

Abstract
Electrocatalysis offers a means of electrochemical signal amplification, yet in DNA-based sensors, electrocatalysis has required high-density DNA films and strict assembly and passivation conditions. Here, we describe the use of hemoglobin as a robust and effective electron sink for electrocatalysis in DNA sensing on low-density DNA films. Protein shielding of the heme redox center minimizes direct reduction at the electrode surface and permits assays on low-density DNA films. Electrocatalysis with methylene blue that is covalently tethered to the DNA by a flexible alkyl chain linkage allows for efficient interactions with both the base stack and hemoglobin. Consistent suppression of the redox signal upon incorporation of a single cytosine-adenine (CA) mismatch in the DNA oligomer demonstrates that both the unamplified and the electrocatalytically amplified redox signals are generated through DNA-mediated charge transport. Electrocatalysis with hemoglobin is robust: It is stable to pH and temperature variations. The utility and applicability of electrocatalysis with hemoglobin is demonstrated through restriction enzyme detection, and an enhancement in sensitivity permits femtomole DNA sampling.
Keywords: DNA charge transport, DNA sensors, mismatch detection
DNA-modified electrodes have been extensively studied in the development of sensitive DNA-based sensors using DNA-mediated charge transport (CT) chemistry (1–18). Sensors based on DNA-mediated CT are exquisitely sensitive to structural perturbations caused by single-base mismatches, lesions, and DNA-binding proteins (2–4). Due to the high specificity, robust nature, and low cost of these DNA-based electrochemical sensors, they are being developed as new cancer diagnostics through the detection of transcription factors and low abundance microRNAs (4, 5, 19, 20). Despite the intrinsic specificity of these devices, these DNA-CT sensors currently lack the required sensitivity to detect biologically relevant levels of cancer markers. Therefore, a means to reliably and robustly amplify the DNA-mediated signal is essential for the application of these technologies to the rapid detection of cancer markers directly from cell lysates.
Different electrochemical strategies have been used to achieve higher sensitivities, including the use of gold nanoparticles, conducting polymers, and catalysis (21–27). Electrocatalysis, however, represents a preferred means of signal amplification in electrochemical sensors (25–27). The degree of electrocatalytic signal amplification depends upon the coupling of the catalysis with the electrode surface and the electrode sink (28). Additionally, robust reporting by electrocatalysis is dependent on the rigorous shielding of the electron sink from the electrode surface, which prevents direct reduction by the electrode and false positive outputs (27). In electrocatalysis, the redox reporter is coupled with a freely diffusing electron sink, typically ferricyanide. Methylene blue (MB) is typically used as the redox reporter for DNA-mediated electrocatalysis as it is well-coupled to the base stack through intercalation (26). Reduction of the reporter leads to the reduction of the electron sink, returning the reporter back to its oxidized form, and allowing for the repeated interrogation of the base stack. To complete this catalytic cycle reporting on DNA-mediated changes, the reporter must interact with both the DNA film and the freely diffusing electron sink (26). Finally, in order for DNA-modified electrodes to be viable for the detection of DNA-binding proteins and the hybridization of oligonucleotides, the surface-bound DNA duplex must be sufficiently accessible (29–32). All three of these constraints must be met for electrocatalysis to be applied as a detection platform in DNA CT-based systems: dual reporter interactions, robust shielding of the electron sink from surface reduction, and DNA accessibility.
Initial electrocatalysis experiments were performed using noncovalently bound DNA reporters and high-density DNA films (2, 3, 25, 26). The freely diffusing noncovalent reporter was able to readily interact with both the electron sink and the base stack while the high-density DNA films limited localization of the reporter to the distal end of the duplex. The high-density films also imposed a strong kinetic barrier to the direct reduction of ferricyanide at the electrode surface (2). This platform demonstrated enhanced sensitivity due to electrocatalytic signal amplification and allowed for efficient detection of single-base mismatches (2, 3, 25, 26). The major limitation of this platform originated from the need for well-packed films to passivate the surface and ensure that the redox probe bound to the top of the DNA film. These high-packing densities can be difficult to achieve reproducibly and lead to poor accessibility of the duplex DNA for analyte binding (29–32).
Low-density DNA films are now the standard means for promoting efficient DNA binding and hybridization events, as they provide higher sensitivity and specificity. Low-density DNA films, however, require covalent tethering of the reporter to the duplex to ensure placement of the reporter near the top of the surface. This covalent tethering of the reporter is furthermore essential for label-free assays of DNA or protein targets. However, low-density films expose the electrode surface, as the negatively charged phosphate backbone of the immobilized DNA is insufficient to impose a kinetic barrier against the direct reduction of positively charged species, such as ferricyanide. A previous attempt at electrocatalysis with low-density films utilized negatively charged functionalized mercapto-alkanes to shield ferricyanide, the electron sink, from the electrode (27). This platform has not been widely employed due to the limited solution accessibility of the reporter and the difficulty imposing sufficient kinetic barriers to the reduction of ferricyanide to drive electrocatalysis. Additionally, this platform did not address the poor interaction between the reporter and the electron sink imposed by the short, rigid covalent tether linking the reporter to the duplex (27). Therefore, a new means to promote efficient interactions between the reporter with the base stack and electron sink, as well as to robustly shield the electron sink from the electrode surface, must be developed for the application of electrocatalytic signal amplification with these low-density DNA films.
Redox-active proteins may be useful as electron sinks for electrocatalysis. Enzymes such as horseradish peroxide and glucose oxidase have been shown to readily interact electronically with phenothiazine dyes, such as MB, and have had wide application as recognition elements for molecular sensing (33–42). Detection platforms based on these enzymes are desirable due to their inherent catalytic activity. The application of a protein as an electron sink is also appealing because the protein shell itself can shield the redox core from the electrode surface (34–41). Hemoglobin exhibits this property and has been shown to couple with Nile blue (42). Additionally, unlike many of the proteins typically employed for signal amplification, the reduction of hemoglobin does not generate any DNA-damaging byproducts like peroxide.
Until now, no single DNA-CT sensor has been compatible both with use of low-density DNA films, required for hybridized DNA or protein targets, and robust electrocatalytic signal amplification, required for sufficient sensitivity. In this work, we report a DNA-CT sensor that robustly and efficiently amplifies the signal from low-density DNA films using electrocatalysis. This advancement is the result of a new flexible alkyl linkage between the reporter and the duplex coupled with a novel electron sink. The new flexible linkage allows for the efficient shuttling of electrons between the duplex and the electron sink (28). The use of hemoglobin allows electronic interaction with MB while inherently being shielded from the electrode surface by the protein shell. No specialized backfilling agents or assembly conditions were required, demonstrating the general applicability of this system. This general technique, electrocatalysis of MB with hemoglobin, can therefore be utilized with current DNA-detection strategies without imposing any new constraints to the platform. Most importantly, the enhanced sensitivity permits femtomole DNA sampling.
Results and Discussion
Electrocatalytic Signal Amplification of MB-Modified DNA by Hemoglobin.
In this study, MB is covalently tethered to DNA with a flexible (CH2)8 linkage, such that, upon reduction, the tethered MB mimics the behavior of a noncovalent reporter (Fig. 1). Hemoglobin is known to interact electrochemically with organic dyes, such as MB, and can function as an electronic sink for catalytically reduced MB (42). The general electrocatalytic cycle developed for this study utilizing MB-DNA and hemoglobin is presented in Fig. 2. The two-electron reduction of MB to leucomethylene blue (LB) significantly decreases the binding affinity of the reporter, resulting in its dissociation from the duplex. LB proceeds to electrochemically interact with, and reduce, two equivalents of freely diffusing hemoglobin, returning the reporter to the oxidized form and completing the electrocatalytic cycle.
Fig. 1.
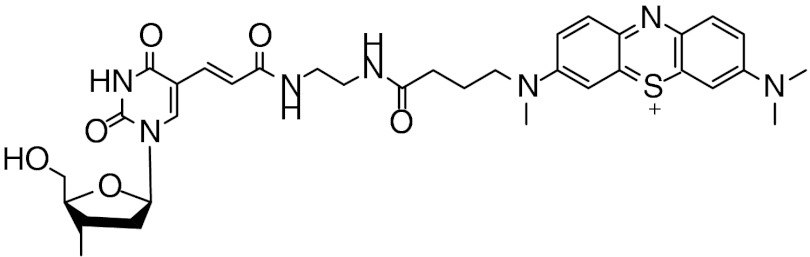
Structure of MB-modified DNA with a flexible (CH2)8 linkage covalently tethering MB to the DNA.
Fig. 2.

Electrocatalytic reduction of MB-modified DNA by hemoglobin. Left: Schematic representation of the electrocatalytic cycle of MB-DNA. In this model, upon reduction of MB to LB, the DNA-binding affinity is significantly decreased, resulting in the dissociation of reporter from the duplex and its subsequent oxidation by freely diffusing hemoglobin. Center: Cyclic voltammetry (scan rate = 30 mV/s) of MB-DNA in the absence (black), DNA without MB in presence (gray) and MB-DNA in the presence (red) of 25 μM hemoglobin in phosphate buffer (5 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7.0). Right: Corresponding chronocoulometry signal (V = -450 mV) acquired over 10 s intervals.
The low-density MB-DNA films employed in this study were assembled without MgCl2 and had typical surface coverages of 1.3 ± 0.2 pmol/cm2. Prior to the addition of hemoglobin, the resulting cyclic voltammograms (CVs) demonstrate a reversible redox couple consistent with a signal generated from MB reduction via DNA-mediated CT: a midpoint potential of -340 mV, peak splitting of -50 mV, and a ratio of near unity for the cathodic and anodic peak areas (Fig. 2). MB-DNA modified electrodes were then examined in the presence of freely diffusing hemoglobin (25 μM). The electrocatalytic signal amplification is apparent in the CV by the characteristic nonreversible signal, where the reductive peak is amplified and the oxidative peak is not observed. Hemoglobin was also added to DNA in the absence of MB, showing that hemoglobin alone has no effect on the observed electrochemistry (Fig. 2). Additionally, the electronic spectrum of hemoglobin showed no significant variation before and after electrocatalysis, indicating that protein integrity was retained (Fig. S1).
In the absence of hemoglobin, chronocoulometry at -450 mV vs. Ag/AgCl, a potential appropriate for reduction MB-DNA, showed a rapid initial accumulation of charge (0.04 μC over 0.3 s), with minimal charge accumulation (0.1 μC) over the subsequent 10 s (Fig. 2). This modest accumulation of charge is consistent with the reduction of surface-bound MB without an electron sink for catalytic cycling. In the presence of hemoglobin, there is the same rapid initial accumulation of charge as in the noncatalytic case, but over the subsequent 10 s, a total of 0.99 ± 0.04 μC of charge is accumulated. A comparison of the accumulated charge as a function of the presence of hemoglobin yields insight into the catalytic behavior of MB. An accumulated charge of 0.99 ± 0.04 μC in the presence of freely diffusing hemoglobin correlates to roughly 10 turnovers per MB-DNA. This electrocatalytic amplification of the accumulated charge due to MB-DNA reduction demonstrates that covalently tethering MB to DNA through a (CH2)8 linkage does not disrupt the ability of hemoglobin to electrochemically couple with MB.
Electrocatalytic Amplification of MB-DNA with Intervening Mismatches.
In order to demonstrate that the electrocatalytically amplified signal is generated via DNA-mediated CT, the signal suppression due to the incorporation of a single-base mismatch was examined. Previously, the degree of signal attenuation upon incorporation of a single-base mismatch indicated that MB covalently tethered to the DNA duplex through a flexible (CH2)12 linkage can be reduced by both DNA CT and direct surface reduction (43). Here, we utilized a shorter (CH2)8 linkage in order to reduce contributions from the direct surface reduction of MB. Using this assembly, Fig. 3 illustrates electrocatalysis with and without an intervening mismatch. Cytosine-guanine (CG)-rich thiol-modified DNA was annealed with complementary well-matched DNA as well as a mismatched sequence containing a single CA mismatched base pair in the middle of the 17-mer duplex. Well-matched and mismatched CG-rich DNAs were each assembled as low-density DNA films on eight electrodes of a multiplexed chip. The use of equivalent thiol strands and multiplexed chips removes variability due to surface quality and film formation.
Fig. 3.

Signal suppression of MB-DNA due to incorporation of a single CA mismatch. Left: Low-density MB-DNA films were assembled on multiplexed chips. Sequence for both well-matched MB-DNA (blue) and mismatched MB-DNA (red) are indicated. Center: In the absence of hemoglobin, CV scans (scan rate = 100 mV/s) were used to demonstrate the characteristic signal attenuation due to incorporation of a single CA mismatch. Right: Chronocoulometry scans (V = -450 mV) were used for electrocatalytically amplified MB-DNA in the presence of 25 μM hemoglobin. All scans were acquired in phosphate buffer (5 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7.0).
As is evident in Fig. 3, signal attenuation is observed in this system with an intervening mismatch. The average reductive peak area from the CVs for well-matched MB-DNA is 5.0 ± 0.4 nC and that of mismatched MB-DNA is 3.2 ± 0.4 nC. This signal attenuation in the presence of a single-base mismatch confirms that the signal observed is generated via DNA-mediated CT. Upon the addition of hemoglobin (25 μM), the MB-DNA signals are electrocatalytically amplified, and the accumulated charge for well-matched and mismatched DNA were compared. The accumulated charge over 10 s from the chronocoulometry data are 0.99 ± 0.04 μC and 0.62 ± 0.02 μC for well-matched and mismatched MB-DNA, respectively (Fig. 3). The accumulated charge obtained for the mismatched MB-DNA was again only 62% of the well-matched MB-DNA signal upon electrocatalytic amplification. The consistency in the signal suppression due to mismatch incorporation indicates that the catalytic reduction of MB-DNA occurs via DNA CT. Therefore, the sensitivity of the observed signal to subtle structural perturbations to the π-stack is still apparent with electrocatalytic amplification.
Variations in pH and Temperature on Amplification.
DNA-modified electrodes are generally stable over the pH range of 5.5–8.5, where minimal damage occurs to DNA. The reduction peak area of MB-DNA, without amplification, is unaffected by changes in pH, while the peak potential is shifted positive with increasing acidity at a rate of 25 mV per pH unit (Fig. S2). This is expected, as MB is protonated upon reduction. Previous work with enzyme-mediated electrocatalytic cascades has demonstrated a nonlinear response to pH changes due to the pH dependence of the enzymatic activity (34–42). In these cases, where catalysis is limited by enzyme activity, constraints are imposed not only on the pH but also on other running conditions relevant to enzyme activity (34–42). In the system presented here, the electrocatalysis is not dependent on hemoglobin reactivity with oxygen, only the capability of hemoglobin to function as an electron sink. The electrocatalytic amplification of MB should therefore be optimal at a pH lower than the isoelectric point of hemoglobin (pI = 6.8) where hemoglobin will be less negatively charged (42). We demonstrated this to be true by examining the accumulated charge in the presence of hemoglobin at a range of pH values (5.5–8.5). All solutions were buffered and had the same salt and hemoglobin concentrations. Between pH 5.5 and 8.5, the accumulated charge increases linearly with increasing acidity at a rate of 0.15 μC per pH unit (Fig. S2). This indicates that the electrocatalytic amplification obtained is not limited by hemoglobin activity and behaves as would be expected for a proton-dependent electrocatalytic cycle. Therefore, hemoglobin activity does not impose new restrictions on the running conditions of DNA-modified electrodes compared to free iron, demonstrating the utility of hemoglobin as an electron sink in electrocatalysis.
The dependence of the total accumulated charge was also examined as a function of the temperature to demonstrate both the stability and necessity for protein activity. The temperature was systematically increased from 24 °C to 38 °C at a rate of 0.5 °C/ min. The accumulation of charge was found to increase at a rate of 20 μC/°C in the range of 24–32 °C (Fig. 4). Within this temperature range, mismatch discrimination was still observed in the catalytic signal. At increased temperatures ranging from 32 °C to 40 °C, the accumulated charge increased at a rate of 60 μC/°C, roughly three times faster than at lower temperatures, and the discrimination due to mismatch incorporation was lost (Fig. 4). While hemoglobin is stable under physiological conditions, the thermal denaturation of hemoglobin is highly pH and concentration-dependent. In the buffer used for this study, a transition was observed by circular dichroism at 32–35 °C (Fig. S3). The accelerated charge accumulation and the loss in mismatch discrimination indicate that upon structural changes to hemoglobin the heme-bound iron is no longer effectively shielded by the protein shell. These low-density DNA films are not designed for passivation against the direct reduction of iron. This temperature profile demonstrates the necessity for intact hemoglobin to shield the iron from the electrode surface in order to observe the electrocatalytically amplified MB-DNA signal and avoid false positives.
Fig. 4.

Temperature dependence of the electrocatalysis of MB-DNA. The temperature was increased at a rate of 0.5 °C/ min by a Peltier device placed beneath the multiplexed chip. The electrocatalytic amplification was monitored by chronocoulometry at two-second intervals. Left: The accumulated charge is plotted as a function of the measured solution temperature. Two different linear regimes can be discerned with rates of 20 μC/°C and 60 μC/°C at moderate (24–32 °C) and high (32–40 °C) temperatures, respectively. Right: In each regime, the effect on mismatch discrimination was examined by chronocoulometry; well-matched (blue) and mismatched (red) MB-DNA are shown at both 25 °C (Bottom) and 38 °C (Top). All scans were acquired in phosphate buffer (5 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7).
Detection of Restriction Enzyme Activity.
Having demonstrated that hemoglobin is an effective electron sink in DNA-mediated electrocatalysis, we can apply this electrocatalysis to the detection of DNA-binding proteins. The restriction enzyme RsaI sequence-specifically cuts duplex DNA that contains the binding site 5′-GTAC-3′ (8). RsaI activity was compared between DNA sequences with and without the binding site (Fig. 5). Half the electrodes of a multiplex chip were treated with RsaI (20 μL of 50 nM) while the other half were left untreated. A comparison between the treated and untreated electrodes was made for each sequence in order to demonstrate the sequence-specificity of RsaI. Additionally, RsaI activity and electrocatalytic signal amplification were compared using both high-density and low-density DNA films to demonstrate the effect of duplex accessibility on the detection of DNA-binding proteins.
Fig. 5.

Electrocatalytic detection of sequence-specific restriction enzyme RsaI activity. Left: Sequences for the control and RsaI binding sequence are indicated. The binding site for RsaI is indicated in purple. All electrodes were incubated with BSA (1 mM for 1 h to minimize any signal attenuation observed from nonspecific protein binding). Electrodes were either Left untreated (red) or treated (green) with RsaI (20 μL of 50 nM) for 2 h. Right: The accumulated charge from the chronocoulometry data (V = -450 mV for 10 s) of MB-DNA in the presence of 25 μM hemoglobin for the control sequence is plotted for both high-density (dark) and low-density (light) DNA films. High-density films were formed by the addition of 100 mM MgCl2 during DNA film self-assembly. The analogous data for the RsaI sequence is also presented.
Low-density films of the RsaI cutting sequence showed accumulated charges of 0.55 ± 0.02 μC for untreated samples and 0.32 ± 0.01 μC for samples treated with RsaI (Fig. 5). With these low-density DNA films, this corresponds to a 40% signal attenuation due to treatment with RsaI. This signal attenuation was sequence-specific, as electrodes modified with low-density DNA films of the control sequence showed no differential from those treated with RsaI (Fig. 5).
Using the traditionally implemented high-density DNA films, no statistically relevant decrease could be discerned from either the RsaI sequence or the control sequence upon RsaI treatment. The RsaI sequence had accumulated charges of 0.36 ± 0.05 μC for untreated and 0.34 ± 0.08 μC for treatment with RsaI, while the control sequence had accumulated charges of 0.38 ± 0.02 μC for untreated and 0.41 ± 0.03 μC for treatment with RsaI (Fig. 5). In addition to the lack of detectable RsaI activity with high-density DNA films, there was significantly higher variability in the electrocatalytically amplified signal with high-density films.
Overall, the low-density DNA films had enhanced electrocatalytic activity and decreased variability compared to the high-density films which can be attributed to the enhanced accessibility of both RsaI and hemoglobin. Thus, electrocatalysis with hemoglobin clearly requires duplex accessibility.
Sensitivity of DNA Detection.
The sensitivity to the reduction of MB-DNA is compared with and without hemoglobin to demonstrate the enhanced sensitivity that is achieved with the overall signal gain of electrocatalysis. The reduction signal obtained from electrodes assembled with decreasing amounts of MB-DNA was compared with and without electrocatalysis. In this experiment, each set of four electrodes on a 16-electrode multiplexed chip was assembled with 20 μL of MB-DNA of varying concentrations. The electrodes were exposed to MB-DNA ranging from 25 μM (500 pmol) down to 250 pM (5 fmol) and allowed to assemble overnight. Once thoroughly washed and backfilled with mercaptohexanol, the electrodes were examined by CV in the absence and chronocoulometry in the presence of electrocatalysis with hemoglobin (25 μM).
The area of the reduction signal from MB-DNA in CV measurements was found to decrease with decreasing amounts of MB-DNA from 500 pmol down to 500 fmol (Fig. 6). This represents a dynamic range of only three orders of magnitude; assembly with less than 500 fmol of MB-DNA results in no discernible peaks from MB-DNA reduction in the CV. Upon addition of hemoglobin, the accumulated charge after 10 s measured by chronocoulometry was examined as a function of the amount of MB-DNA. Electrocatalytic signal amplification with hemoglobin resulted in a significantly enhanced dynamic range, where the decrease in the accumulated charge due to decreasing moles of MB-DNA during monolayer formation was discernible from 500 pmol down to 5 fmol, corresponding to an enhanced dynamic range of five orders of magnitude (Fig. 6). This enhancement corresponds to a 100-fold improvement in the sensitivity of the device and a dynamic range down to femtomoles.
Fig. 6.

Enhanced sensitivity for MB-DNA reduction upon electrocatalytic signal amplification. Electrodes were incubated with 20 μL of MB-DNA of varying concentrations, ranging from 25 μM down to 250 pM, resulting in the electrodes being exposed to 500 pmol down to 5 fmol of MB-DNA during monolayer formation. The surfaces were thoroughly washed and backfilled with mercaptohexanol prior to measurement. Scans were performed in phosphate buffer (5 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7) with and without hemoglobin (25 μM). The reduction of MB-DNA was quantified from the reductive peak area in the CV (black), without hemoglobin, and the accumulated charge after 10 s in the chronocoulometry (red), with hemoglobin.
Implications.
In this study, a new means to improve the sensitivity of DNA-modified electrodes by electrocatalysis is presented using the novel electron sink hemoglobin. The use of low-density films is integral for current DNA-binding protein and oligonucleotide detection strategies, and electrocatalysis with hemoglobin is shown to be the first compatible means of enhancing the overall signal gain of these systems. Using a new flexible linkage between MB and the DNA, and hemoglobin as an electron sink, DNA-mediated CT in low-density DNA films was electrocatalytically amplified. As an electron sink, hemoglobin is effective through physiologically relevant ranges of pH and temperature and, as a compact protein, does not carry out direct reduction at the surface, minimizing false positives. Restriction enzyme reaction can be readily detected with high sensitivity, given the duplex accessibility of the low-density films and high efficiency of MB reaction with hemoglobin. Most importantly, the sensitivity for MB-DNA reduction was demonstrated to be improved ≥100-fold upon electrocatalytic signal amplification. This enhanced sensitivity, which allows a high signal-to-noise ratio from an electrode assembled with femtomoles of MB-DNA, should provide the response necessary to detect the unlabeled cellular markers, both DNA and protein targets, at low concentrations (19, 20). As electrocatalytic signal amplification with hemoglobin is a general means of enhancing the sensitivity of current DNA-modified electrode technologies, it should be applicable to many DNA electrochemical strategies based on redox reporting of MB.
Materials and Methods
All chemicals were purchased from Sigma Aldrich and used without further purification. All proteins were purchased from New England Biolabs. Oligonucleotides without unnatural modifications were purchased from Integrated DNA Technologies while modified oligonucleotides were synthesized on a 3400 Applied Biosystems DNA synthesizer. Modified phosphoramidites were purchased from Glen Research. Hemoglobin was obtained from Sigma Aldrich as a lyophilized powder from Bovine Blood.
Oligonucleotides Preparation.
MB-modified DNA was prepared by the synthesis of amino-modified DNA utilizing an Amino-(CH2)2-dT phosphoramidite (Fig. 1). Amino-modified DNA, as well as thiol-modified DNA, was purified through standard procedures, as previously reported (6). MB modified with a terminal carboxylic acid was synthesized and coupled to the amino modified DNA (43). All single-stranded DNA was purified by reverse-phase HPLC and characterized with matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry.
Single-stranded DNA stock solutions were prepared in phosphate buffer (5.0 mM phosphate, 50 mM NaCl, and pH 7) and quantified by UV–Vis spectroscopy based on their absorbance at 260 nm. The extinction coefficients for the single-strand DNA were estimated using SciTools from IDT. The extinction coefficient for MB-modified DNA at 260 nm was corrected for the contribution of MB to the absorbance by adding 10,300 M-1 cm-1 to the IDT-calculated extinction coefficient. All DNA solutions were thoroughly deoxygenated with argon prior to annealing. Equimolar amounts of single-stranded stocks were combined and annealed by heating to 90 °C and cooling to ambient temperature over 90 min.
DNA-Modified Electrodes.
Multiplex chips were employed for the electrochemical experiments and were fabricated as previously reported (6). Chips consisted of 16 gold electrodes (2 mm2 area) that were divided into four quadrants of four electrodes. Duplex DNA (25 μL of 25 μM, except for concentration dependence experiment) was assembled overnight (20–24 h) in a humid environment, allowing for monolayer formation. Low-density films were assembled in the absence of MgCl2 while high-density films were assembled with the addition of 100 mM MgCl2. Once DNA films were assembled and thoroughly washed with phosphate buffer, the electrodes were backfilled with 1 mM 6-mercaptohexanol (MCH) for 45 min in phosphate buffer with 5% glycerol. The electrodes were scanned in a common running buffer (5.0 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7). Electrocatalysis data was acquired following the addition of 25 μM hemoglobin (5 mM phosphate, 5 mM NaCl, 40 mM MgCl2, 5 mM spermidine, and pH 7) to the central well.
Electrochemical Measurements.
Electrochemical measurements were performed with a CHI620D Electrochemical Analyzer and a 16-channel multiplexer from CH Instruments. A three-electrode setup was used with a common Ag/AgCl reference and a Pt wire auxiliary electrodes placed in the central buffer solution. Cyclic voltammetry data were collected at 100 mV/s unless otherwise indicated, and chronocoulometry data were acquired at an applied potential of -450 mV for an interval of 10 s unless otherwise indicated. Nonelectrocatalytic CV data were quantified by the area of the reduction peak for MB while electrocatalytic chronocoulometry data were quantified by the accumulated charge after 10 s. The surface coverage of the MB-DNA was based on the area of the reduction peak observed for the distally bound MB and calculated as previously reported (6). The catalytic turnover was calculated based on the ratio of the accumulated charge in the presence and absence of hemoglobin.
Temperature Dependence.
Temperature dependence experiments were performed by assembling the chip in a holder with a Peltier (thermoelectric) device (Melcor Corp.) placed underneath the chip. The temperature was measured using a digital probe placed in the central well containing the running buffer. Chronocoulometry data were repeatedly acquired at pulse widths of two seconds, while heating the chip at a rate of 0.5 °C/ min over a temperature range of 24–38 °C.
Restriction Enzyme Detection.
DNA-modified electrodes used for RsaI detection were incubated with BSA (1 mM in 5.0 mM phosphate, 50 mM NaCl, 4.0 mM MgCl2, 4.0 mM spermidine, 50 μM EDTA, 10% glycerol, and pH 7) for 1 h to minimize any signal attenuation observed from nonspecific protein binding. The electrodes were then washed and scanned in the running buffer (8). Half of the electrodes were then incubated for 2 h with RsaI restriction enzyme (20 μL of 50 nM) in RsaI reaction buffer (10 mM Tris, 50 mM NaCl, 4 mM spermidine, 10 mM MgCl2, and pH 7.9) and then washed and scanned in phosphate buffer.
Supplementary Material
ACKNOWLEDGMENTS.
This research was supported by the National Institute of Health (GM61077) and ONR (N00014-09-1-1117). We would like to thank Donald S. Clark for an undergraduate fellowship to L.F.G. The authors thank N. Muren for discussions and contributions in fabricating the multiplexed chips. This work was completed in part in the Caltech Micro/Nano Fabrication Laboratory.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201551109/-/DCSupplemental.
References
- 1.Kelley SO, Barton JK. Electrochemistry of methylene blue bound to a DNA-modified electrode. Bioconjug Chem. 1997;8:31–37. doi: 10.1021/bc960070o. [DOI] [PubMed] [Google Scholar]
- 2.Kelley SO, Boon E, Barton JK, Jackson N, Hill M. Single-base mismatch detection based on charge transduction through DNA. Nucleic Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon E, Ceres D, Drummond T, Hill M, Barton JK. Mutation detection by electrocatalysis at DNA-modified electrodes. Nat Biotechnol. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 4.Boon E, Salas J, Barton JK. An electrical probe of protein–DNA interactions on DNA-modified surfaces. Nat Biotechnol. 2002;20:282–286. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 5.Gorodetsky A, Buzzeo MC, Barton JK. DNA-mediated Electrochemistry. Bioconjug Chem. 2008;19:2285–2296. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slinker J, Muren N, Gorodetsky A, Barton JK. Multiplexed DNA-modified electrodes. J Am Chem Soc. 2010;132:2769–2774. doi: 10.1021/ja909915m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelley SO, Jackson NM, Hill MG, Barton JK. Long-range electron transfer through DNA films. Angew Chem Int Ed. 1999;38:941–945. doi: 10.1002/(SICI)1521-3773(19990401)38:7<941::AID-ANIE941>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Slinker J, Muren N, Renfrew S, Barton JK. DNA charge transport over 34 nm. Nat Chem. 2011;3:230–235. doi: 10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy CJ, et al. Long-range photoinduced electron transfer through a DNA helix. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 10.Cai Z, Sevilla MD. Long range charge transfer in DNA II. In: Schuster GB, editor. Topics in Current Chemistry. Vol 237. Berlin, Heidelberg: Springer; 2004. p. 103. [Google Scholar]
- 11.Wagenknecht HA, editor. Charge Transfer in DNA: From Mechanism to Application. Weinheim: Wiley-VCH Verlag GmbH & Co KGaA; 2005. [Google Scholar]
- 12.Holmlin ER, Dandliker PJ, Barton JK. Charge transfer through the DNA base stack. Angew Chem Int Ed. 1997;36:2714–2730. [Google Scholar]
- 13.Genereux JG, Barton JK. Mechanisms for DNA charge transport. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 15.Kelley SO, Barton JK. Electron transfer between bases in double helical DNA. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 16.Kelley SO, Holmlin RE, Stemp EDA, Barton JK. Photoinduced electron transfer in ethidium-modified DNA duplexes: Dependence on distance and base stacking. J Am Chem Soc. 1997;119:9861–9870. [Google Scholar]
- 17.Delaney S, Barton JK. Long-range DNA charge transport. J Org Chem. 2003;68:6475–6483. doi: 10.1021/jo030095y. [DOI] [PubMed] [Google Scholar]
- 18.Genereux JC, Boal AK, Barton JK. DNA-mediated charge transport in redox sensing and signaling. J Am Chem Soc. 2010;132:891–905. doi: 10.1021/ja907669c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Husale S, Persson H, Sahin O. DNA nanomechanics allows direct digital detection of complementary DNA and microRNA targets. Nature. 2009;462:1075–1079. doi: 10.1038/nature08626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang Z, et al. Direct profiling of cancer biomarkers in tumor tissue using a multiplexed nanostructured microelectrode integrated circuit. ACS Nano. 2009;3:3201–3213. doi: 10.1021/nn900733d. [DOI] [PubMed] [Google Scholar]
- 21.Shao Z, Liu Y, Xiao H, Li G. PCR-free electrochemical assay of telomerase activity. Electrochem Commun. 2008;10:1502–1504. [Google Scholar]
- 22.Xia F, et al. An electrochemical supersandwich assay for sensitive and selective DNA detection in complex matrices. J Am Chem Soc. 2010;132:14346–14348. doi: 10.1021/ja104998m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe A, et al. Electrochemical biosensors employing an internal electrode attachment site and achieving reversible, high gain detection of specific nucleic acid sequences. Anal Chem. 2011;83:9462–9466. doi: 10.1021/ac202171x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das J, Yang H. Enhancement of electrocatalytic activity of DNA-conjugated gold nanoparticles and its application to DNA detection. J Phys Chem C. 2009;113:6093–6099. [Google Scholar]
- 25.Boon E, et al. An electrochemical probe of DNA stacking in an antisense oligonucleotide containing a C3′-endo-locked sugar. Angew Chem Int Ed. 2002;41:3402–3405. doi: 10.1002/1521-3773(20020916)41:18<3402::AID-ANIE3402>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 26.Boon E, et al. Reduction of ferricyanide by methylene blue at a DNA-modified rotating-disk electrode. Langmuir. 2003;19:9255–9259. [Google Scholar]
- 27.Gorodetsky A, Hammond W, Hill M, Slowinski K, Barton JK. Scanning electrochemical microscopy of DNA monolayers modified with Nile blue. Langmuir. 2008;24:14282–14288. doi: 10.1021/1a8029243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anson F. Kinetic behavior to be expected from outer-sphere redox catalysts confined within polymeric films on electrode surfaces. J Phys Chem. 1980;84:3336–3338. [Google Scholar]
- 29.Zhang J, Wang L, Pan D, Song S, Fan C. DNA hybridization “turns on” electrocatalysis at gold electrodes. Chem Commun. 2007;43:1154–1156. doi: 10.1039/b615742d. Cambridge. [DOI] [PubMed] [Google Scholar]
- 30.Lapierre M, O’Keefe M, Taft B, Kelley S. Electrocatalytic detection of pathogenic DNA sequences and antibiotic resistance markers. Anal Chem. 2003;75:6327–6333. doi: 10.1021/ac0349429. [DOI] [PubMed] [Google Scholar]
- 31.Lapierre-Devlin M, et al. Amplified electrocatalysis at DNA-modified nanowires. Nano Lett. 2005;5:1051–1055. doi: 10.1021/nl050483a. [DOI] [PubMed] [Google Scholar]
- 32.Biagiotti V, et al. Probe accessibility effects on the performance of electrochemical biosensors employing DNA monolayers. Anal Bioanal Chem. 2012;402:413–421. doi: 10.1007/s00216-011-5361-0. [DOI] [PubMed] [Google Scholar]
- 33.Pelossof G, Tel-Vered R, Elbaz J, Willner I. Amplified biosensing using the horseradish peroxidase-mimicking DNAzyme as an electrocatalyst. Anal Chem. 2010;82:4396–4402. doi: 10.1021/ac100095u. [DOI] [PubMed] [Google Scholar]
- 34.Du P, Zhou B, Cai C. Development of an amperometric biosensor for glucose based on electrocatalytic reduction of hydrogen peroxide at the single walled carbon nanotube/Nile blue A nanocomposite modified electrode. Electroanal Chem. 2008;614:149–156. [Google Scholar]
- 35.Qian J, Liu Y, Liu H, Yu T, Deng J. An amperometric new methylene blue N-mediating sensor for hydrogen peroxide based on regenerated silk fibroin as an immobilization matrix for peroxidase. Anal Biochem. 1996;236:208–214. doi: 10.1006/abio.1996.0158. [DOI] [PubMed] [Google Scholar]
- 36.Ni F, Feng H, Gorton L, Cotton T. Electrochemical and SERS studies of chemically modified electrodes: Nile blue A, a mediator for NADH oxidation. Langmuir. 1990;6:66–73. [Google Scholar]
- 37.Ju H, Shen C. Electrocatalytic reduction and determination of dissolved oxygen at a poly(Nile blue) modified electrode. Electroanalysis. 2001;13:789–793. [Google Scholar]
- 38.Xu J, Zhu J, Wu Q, Hu Z, Chen H. An amperometric biosensor based on the coimmobilization of horseradish peroxidase and methylene blue on a carbon nanotubes modified electrode. Electroanalysis. 2003;15:219–224. [Google Scholar]
- 39.Liu H, Lu J, Zhang M, Pang D. Electrochemical properties of Nile blue covalently immobilized on a self-assembled thiol-monolayer modified gold electrodes. Anal Sci. 2002;18:1339–1344. doi: 10.2116/analsci.18.1339. [DOI] [PubMed] [Google Scholar]
- 40.Du P, Wu P, Cai C. A glucose biosensor based on electrocatalytic oxidation of NADPH at single-walled carbon nanotubes functionalized with poly(nile blue A) J Electroanal Chem. 2008;624:21–26. [Google Scholar]
- 41.Evtugyn G, Porfierva A, Hianik T, Cheburova M, Budnikov H. Potentiometric DNA sensor based on electropolymerized phenothiazines for protein detection. Electroanalysis. 2008;20:1300–1308. [Google Scholar]
- 42.Kuramitz H, et al. Electrocatalytic reduction of hemoglobin at a self-assembled monolayer electrode containing redox dye, Nile blue as an electron-transfer mediator. Anal Sci. 1999;15:589–592. [Google Scholar]
- 43.Pheeney CG, Barton JK. DNA electrochemistry with tethered methylene blue. Langmuir. 2012;28:7063–7070. doi: 10.1021/la300566x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.