput. 2010;6:1990–2001. doi: 10.1021/ct1001494. [DOI] [PubMed] [Google Scholar]
- 30.Steinmann SN, Corminboeuf C. A generalized-gradient approximation exchange hole model for dispersion coefficients. J Chem Phys. 2011;134:044117. doi: 10.1063/1.3545985. [DOI] [PubMed] [Google Scholar]
- 31.Dunning TH., Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 32.Balabanov NB, Peterson KA. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc-Zn. J Chem Phys. 2005;123:064107. doi: 10.1063/1.1998907. [DOI] [PubMed] [Google Scholar]
- 33.Steinmann SN, Corminboeuf C. Comprehensive benchmarking of a density-dependent dispersion correction. J Chem Theory Comput. 2011;7:3567–3577. doi: 10.1021/ct200602x. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Truhlar DG. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys. 2006;125:194101. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- 35.Frisch MJ, et al. Gaussian 09. Wallingford, CT: Gaussian, Inc; 2009. [Google Scholar]
- 36.Runge E, Gross EKU. Density-functional theory for time-dependent systems. Phys Rev Lett. 1984;52:997–1000. [Google Scholar]
- 37.Gross EKU, Kohn W. Local density-functional theory of frequency-dependent linear response. Phys Rev Lett. 1985;55:2850–2852. doi: 10.1103/PhysRevLett.55.2850. [DOI] [PubMed] [Google Scholar]
- 38.Cramer CJ. Essentials of Computational Chemistry. 2nd Ed. Chichester, U.K.: Wiley; 2004. [Google Scholar]
- 39.Ditchfield R. Self-consistent perturbation-theory of diamagnetism. 1. Gauge-invariant LCAO method for NMR chemical-shifts. Mol Phys. 1974;27:789–807. [Google Scholar]
- 40.Huzinaga S, Klobukowski M. Well-tempered Gaussian basis sets for the calculation of matrix Hartree–Fock wave functions. Chem Phys Lett. 1993;212:260–264. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.