

Abstract
Monoclonal antibodies can specifically bind or even inhibit drug targets and have hence become the fastest growing class of human therapeutics. Although they can be screened for binding affinities at very high throughput using systems such as phage display, screening for functional properties (e.g., the inhibition of a drug target) is much more challenging. Typically these screens require the generation of immortalized hybridoma cells, as well as clonal expansion in microtiter plates over several weeks, and the number of clones that can be assayed is typically no more than a few thousand. We present here a microfluidic platform allowing the functional screening of up to 300,000 individual hybridoma cell clones within less than a day. This approach should also be applicable to nonimmortalized primary B-cells, as no cell proliferation is required: Individual cells are encapsulated into aqueous microdroplets and assayed directly for the release of antibodies inhibiting a drug target based on fluorescence. We used this system to perform a model screen for antibodies that inhibit angiotensin converting enzyme 1, a target for hypertension and congestive heart failure drugs. When cells expressing these antibodies were spiked into an unrelated hybridoma cell population in a ratio of 1∶10,000 we observed a 9,400-fold enrichment after fluorescence activated droplet sorting. A wide variance in antibody expression levels at the single-cell level within a single hybridoma line was observed and high expressors could be successfully sorted and recultivated.
Keywords: single-cell screening, high-throughput screening, cell-based assay, monoclonal antibody, angiotensin converting enzyme 1
Antibodies are powerful research and diagnostic tools and have proven to be potent therapeutics against infectious, autoimmune, and neoplastic diseases. Indeed, the number of therapeutic monoclonal antibodies (mABs) reaching the market is increasing exponentially (1) and the global monoclonal antibody market for therapeutic use was $38 billion in 2009 (2). Monoclonal antibodies can be screened very efficiently for binding using phage display and related technologies (3). However, binding alone is not sufficient; therapeutic antibodies must also modulate (typically inhibit) the activity of the target whereas these methods select only for binding to a drug target and not for inhibition of its function. To overcome this limitation, functional antibody screens are typically carried out using hybridoma cell technology (4). In this approach, laboratory animals are immunized with the antigen of interest before antibody-releasing B-cells are isolated from spleen. These B-cells are then rendered immortal by fusion with myeloma cells, diluted to generate microtitre plate wells containing single cells and expanded to form clonal populations. Subsequently, the supernatant of each population can be tested to screen for the desired activity. However, the need for clonal cell expansion (to obtain detectable concentrations of antibodies), and hence cell immortalization, typically limits the number of clones that can be screened to no more than a few thousand. Improved techniques have been described facilitating the screening of > 105 clones in < 12 h, based on, for example, antigen-based microarrays (5), or compartmentalization of individual cells in 0.1–1 nL lithographically fabricated microwells (6). Still, these methods can only be used to screen for binding activity and do not allow functional assays.
Droplet-based microfluidics (7) holds great potential for functional high-throughput screening at the single-cell level. In these systems, cells are encapsulated into aqueous droplets surrounded by an immiscible carrier phase (e.g., fluorinated oil) (8). Each droplet serves as a miniaturized assay vessel of picoliter-nanoliter volume, and up to several hundred droplets can be generated per second. Furthermore, fluorescence assays and fluorescence-activated droplet sorting (FADS) can be carried out at a similar throughput (9). FADS is similar to fluorescence-activated cell sorting (FACS), but is not limited to sorting based on intracellular or cell-surface markers: With FADS the entire microvessels are sorted rather than cells, allowing screening of secreted proteins (such as antibodies) as well. Typically, all components of a fluorescence assay are added directly during encapsulation, or at a later time point upon fusion with a second droplet species hosting the assay reagents. Subsequently, the droplets pass through a laser beam, the emitted light is measured, and droplets with particular fluorescence intensities are diverted into a collection channel using electric fields (7). Although these steps have been demonstrated individually for small droplets (approximately 30 μm), an integrated chip combining all required modules in a single platform, as well as fusion and sorting modules allowing droplets big enough for the cultivation of mammalian cells (approximately 100 μm in diameter) to be manipulated, have not previously been described. In consequence, single mammalian cells have been analyzed in droplets (8) and it had also been shown that detectable antibody concentrations can be obtained from individually encapsulated hybridoma cells (10), but the sorting of encapsulated mammalian cells based on a functional screen has not yet been achieved. We present here a fully integrated system that can overcome this limitation and demonstrate the screening and sorting of hybridoma cells for the release of antibodies inhibiting angiotensin converting enzyme 1 (ACE-1; Fig. 1A).
Fig. 1.

Microfluidic setup. (A) A mixed population of hybridoma cells either expressing the ACE-1 inhibitory antibody 4E3 or the noninhibitory antibody Elec-403 is encapsulated into droplets together with recombinant ACE-1. (B) Fluorogenic ACE-1 substrate. The ACE-1 cleavage site is indicated by an arrow. (C) Microfluidic chip for cell encapsulation. (D) Integrated microfluidic chip for the reinjection of droplets after a 6 h off-chip incubation period. Droplets hosting cells are fused with droplets containing the fluorogenic ACE-1 substrate and subsequently incubated for 30 min in a delay line. The final sorting module allows specific collection of droplets with low fluorescence intensity (indicating inhibition of ACE-1).
ACE-1 plays a key role in the regulation of blood pressure and the development of vascular pathology and remodeling. It is a type I integral membrane protein which is converted into a soluble circulating form by membrane protein secretases (11). ACE-1 has two catalytic domains, the so-called N- and C-domain, both having the capacity to hydrolyze the same peptides (angiotensin I and bradykinin). However, the two catalytic sites have different substrate specificities and catalytic properties as well as different affinities for competitive inhibitors (12, 13). Monoclonal antibodies against this enzyme have been used in structural and functional studies of ACE-1 (14, 15) and mAbs against ACE-1 have both diagnostic and therapeutic potential: ACE-1 is a target for drugs to treat hypertension and congestive heart failure (16). Here we demonstrate the enrichment of hybridoma cells secreting the ACE-1 inhibitory mAb 4E3 (14, 15) from a large excess of unrelated hybridoma cells.
Results
Design of the Microfluidic Platform.
We developed here an integrated microfluidic system consisting of a previously described drop maker for the encapsulation of cells into droplets with a volume of 660 pL (8) (Fig. 1C) and a unique device for the manipulation of these droplets subsequent to an off-chip incubation period (drops are collected outside the microfluidic device in a separate syringe). During this incubation period detectable concentrations of antibodies are released from encapsulated hybridoma cells into the droplets (Fig. 1A). In the next step, the syringe in which the droplets were incubated is used to reinject them into a second microfluidic chip (Fig. 1D). This second chip allows the addition of further substrates by droplet fusion, as well as incubation and sorting based on fluorescence. The droplet fusion module (Fig. S1 and Movie S1) is a further development of a pillar-induced passive fusion device (17), into which we integrated a second drop maker: Large droplets (660 pL) hosting single hybridoma cells slow down in the pillar chamber (due to the drainage of oil) and are brought into contact with small (25 pL volume) droplets containing a custom-made FRET peptide (Fig. 1B) mediating a green fluorescence signal upon hydrolysis by recombinant ACE-1. These 25 pL droplets are generated in the absence of any surfactant (amphiphilic compounds usually added to stabilize the droplets and to reduce their adherence to the channel walls; also referred to as wetting) and stop when reaching the pillar chamber due to wetting and the drainage of oil. Furthermore, they fuse spontaneously with any incoming large droplets, even if these are stabilized with surfactant (as required to allow off-chip incubation). A high level of synchronization between the small and the large droplets can be achieved, resulting in the reliable addition of specific amounts of substrates.
Efficiency of the Droplet Fusion Module.
To analyze the fusion efficiency at different flow rates, we generated small (25 pL) and large (660 pL) droplets on the same chip and labeled them with different fluorescein concentrations. Subsequently, we varied the aqueous flow rates of both types of droplets and monitored the fusion efficiency by video analysis and fluorescence measurements. Precise 1∶1 fusion events were obtained for a range of different flow rates at frequencies of up to 50 Hz and with an efficiency of more than 99% (Fig. S2A, regime I and Movie S1). Failure of the fusion process occurred only if either the frequency of the small droplets was too high (resulting in the release of nonfused small droplets from the pillar chamber; Fig. S2A, regime II), or if the frequency of the large droplets was very high in comparison to that of the small droplets (Fig. S2A, regime III), in which case the large droplets passed the small droplets without fusion.
Screening of a Heterogeneous Hybridoma Cell Population.
Next, we performed a model screen. For this purpose we stained 4E3-hybridoma cells, producing an mAB inhibiting ACE-1, with calcein-red/orange and diluted them in a 1∶75 excess of nonstained hybridoma cells releasing an unrelated antibody [Elec-403 antibody inhibiting Electrophorus electricus acetylcholinesterase (18)]. This cell suspension was subsequently encapsulated into 660 pL droplets together with recombinant ACE-1. The average number of cells per droplet was approximately 0.3, as measured by video analysis of the cell encapsulation process (1,000 droplets in total) (Movie S2: 65.7% empty drops; 29.5% drops with single cells; 4.8% drops with more than one cell). These results are in good agreement with previous studies showing that the number of cells per droplet follows a Poisson distribution when encapsulating human cell lines in this device (8). These experiments also demonstrated that adherent as well as suspension cells showed a viability of 90% and above during the first two days in drops of the same volume.
After encapsulating hybridoma cells, we incubated the resulting emulsion for 6 h off-chip to obtain significant antibody concentrations (around 20 μg/mL). Longer incubation times resulted in even higher 4E3 antibody concentrations (> 30 μg/mL; Fig. S3A), but turned out to be incompatible with the downstream assay for ACE-1 activity: We observed that with increasing incubation times the supernatants showed higher levels of unspecific conversion of the ACE-1 FRET substrate (Fig. S3A and B). Following off-chip incubation, the droplets were reinjected into the second device (Fig. 1D and Movie S3), fused with droplets containing the fluorogenic ACE-1 substrate and incubated in a delay line for another 30 min (to facilitate generation of the fluorescent product). Finally, the droplets were analyzed and sorted, triggered on fluorescence (19) (Fig. 1D and Movie S4). When the green fluorescence intensity was plotted against the droplet width [used to measure droplet coalescence (8)], three populations were observed (Fig. 2A). One main population showing strong green fluorescence corresponded to droplets in which no inhibition of ACE-1 occurred. Another very sharp, but much smaller population exhibiting almost no green fluorescence signal corresponded to droplets that did not obtain any fluorogenic substrate (due to failed fusion). In between these populations a third population of droplets with intermediate green fluorescence signals could be observed. This last population corresponds to droplets in which inhibition of ACE-1 occurred and was characterized by a high variance in green fluorescence. As with FACS, we set gates for different green fluorescence intensities (Fig. 2A) and collected the corresponding droplets separately. After breaking the emulsion, the recovered cells were analyzed for calcein-red/orange staining. Whereas before the sort only 3% of the mixed cell population were calcein-red/orange-positive (corresponding to 4E3-expressing cells), this value increased to approximately 83% for cells recovered from droplets with intermediate fluorescence signals (Fig. 2B). In contrast, the cell populations recovered from droplets with high fluorescence intensity (indicating no inhibition of ACE-1) included only 0.8–6% stained cells. The presence of some stained cells in the noninhibited population is not surprising, as dead 4E3 hybridoma cells or cells expressing only low levels of 4E3 antibodies inevitably end up in this population, too.
Fig. 2.

Sorting of 4E3 hybridoma cells mixed with a 75-fold excess of unrelated control cells. A population of calcein-red/orange-stained hybridoma cells expressing 4E3 antibody and nonstained hybridoma cells expressing Elec-403 antibody was mixed in a 1∶75 ratio and sorted. The fluorescence signals (green channel corresponding to ACE-1 activity; x axis) of the drops at the sorting junction were plotted against the droplet width (y axis) and gates for the collection of droplets with specific fluorescence intensity were set (green and red rectangles). The relative frequency of all events is color coded as indicated on the right. Bright field (Top) and red/orange fluorescence (Bottom) images shows the nonsorted cell population as well as cells recovered from droplets within the specific gates. The percentage of calcein-red/orange-stained hybridoma cells is indicated.
Biochemical Characterization of Cell Culture Supernatants from Recovered Sorted Cells.
Next, we repeated the experiment using multiple narrow gates for the sorting of droplets with intermediate fluorescence intensity (indicating ACE-1 inhibition) and analyzed the secretion of 4E3 antibody from cultured recovered cells, as well as the ACE-1-inhibitory activity of the supernatants (Fig. 3). Compared to the nonsorted population, cultures of cells recovered from droplets within all of these gates showed strongly increased yields of 4E3 antibody (up to 12.5-fold), as determined by ELISA. In contrast, cultures of cells recovered from the two other droplet populations (nonfused droplets and droplets with high fluorescence intensity) did not show elevated levels of 4E3 antibody. Analysis of cell culture supernatants for ACE-1 inhibition showed similar results: Whereas supernatants from the nonsorted population did not significantly decrease ACE-1 activity, supernatants of cells recovered from droplets with intermediate fluorescence showed a strong inhibitory effect (approximately 50% decrease in ACE-1 activity). In fact, some of these nonpurified supernatants lowered ACE-1 activity even more than purified antibodies (200 μg/mL) from the 4E3 cell line.
Fig. 3.

Biochemical characterization of supernatants from recovered cells after sorting a 1∶75 mixture of 4E3 and Elec-403 hybridoma cells. (A) The fluorescence signals (green channel corresponding to ACE-1 activity; x axis) of the drops at the sorting junction were plotted against the droplet width (y axis). The relative frequency of all events is color coded as indicated on the right. Cells were recovered from droplets within gates for specific fluorescence intensities (colored rectangles labeled with capital letters), expanded and characterized biochemically (B and C). The percentage of total droplets in each gate is indicated. (B) Concentration of 4E3 antibody in the supernatant of the recovered, expanded, cells determined by ELISA. Error bars correspond to ± 1 standard deviation. (C) Activity of recombinant ACE-1 in the presence of the corresponding cell culture supernatants. Control samples refer to samples without ACE-1 (negative) or without antibodies (positive).
Mimicking the Selection of Individual Clones from Large Heterogeneous Populations.
To mimic the selection of individual hybridoma cell clones from large heterogeneous populations, we repeated the experiments using higher dilutions of the 4E3 hybridoma cells (4E3 and Elec-403 hybridoma cells in ratios of 1∶1,000 and 1∶10,000) and additionally performed clonal expansion of individually sorted cells. We again stained the 4E3 hybridoma cells prior to the sort to allow direct measurement of the sorting efficiency. The scatter plot of the fluorescence signals of drops containing these cell mixtures versus the width showed similar results compared to the 1∶75 cell mixture (Figs. 2 and 3). Because of the much lower absolute number of 4E3 cells we set only two gates (1∶1,000 sample) or one gate (1∶10,000 sample) for the collection of droplets showing decreased fluorescence signals (indicating ACE-1 inhibition), plus an additional gate for the main high fluorescence droplet population (Fig. 4).
Fig. 4.

Selection of individual hybridoma cell clones from large heterogeneous populations. Mixed populations of calcein-red/orange-stained hybridoma cells expressing 4E3 antibody and nonstained hybridoma cells expressing Elec-403 antibody in ratios of 1∶1,000 (A) and 1∶10,000 (C) were sorted. The fluorescence signals (green channel corresponding to ACE-1 activity; x axis) of the drops at the sorting junction were plotted against the droplet width (y axis) and gates for the collection of droplets with specific fluorescence intensity were set (green and red rectangles). The percentage of total droplets in each gate is indicated. The relative frequency of all events is color coded as indicated on the right. (B and D) Bright field (Top) and red/orange fluorescence (Bottom) images of the 1∶1,000 and 1∶10,000 cell dilutions before the sort and after recovery from droplets within the specific gates. The percentage of calcein-red/orange-stained hybridoma cells is indicated.
The number of stained hybridoma cells recovered from the inhibited population indicated an enrichment factor of 700-fold for the 1∶1,000 mixture: Before sorting only 0.11% of the mixed cell population were calcein-red/orange-positive (corresponding to 4E3-expressing cells), whereas after the sorting approximately 78% of the cells recovered from droplets with decreased fluorescence signals were calcein-red/orange positive. An even higher enrichment factor of around 9,400-fold was achieved for the 1∶10,000 mixture for which the percentage of stained 4E3 hybridoma cells increased from 0.01% before sorting to 94% after sorting. This higher enrichment factor is consistent with the fact that the main source of false positives is the cocompartmentalization of two cells (one positive and one negative) in the same droplet. With a Poisson distribution of cells in droplets, the maximally achievable enrichment factor inversely correlates with both the initial ratio of positive to negative cells (ε0) and the average number of cells per droplet (λ) (9).
Interestingly the percentage of stained hybridoma cells isolated from the two nonoverlapping gates of the inhibited population in the 1∶1,000 mixture was highly similar (75% in gate A and 78% in gate B), indicating that the higher inhibition of ACE-1 activity in drops from gate A was not a consequence of an increased enrichment of 4E3 hybridoma cells. To further assess this point, we broke the pools of drops sorted using the different gates to directly measure the concentrations of 4E3 antibodies and total IgG levels in the drops (Fig. S4A). This analysis revealed a higher concentration of 4E3 antibodies in drops from gate A compared to gate B (300 μg/mL versus 200 μg/mL), thus indicating that the difference in the inhibition rate of ACE-1 resulted from an altered production rate of the 4E3 antibody inhibiting ACE-1. As expected, drops from gate C contained almost undetectable amounts of 4E3 antibody (0.3 μg/mL) as well as lower amounts of total IgG (30 μg/mL) than drops from gate A and B. In parallel, cells were recovered directly from the pool of broken drops sorted using each gate and 20 single cells were individually pipetted into 96-well plates (one cell per well). After expansion for 14 days, 500 cells for each well were seeded again in fresh medium and incubated for 5 d (Fig. S5). The supernatants were then analyzed to measure the concentration of total IgG and 4E3 antibody, as well as for their inhibitory effect on ACE-1. To obtain average values for each population, the same procedure was carried out in parallel using 500 pooled cells from each gate. Overall, 590 cells were isolated from 657 sorted droplets within gate A and 640 cells were isolated from 723 sorted droplets within gate B. From these pools all the 20 randomly picked clones seeded into the microtiter plates could be successfully recultivated, indicating that the cell recovery rate does not significantly limit the efficiency of the system. In the experiment based on a 1∶1,000 dilution, high 4E3 antibody concentrations (between 168 μg/mL and 350 μg/mL, equaling the total IgG concentration) were observed in all supernatants of the cells isolated from gate A (Fig. 5) along with elevated ACE-1 inhibition (from 42% to 72% decreased ACE-1 activity). Supernatants of cells isolated from gate B (corresponding to lower ACE-1 inhibition in drops) also showed high concentrations of 4E3 antibodies (between 61 μg/mL and 207 μg/mL), but significantly lower than those from gate A. Furthermore, they reduced ACE-1 activity to a lower extent (by about 22–52%).
Fig. 5.

Biochemical characterization of supernatants from individually expanded hybridoma cell clones before sorting and after recovery from droplets within the specific gates. After clonal expansion of single hybridoma cells isolated from droplets within the specific gates, 500 cells of each clone were seeded in 200 μL of fresh media and the supernatants were analyzed after five days of incubation. In parallel 500 cells from each gate were pooled and characterized the same way (except for the 1∶10,000 sorting where the total number of cells isolated from the inhibited population was 18). The heat maps show the total concentration of antibodies and the concentration of 4E3 antibodies (color coded from green to red), as well as the relative ACE-1 activity of the same supernatants (color coded from dark blue to light blue). The dilution refers to the ratio of 4E3 cells to Elec-403 cells before the sort. Capital letters (A–E) correspond to the gates used during droplet sorting as specified in Fig. 4.
Similar results were obtained in the experiment based on the 1 to 10,000 dilution. However, because of the high dilution factor only 21 droplets fell within gate D and were sorted. Nevertheless, 18 cells could be recovered, indicating a high recovery rate. All but one of the recovered cells were stained with calcein-red/orange indicating that they were 4E3 cells. After clonal expansion, the supernatants contained 4E3 antibody concentrations ranging from 152–330 μg/mL, (corresponding to the total IgG concentration) and mediated an inhibition of ACE-1 of 41–73% (Fig. 5). Cells isolated from gate E (corresponding to drops in which ACE-1 activity was not decreased) neither showed detectable expression of 4E3 antibodies nor any inhibitory effect on ACE-1 activity (Fig. 5). Furthermore, the total IgG levels were comparable to those observed for supernatants of nonsorted 4E3 cells. The correlation between ACE-1 inhibition and 4E3 antibody concentrations for all samples was in very good agreement with control experiments based on different concentrations of purified 4E3 antibody (Fig. S6), hence demonstrating the accuracy of the experiments.
All the 98 sorted cells individually seeded (all gates, 1∶1,000 and 1∶10,000 dilution) were successfully expanded indicating a very high survival rate [more comprehensive data on cell survival in equally sized droplets has been published previously (8)].
Taken together, these results prove that our approach allowed the isolation of hybridoma cells that, over a period of at least three weeks, released up to 10-fold higher 4E3 antibody concentrations, and showed up to 5.6-fold increased inhibition of ACE-1 compared to the supernatant of the nonsorted 4E3 hybridoma cell line.
Discussion
We describe here a droplet-based microfluidic platform integrating modules for the generation, incubation, fusion, and sorting of relatively large droplets (660 pL) well-adapted for assays involving mammalian cells. This technology should open the way for a variety of applications, such as the functional characterization of cell libraries at high throughput. Because the cells are compartmentalized in droplets, even molecules secreted or converted by individual cells are obtained in relatively high concentrations and can hence be assayed efficiently. This feature is a major advantage compared to FACS (20, 21) and should facilitate metabolic studies as well as the functional screening of antibodies. Here, we successfully used this microfluidic platform to sort hybridoma cells for the release of antibodies inhibiting a clinically relevant drug target at a sorting rate of 5 × 104 cells per hour. Noteworthy, the 6 h incubation step, required to allow secretion of antibodies from single, compartmentalized cells, is independent of the total sample number and further droplets can be generated and sorted in the meantime (stored in separate reservoirs for incubation).
Performing the screens on the single-cell level revealed a wide variance in antibody expression levels within a single hybridoma line and cells could be efficiently sorted and recovered based on the level of ACE-1 inhibitory activity. In fact, the antibody secretion rate of 4E3 hybridoma cells (releasing ACE-1 inhibiting antibodies) recovered from droplets with low fluorescence intensity, indicating efficient ACE-1 inhibition, was significantly higher than that of the unsorted 4E3 cell line. ELISA data indicated approximately eightfold higher concentrations of the anti-ACE-1 antibody on average and up to 10-fold higher concentrations for some individual clones. It is well-known that point mutations and chromosome rearrangements frequently occur in hybridoma cell populations and change the expression level of individual cells (22). Over time, this effect can result in a percentage of nonantibody producing cells (within a hybridoma cell population derived from one and the same clone) between 40% and 85% (22) and illustrates the need for frequent single-cell sorting of existing hybridoma cell lines. As demonstrated here, our approach allows the specific selection and expansion of cells releasing higher amounts of antibodies (over a period of at least three weeks) compared to the nonsorted hybridoma cell population. Moreover, the sensitivity of the system should even facilitate the functional screening of large cell libraries subsequent to immunization experiments, because we successfully demonstrated the selection and expansion of individual positive cells (releasing antibodies with desired properties) from a 10,000-fold excess of negative cells. As no cell proliferation is required, this technique could also open the way to directly screen nonimmortalized primary B-cells or plasma cells, which might be particularly useful for the cloning of antibodies from human donors, such as disease survivors expressing therapeutically relevant antibodies of unknown identity. For example, the immune system of HIV-infected individuals sometimes evolves HIV-neutralizing antibodies, but their identification and characterization is still difficult and very time consuming (23). Just recently, a method for the selection of primary B-cells releasing antibodies binding influenza virus hemagglutinin A has been developed in a microtiter plate format and even enabled the identification of a neutralizing antibody (24). However, the screen itself was still based on binding activities and did not allow the direct selection for functional properties. In contrast, the approach described here can overcome this limitation, even though distinguishing between highly inhibitory antibodies secreted at low concentrations and less inhibitory antibodies secreted at high concentrations might be difficult. Nonetheless the results obtained here clearly show an up to 9,400-fold enrichment of cells expressing antibodies with desired properties using fluorescence activated droplet sorting. The false positives observed occasionally are most likely due to the co-encapsulation of a negative and a positive cell into the same droplet and can be reduced by starting with a lower cell density during compartmentalization (at the price of a lower overall throughput) (19). Alternatively hydrodynamic cell encapsulation modules (25–27) allowing the specific generation of droplets hosting single cells could be used. False positives can also be ruled out during downstream biochemical characterizations of sorted cells, at which stage truly quantitative data on the inhibitory potency of the released antibodies can be obtained. Compared to conventional approaches our system also requires fewer cells and should allow screening of a much larger fraction of the immune repertoire: In a conventional hybridoma experiment, typically no more than a few thousands of hybridoma clones are screened which represents only a tiny fraction (approximately 1/104) of the available antibody repertoire in a mouse, and an even smaller fraction (approximately 1/106) of the available human repertoire. Hence the technique described here the selection of antibodies against less immunogenic, but functionally more relevant, epitopes.
Materials and Methods
Heterogeneous hybridoma cell populations [4E3 and Elec-403 (13–15, 18) cells in a ratio of 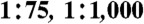 or 1∶10,000] were encapsulated into 660 pL drops at a density of 1.25 × 106 cells/mL together with 1.6 ng/mL ACE-1 (R&D Systems). The resulting emulsion was incubated off-chip for 6 h at 37 °C under a 5% CO2 atmosphere, followed by reinjection into the integrated microfluidic chip (Fig. 1D), where drops containing the hybridoma cells were fused with a second drop species containing a fluorogenic ACE-1 substrate (Fig. 1B). Drops showing low fluorescence intensities (indicating a low ACE-1 activity) were sorted by applying an electrical field via embedded electrodes adjacent to the channels. Sorted drops were broken by adding an equal volume of 1H, 1H, 2H, 2H-Perfluoro-1-octanol (Aldrich) and subsequently hybridoma cells were isolated and seeded into 96-well plates (Fig. S5). After expansion, the supernatants were characterized for their antibody concentration and ACE-1 inhibiting activity. For detailed experimental procedures, see SI Materials and Methods.
or 1∶10,000] were encapsulated into 660 pL drops at a density of 1.25 × 106 cells/mL together with 1.6 ng/mL ACE-1 (R&D Systems). The resulting emulsion was incubated off-chip for 6 h at 37 °C under a 5% CO2 atmosphere, followed by reinjection into the integrated microfluidic chip (Fig. 1D), where drops containing the hybridoma cells were fused with a second drop species containing a fluorogenic ACE-1 substrate (Fig. 1B). Drops showing low fluorescence intensities (indicating a low ACE-1 activity) were sorted by applying an electrical field via embedded electrodes adjacent to the channels. Sorted drops were broken by adding an equal volume of 1H, 1H, 2H, 2H-Perfluoro-1-octanol (Aldrich) and subsequently hybridoma cells were isolated and seeded into 96-well plates (Fig. S5). After expansion, the supernatants were characterized for their antibody concentration and ACE-1 inhibiting activity. For detailed experimental procedures, see SI Materials and Methods.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Christophe Créminon for kindly providing Elec-403 hybridoma cells and Alan Sawyer for valuable comments on the manuscript.
Footnotes
Conflict of interest statement: C.A.M and A.D.G are inventors on patent applications including some of the ideas described in this manuscript.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204514109/-/DCSupplemental.
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 2.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 3.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 4.Karsunke XY, et al. Screening and characterization of new monoclonal anti-benzo[a]pyrene antibodies using automated flow-through microarray technology. J Immunol Methods. 2011;371:81–90. doi: 10.1016/j.jim.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Sawyer A, et al. High throughput production of mouse monoclonal antibodies using antigen microarrays. Proteomics. 2005;5:4070–4081. doi: 10.1002/pmic.200401279. [DOI] [PubMed] [Google Scholar]
- 6.Ogunniyi AO, Story CM, Papa E, Guillen E, Love JC. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat Protoc. 2009;4:767–782. doi: 10.1038/nprot.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theberge AB, et al. Microdroplets in microfluidics: An evolving platform for discoveries in chemistry and biology. Angew Chem Int Ed Engl. 2010;49:5846–5868. doi: 10.1002/anie.200906653. [DOI] [PubMed] [Google Scholar]
- 8.Clausell-Tormos J, et al. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem Biol. 2008;15:427–437. doi: 10.1016/j.chembiol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Baret JC, et al. Fluorescence-activated droplet sorting (FADS): Efficient microfluidic cell sorting based on enzymatic activity. Lab Chip. 2009;9:1850–1858. doi: 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- 10.Koster S, et al. Drop-based microfluidic devices for encapsulation of single cells. Lab Chip. 2008;8:1110–1115. doi: 10.1039/b802941e. [DOI] [PubMed] [Google Scholar]
- 11.Parvathy S, et al. Angiotensin-converting enzyme secretase is inhibited by zinc metalloprotease inhibitors and requires its substrate to be inserted in a lipid bilayer. Biochem J. 1997;327:37–43. doi: 10.1042/bj3270037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei L, Clauser E, Alhenc-Gelas F, Corvol P. The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J Biol Chem. 1992;267:13398–13405. [PubMed] [Google Scholar]
- 13.Danilov S, et al. Structure-function analysis of angiotensin I-converting enzyme using monoclonal antibodies. Selective inhibition of the amino-terminal active site. J Biol Chem. 1994;269:26806–26814. [PubMed] [Google Scholar]
- 14.Skirgello OE, et al. Inhibitory antibodies to human angiotensin-converting enzyme: Fine epitope mapping and mechanism of action. Biochemistry. 2006;45:4831–4847. doi: 10.1021/bi052591h. [DOI] [PubMed] [Google Scholar]
- 15.Naperova IA, et al. [Characteristics of monoclonal antibody binding with the C domain of human angiotensin converting enzyme] Bioorg Khim. 2008;34:358–364. doi: 10.1134/s1068162008030126. [DOI] [PubMed] [Google Scholar]
- 16.Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1:621–636. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- 17.Niu X, Gulati S, Edel JB, deMello AJ. Pillar-induced droplet merging in microfluidic circuits. Lab Chip. 2008;8:1837–1841. doi: 10.1039/b813325e. [DOI] [PubMed] [Google Scholar]
- 18.Remy MH, Frobert Y, Grassi J. Characterization of monoclonal antibodies that strongly inhibit Electrophorus electricus acetylcholinesterase. Eur J Biochem. 1995;231:651–658. doi: 10.1111/j.1432-1033.1995.0651d.x. [DOI] [PubMed] [Google Scholar]
- 19.Baret JC, et al. Fluorescence-activated droplet sorting (FADS): Efficient microfluidic cell sorting based on enzymatic activity. Lab Chip. 2009;9:1850–1858. doi: 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- 20.Pierzchalski A, Mittag A, Tarnok A. Introduction A: Recent advances in cytometry instrumentation, probes, and methods—review. Methods Cell Biol. 2011;102:1–21. doi: 10.1016/B978-0-12-374912-3.00001-8. [DOI] [PubMed] [Google Scholar]
- 21.Mouquet H, et al. Memory B cell antibodies to HIV-1 gp140 cloned from individuals infected with clade A and B viruses. PLoS One. 2011;6:e24078. doi: 10.1371/journal.pone.0024078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kromenaker SJ, Srienc F. Stability of producer hybridoma cell lines after cell sorting: A case study. Biotechnol Prog. 1994;10:299–307. doi: 10.1021/bp00027a010. [DOI] [PubMed] [Google Scholar]
- 23.Pietzsch J, et al. Anti-gp41 antibodies cloned from HIV-infected patients with broadly neutralizing serologic activity. J Virol. 2010;84:5032–5042. doi: 10.1128/JVI.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 25.Chabert M, Viovy JL. Microfluidic high-throughput encapsulation and hydrodynamic self-sorting of single cells. Proc Natl Acad Sci USA. 2008;105:3191–3196. doi: 10.1073/pnas.0708321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edd JF, et al. Controlled encapsulation of single-cells into monodisperse picolitre drops. Lab Chip. 2008;8:1262–1264. doi: 10.1039/b805456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abate AR, Chen CH, Agresti JJ, Weitz DA. Beating Poisson encapsulation statistics using close-packed ordering. Lab Chip. 2009;9:2628–2631. doi: 10.1039/b909386a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.