

The crystal structure of CYP38, a proposed photosystem II assembly factor, is reported at 2.39-Å resolution. The structure, along with functional assays, suggests that CYP38 is not an active peptidyl-prolyl cis/trans isomerase, and interaction of the N-terminal helical domain with the putative C-terminal cyclophilin domain implies that CYP38 functions as a chaperone via an intramolecular autoinhibition mechanism.
Abstract
Cyclophilin38 (CYP38) is one of the highly divergent cyclophilins from Arabidopsis thaliana. Here, we report the crystal structure of the At-CYP38 protein (residues 83 to 437 of 437 amino acids) at 2.39-Å resolution. The structure reveals two distinct domains: an N-terminal helical bundle and a C-terminal cyclophilin β-barrel, connected by an acidic loop. Two N-terminal β-strands become part of the C-terminal cyclophilin β-barrel, thereby making a previously undiscovered domain organization. This study shows that CYP38 does not possess peptidyl-prolyl cis/trans isomerase activity and identifies a possible interaction of CYP38 with the E-loop of chlorophyll protein47 (CP47), a component of photosystem II. The interaction of CYP38 with the E-loop of CP47 is mediated through its cyclophilin domain. The N-terminal helical domain is closely packed together with the putative C-terminal cyclophilin domain and establishes a strong intramolecular interaction, thereby preventing the access of the cyclophilin domain to other proteins. This was further verified by protein–protein interaction assays using the yeast two-hybrid system. Furthermore, the non-Leucine zipper N-terminal helical bundle contains several new elements for protein–protein interaction that may be of functional significance. Together, this study provides the structure of a plant cyclophilin and explains a possible mechanism for autoinhibition of its function through an intramolecular interaction.
INTRODUCTION
Immunophilins mediate immune suppression and vary considerably in both form and function. They are classified into two major families (according to their immunosuppressant ligand partners): the FK-506 binding proteins (FKBPs) and the cyclosporin A binding proteins (cyclophilins [CYPs]). Despite little sequence similarity, most immunophilins possess peptidyl-prolyl cis/trans isomerase (PPIase) enzymatic activity, which is important for proper protein folding. However, not all immunophilin functions are explained by the PPIase activity or cyclosporin A binding alone.
Over 300 cyclophilins have been identified from a wide variety of organisms, ranging from archaea to human (Andreeva et al., 1999; Ivery, 2000; Galat, 2003). The abundance and diversity of single and multidomain immunophilins identified to date underline the functional versatility of this family and are further exemplified by the presence of multiple immunophilins within an organism. Compared with other organisms, plants are known to possess a much larger number of immunophilin isoforms (Vallon, 2005; Ahn et al., 2010). The Arabidopsis thaliana genome alone consists of 29 CYP isoforms and 23 FKBP isoforms (He et al., 2004; Romano et al., 2004). The discovery of plant cyclophilins has not only demonstrated conservation of these proteins in a full spectrum of biological systems, but has also provided clues to their potential functions in plants. The early works that proposed the distribution of cyclophilins throughout the plant cell (Breiman et al., 1992; Luan et al., 1994) have been confirmed and expanded by genomic and proteomic approaches, which have provided detailed subcellular localization data for these large gene families (Peltier et al., 2002; Kleffmann et al., 2004).
In Arabidopsis, up to five cyclophilins and 11 FKBPs are predicted to reside in the thylakoid lumen (Edvardsson et al., 2007). The immunophilin with the highest PPIase activity in the thylakoid lumen is FKBP13, whose crystal structure has already been reported (Gopalan et al., 2004, 2006), followed by CYP20-2. The spinach (Spinacia oleracea) homolog thylakoid lumen protein40 (TLP40) (82% sequence identity to At-CYP38) is known to be an active thylakoid lumenal PPIase with enzymatic activity observed in vitro (Fulgosi et al., 1998). The spinach thylakoid lumen has TLP20 (a homolog of At-CYP20-2) and TLP40 as the active PPIases. However, PPIase activity of the Arabidopsis thylakoid lumen has been shown to be restricted to FKBP13 and CYP20-2 (Shapiguzov et al., 2006; Edvardsson et al., 2007; Ingelsson et al., 2009). Hence, it is very likely that the remaining 14 immunophilins in the thylakoid lumen have developed functions other than PPIase activity. Sequence analysis reveals that some of the lumenal immunophilins are so divergent that they have lost most of the conserved active site residues that are essential for PPIase activity (He et al., 2004; Lima et al., 2006). It is known that CYP38 plays a critical role in the assembly and maintenance of photosystem II (PSII) supercomplexes in Arabidopsis. Mutant plants lacking CYP38 display stunted growth and are hypersensitive to light due to defective PSII supercomplexes (Fu et al., 2007). This protein may be essential for the correct folding of the D1 protein and CP43 of PSII and successful assembly of the oxygen evolving complex, whereas the absence of CYP38 renders the PSII complexes extremely susceptible to photoinhibition (Sirpiö et al., 2008). Even though it has been predicted that CYP38 might function as a totally new entity of chaperone or might have developed some other enzymatic activity, it is not clear why it does not have PPIase activity in spite of its having a putative CYP domain. To address this question, we determined the crystal structure of CYP38. We tested and confirmed that CYP38 does not possess PPIase activity, and the structure presented in this article clearly explains why CYP38 is not an active PPIase.
RESULTS AND DISCUSSION
Overall Structure
The crystal structure of the Arabidopsis CYP38 protein (residues 83 to 437 of 437 amino acids) is reported here at 2.39-Å resolution. The structure reveals two distinct domains (Figure 1A). The N terminus consists of a short helix (α1), followed by a helical bundle domain (residues 102 to 216), made up of four helices (α2 to α5) of varying lengths. This domain is followed by a typical cyclophilin domain (residues 238 to 423), made up of a β-barrel that is capped by an α-helix at each end. The two domains are connected by a loop, which has an excess of negatively charged residues. Most interestingly, the extreme N terminus of the protein (residues 83 to 96) gets into the C-terminal cyclophilin domain and forms part of the β-barrel. This feature has not been observed before in a cyclophilin. The loops in the cyclophilin domain are quite disordered. Also, the first 10 of the 14 amino acids of the N-terminal tag linker and the last four C-terminal residues are not observed in the electron density map.
Figure 1.

Structure of CYP38.
(A) Overall structure of CYP38 (83 to 437) with β-helices shown in red, β-strands in yellow, and loops in green. Two N-terminal β-strands that form part of the C-terminal β-barrel are shown in cyan.
(B) CYP domain organization in CYP38 (i) and hCYPA (ii).
(C) Structural overlap of the CYP domain of CYP38 (residues 232 to 433; orange) and hCYPA (residues 1 to 165; lemon green). The N-terminal β-strands of CYP38 that form part of the CYP domain are shown in blue. The structure figures were prepared using PyMOL.
Helical Domain
Helix α1 lies close to the cyclophilin domain. Helices α2 and α3 are split into two shorter fragments each, and the fragments are connected by very short loops. Helices α4 and α5 are long and are connected to each other by a long loop. The α2 and α3 region (residues 125 and 161) was predicted to be a Leu zipper (He et al., 2004), based on the fact that Leu or Ile residues are present almost at every 7th position. However, the present structure does not support this prediction. The helices do not intertwine to form a coiled coil and do not show a well-defined hydrophobic surface with the Leu/Ile residues facing on one side, which is the characteristic feature of the Leu zipper architecture. Also, some of the 7th position Leu residues fall in the loop region between the helices, which is unlikely for a Leu zipper. Even the low-resolution (3.5 Å) electron density map obtained for the wild-type CYP38 clearly indicated that the helical region does not form a zipper domain. This alleviates the doubt that the mutation of three Leu residues (which was needed for structure determination) could have disturbed the Leu zipper arrangement. The four helices of the helical bundle are packed together mainly through hydrophobic interactions. The internal surface of the bundle is highly hydrophobic and is mainly occupied by Leu, Ile, Ala, and Val residues. Furthermore, the helices are rich in charged residues, and all charged and hydrophilic residues protrude toward solvent. The four-helix bundle motif is relatively common in proteins. A DALI search (Holm and Sander, 1993) with the helical bundle (residues 115 to 217) shows that the closest structural homologs are spinach photosystem b Q (PsbQ) (PDB code 1NZE; Z score, 10.1) and Escherichia coli cytochrome b562 (PDB code 256B; Z score, 10.0) (see Supplemental Figure 1 online). PsbQ is a 16-kD oxygen evolving subunit of PSII, and cytochrome b562 is a heme binding protein involved in electron transport as well as DNA-dependent transcription regulation. These structures include a four-helix bundle with up-and-down topology. However, no region of the above two proteins shows any strong sequence similarity to CYP38. PsbQ is only ∼11% identical and aligns with the helical bundle of CYP38 with a root mean square deviation of 1.71 Å over a stretch of 88 residues. Cytochrome b562 is ∼10% identical and aligns with a root mean square deviation of 1.73 Å for 83 residues.
A Previously Undiscovered Cyclophilin Domain
The cyclophilin domain of CYP38 has the typical β-barrel structure, closed at both ends by α-helices. Eight antiparallel β-strands make the barrel. There are differences between the cyclophilin domain of CYP38 and other known cyclophilin structures. Divergent cyclophilins, which form a separate class of cyclophilins, have an additional loop of five to several amino acids at about residue 50 (human cyclophilin A [hCYPA] numbering). Also, another common feature of this group of proteins is the presence of two reduced Cys residues, which come together in close proximity and hence might be involved in a redox signaling process. Even though the CYP38 cyclophilin domain has extensive loops, they do not have any conformation similar to the loops of other known divergent cyclophilins. In fact, this domain does not even have the conserved Cys residues that are found in other divergent cyclophilins, making it a new kind of divergent cyclophilin. CYP38 got its name due to the presence of a putative CYP domain in the C terminus, and cyclophilins are known to be active PPIases. However, the current structure does not quite support CYP38 being a PPIase, even though it satisfies the requirements of a CYP fold.
CYP38 and PPIase Activity
When the CYP38 cyclophilin sequence (residues 232 to 437) is aligned with that of the well-studied hCYPA and other known cyclophilins (see Supplemental Figure 2 online), the residues corresponding to the β5 and β6 region align well, suggesting that CYP38 could also be a PPIase. At least three of the critical residues for PPIase activity are located in this region of the active site. But when compared structurally, the β1-β2 region of the current structure occupies the β5-β6 region of hCYPA (Figure 1B), and the residues that are required for PPIase activity become part of a long loop. There are also variations in the lengths of the secondary structural elements constituting the capped barrel compared with those of hCYPA (Figure 1C). The current CYP38 structure contains an additional β-strand (β7) that lies above the putative active site. This could further block the access of any PPIase substrate to the cyclophilin barrel. In other cyclophilins, the corresponding region forms a 310 helix and also contributes to form the active site. This β-strand (β7) of CYP38 is so oriented that it makes a cavity, perpendicular to the β-barrel in the active site region. Unlike other cyclophilins, the active site groove of CYP38 is not well defined (Figure 2) because of some overhanging loops and the additional out of barrel β-strand. Structure-based sequence alignment (see Supplemental Figure 2 online) shows that the residues forming β6 in CYP38 are actually in a loop in other known cyclophilin structures and precede their active site β-strands (β5 and β6). Moreover, the N-terminal helical bundle, which is held tightly to the cyclophilin domain through the N-terminal interaction, could also block the CYP domain from binding to any functional targets.
Figure 2.

Electron Density around the β1-β2 Region of CYP38.
Stereo view of the 2Fo-Fc density map around the β1-β2 region of CYP38 at the 1.0σ contour level. Note the two strands come from the N-terminal helical bundle. The neighboring two strands β6 and β8 belong to the cyclophilin domain. The drawing was prepared in O.
Indeed, in vitro PPIase assays revealed that the full-length CYP38 protein had no PPIase activity (Figure 3). We also tested a truncated version of CYP38, spanning residues 100 to 437, which avoids the N-terminal residues that get into the CYP domain. In addition, we included the glutathione S-transferase (GST) protein as a negative control and FKBP13 as a positive control. FKBP13 was found to be a very active PPIase, and GST alone did not show any PPIase activity (the curve being the same as a spontaneous reaction curve). FKBP13 showed significant PPIase activity even after 1000 times dilution. CYP38 showed no measurable PPIase activity, and the catalytic curves were essentially the same as the GST curve. We also included additional GST fusion CYP38 proteins of varying N-terminal truncations in the PPIase assay. The additional truncated proteins were not stable upon cleavage from the GST tag, suggesting inherent instability of these proteins. We measured PPIase activity of GST-FKBP13, GST-CYP38, GST-CYP38 100-437, GST-CYP38 111-437, and GST-CYP38 236-437. None of the GST-CYP38 proteins showed PPIase activity as opposed to the highly active GST-FKBP13 (see Supplemental Figure 3 online). Even though the PPIase-dependent cis-trans transition of N-succinyl-AAPF-p-nitroanilide occurs in a very short time period (1 to 2 min), it is possible that the CYP38 proteins get degraded in such an assay where a protease is included. Therefore, we monitored the CYP38 proteins before and after the PPIase assay using SDS-PAGE coupled with Coomassie blue staining, and we found that about half of the protein remained undigested after the assay and FKBP13 also behaved the same way (see Supplemental Figure 4 online). These results suggest that the CYP38 protein does not possess any PPIase activity and may thus represent a previously uncharacterized cyclophilin with other functions.
Figure 3.

Kinetic Traces for PPIase Activity Measurement of FKBP13, CYP38, and CYP38 100-437.
In the panel for FKBP13, 1 mL reaction mixture contained 20 (blue), 2 (brown), 0.2 (red), and 0.02 (gray) µg protein and 20 µg GST (black). In the panels for CYP38 and CYP38 100-437, 1 mL reaction mixture contained 50 (purple), 20 (blue), and 2 (brown) µg protein and 20 µg GST (black).
Intramolecular Interaction: A Possible Autoinhibitory Mechanism of CYP38 Function
The overall structure of CYP38 reveals two distinct domains. The N-terminal helical bundle, a previously undescribed protein–protein interaction module, is held tightly to the cyclophilin domain possibly through intramolecular interaction. We tested whether physical interaction between the two domains indeed takes place using a yeast two-hybrid system (Figure 4). The putative CYP domain of CYP38 interacts directly with the various N-terminal elements. These results show that the CYP domain of CYP38 (residues 236 to 437) interacts equally well with N-terminal domains that span amino acids 78 to 217 and 92 to 217, whereas the interaction between the putative CYP domain (residues 236 to 437) and 100 to 217 is relatively weak. Furthermore, the peptide fragments without the N-terminal helical domain, residues 111 to 217 and 135 to 217, did not interact with the CYP domain (residues 236 to 437). These results show that the N-terminal stretch from 92 to 110 is required for the interdomain interaction. We hypothesized that intramolecular interaction in the CYP38 protein may be functionally important for regulation of its function in vivo. In an initial effort to test this possibility, we began to identify potential interaction partners for CYP38 in the thylakoid membrane and lumen by screening a yeast two-hybrid library harboring a large majority of the previously identified lumenal proteins and lumenal domains of various thylakoid membrane proteins. The library was screened against baits including mature full-length CYP38, CYP38 N-terminal stretch 92 to 232, and the CYP domain (residues 236 to 437). Interestingly, the mature protein and the N-terminal stretch did not interact with any of the proteins in the library, whereas the putative CYP domain was found to interact with the E-loop of PSII subunit chlorophyll protein47 (CP47). This interaction is consistent with the in vivo function of CYP38 in the assembly of PSII supercomplex (Fu et al., 2007; Sirpiö et al., 2008). We further tested and confirmed that the E-loop of CP47 can interact with CYP38 236-437 and CYP38 111-437 but not with CYP38 78-437, CYP38 92-437, or CYP38 100-437 (see Supplemental Figure 5 online). The presence of the N-terminal helical domain required for intramolecular interaction in CYP38 blocks the interaction with its potential target CP47, implying its function as an inhibitory domain in CYP38–CP47 interaction. Although the functional relevance of the CYP38–CP47 interaction awaits further study, it provides a relevant model to establish the autoinhibitory mechanism that appears to operate in CYP38 function. Our attempts to crystallize the CYP domain or other N-terminal truncated forms of CYP38 were not successful. We do not know whether the CYP domain by itself can arrange into a discernable structure in the absence of the interacting N-terminal stretch.
Figure 4.

Yeast Two-Hybrid Analysis of Domain Interaction in CYP38.
Several N-terminal helical bundle truncation fragments and the CYP38 CYP domain were cloned into the BD- and AD-containing vectors, as labeled. The growth of yeast, when transformed with these constructs, on selection media indicates protein–protein interaction of the corresponding domains.
Additional Functional Implications
Based on the structure, we propose that the very N-terminal domain may cause the inhibition of CYP38 function by puncturing the β-barrel structure of the CYP domain. This structural element also provides a physical link for the tight packaging between the CYP domain and the N-terminal helical domain. This intramolecular interaction could keep the protein inactive when a specific activity is not needed. Although this autoinhibition is known as a regulatory mechanism for other plant proteins, such as salt-overly-sensitive1 (Quintero et al., 2011) and calcium-dependent protein kinase (Harper et al., 2004), it appears to be a unique mechanism among immunophilins.
It is puzzling to note that spinach TLP40 retains strong PPIase activity, despite a high overall sequence identity with Arabidopsis CYP38 (see Supplemental Figure 6 online). Some differences do exist in β6 of CYP38 and its corresponding stretch in TLP40 and also in the extreme N terminus. We can only hypothesize, in the absence of a crystal structure for TLP40, that these subtle differences may have caused a different organization of the PPIase domain in TLP40, thereby explaining the difference between the two homologs with respect to their PPIase activity.
We found that the sequence 102DPEALLR108 of the short helix (α1) at the N terminus is ∼57% identical to 492DPNYLHR498 and 531DPVANVR537 of the regulatory subunit A of protein phosphatase 2A (PP2A). These regions of PP2A A subunits bind to PP2A catalytic subunit C (Ruediger et al., 1994). Phosphatase binding proteins are known to interact via such short stretches. It has been predicted that CYP38 may also interact with a phosphatase using the similar stretch of amino acid sequence. However, this speculation does not agree well with the current structure in which the putative phosphatase binding region is sandwiched between the helical bundle and the CYP domain and may be difficult for phosphatases to access. The yeast two-hybrid studies also indicate the significance of the stretch DPEALLR in the interdomain interaction. Thus any plausible interaction of CYP38 with other proteins must be mediated by other regions. Perhaps, this short stretch is important in accomplishing the helical domain’s regulatory function on the CYP domain.
In the CYP38 structure, the region between Glu-167 and Asp-255 that forms part of the helical bundle has clusters of charged residues (21 acidic and 16 basic residues) with a net surplus of acidic residues. In other high molecular weight immunophilins, such as CYP40 and FKBP52, similar clusters of charged residues are involved in protein–protein interactions, such as interaction with heat shock protein90 (Ratajczak and Carrello, 1996). From our current structure, as well as from the yeast two-hybrid studies, it is not clear whether CYP38 could perform any such interactions within the thylakoid lumen. In fact, the overall surface of the CYP38 protein is highly negatively charged (Figure 5). This property could be of significance in the context of its location within the thylakoid lumen where pH dramatically fluctuates during the day–night cycle (Holt et al., 2004). It is interesting to note that the high concentration of negative charges on the surface of the CYP domain is mostly on the opposite side of the putative PPIase active site. This could greatly influence the interactions between CYP38 and its target proteins within the thylakoid lumen.
Figure 5.

Surface Charge Distribution in CYP38.
The views (i) and (ii) are rotated by 180°. Positive and negative potentials are shown in blue (>10 kT/e) and red (< −10 kT/e), respectively. The figures were created using GRASP.
In summary, this article presents the structure of a plant cyclophilin. The structure of CYP38 reveals a unique and previously uncharacterized CYP domain with an interesting intramolecular interaction, which probably represents an autoinhibition mechanism of CYP38 function. CYP38 is a multidomain cyclophilin that is known to be localized in the thylakoid lumen and functions in the assembly and maintenance of PSII supercomplex (Fu et al., 2007). Because CYP38, unlike typical cyclophilins, is not a PPIase, we hypothesize that the C-terminal CYP domain is still the catalytic domain (possibly for chaperone activity), and the N-terminal helical domain is likely to be involved in a regulatory function. As a resident of the lumen, At-CYP38 functions in PSII assembly and the C-terminal domain is likely to interact with the PSII subunits, aiding the folding and assembly of the subunits. Further work will be directed to identifying the direct targets of CYP38 and its mode of action in the thylakoid lumen.
METHODS
Protein Expression and Purification
In our attempts to express several fragments of At-CYP38, including the mature protein (residues 92 to 437; Schubert et al., 2002), only the gene encoding residues 83 to 437 in the pGEX-KG vector gave soluble expression. In addition, for structure determination, the codons for three Leu residues at positions 111, 125, and 154 of the protein were mutated to code for Met by site-directed mutagenesis. The GST fusion proteins were overexpressed in Escherichia coli BL21 (DE3) cells using the protocol described earlier (Vasudevan et al., 2005). In brief, the cells were grown in Luria-Bertani medium to an OD600 of 0.6 at 30°C, and expression of the recombinant protein was induced with 0.4 mM isopropylthio-β-galactoside at 25°C and 220 rpm for 4 h. The selenomethionine derivatives of the wild type and mutant CYP38 proteins were produced in E. coli BL21 (DE3) cells in Minimal M9 medium (Doublié, 1997). Cells were harvested by centrifugation at 4200g for 10 min at 4°C and were resuspended in ice-cold medium containing 20 mM Tris HCl buffer, pH 7.5, 0.5 M NaCl, 1 mM EDTA, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride. After sonication, the crude lysate was centrifuged at 42,400g for 45 min at 4°C, and the supernatant fraction was applied to a GST affinity column. The contaminant proteins that loosely bound to the affinity column were removed by washes with a high salt wash medium containing 20 mM Tris HCl buffer, pH 7.5, 0.9 M NaCl, 1 mM DTT, and 1 mM EDTA and a low salt wash medium containing 20 mM Tris HCl buffer, pH 7.5, 0.15 M NaCl, 1 mM DTT, and 1 mM EDTA. GST fusion forms of CYP38 100-437, CYP38 111-437, and CYP38 236-437 for PPIase assays were also expressed and purified using a similar protocol. To obtain a nonfusion form of CYP 83-437 and CYP 100-437, the GST tag of the fusion protein was cleaved on-column with thrombin in the low salt wash medium, and the CYP38 protein was obtained in the flow-through. For protein used in crystallization experiments, the final purity was achieved by gel filtration on a HiLoad 16/60 Superdex-75 column (GE Healthcare) previously equilibrated with the low salt wash medium containing 1 mM phenylmethylsulfonyl fluoride. The purified protein was analyzed by SDS-PAGE and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry. The molecular weight was determined to be 40,389.69 ± 1.217, and the protein used for crystallography was >95% pure. Our attempts to express the mature protein (residues 92 to 437) were not successful. Details of the various primers used in this work can be found in Supplemental Table 1 online.
Crystallization, Data Collection, and Structure Determination
The CYP38 protein crystals that were grown at 20°C by the hanging drop vapor diffusion method using a condition comprising of 20% polyethylene glycol 6000, 2.5% t-butanol, and 0.1 M sodium citrate, pH 5.5, did not diffract x-rays. The crystals that were obtained in the same condition from the vapor batch method (Mortuza et al., 2004) diffracted well (Vasudevan et al., 2005). The crystals were rapidly swept through crystallization solution containing an additional 1.25% t-butanol and 20% glycerol as cryoprotectants and were flash-cooled in liquid nitrogen. Native and multi-wavelength anomalous dispersion (MAD) diffraction data sets were collected at the X12C beamline of National Synchrotron Light Source with an ADSC Q210 charge-coupled device detector. Data collection was performed at 100K. For the native CYP38 protein crystal, a data set of 180 images with 1° oscillation was collected at the wavelength of 0.95 Å. MAD data for the triple L to M mutant CYP38 selenomethionine derivative crystal (180 images, 1° oscillation) were collected at three different wavelengths, based on the selenium absorption spectrum. The data were indexed, integrated, and scaled using the HKL2000 program package (Otwinowski and Minor, 1997). The structure was solved by the MAD method using BnP (Weeks et al., 2002). The resulting phases were used for density modification and automated model building using ARP/wARP (Perrakis et al., 1999). Whereas the program could trace short stretches of the structure, all remaining residues were manually traced using the O program (Jones et al., 1991). Refinement of the structure was performed using CNS (Brünger et al., 1998) as well as Refmac5 of the CCP4 suite (Murshudov et al., 1997). PROCHECK (Laskowski et al., 1993) was used for structure validation. Figures were prepared using the program PyMOL (DeLano, 2002). The surface charge distribution figures were prepared with GRASP (Nicholls et al., 1991). The data collection and refinement statistics are presented in Table 1.
Table 1. Crystal, Data Collection, and Refinement Statistics for CYP38.
| Parameters | Native | Inflection | Peak | Remote |
| Unit-cell (native and Se-Met crystals) | ||||
|---|---|---|---|---|
| Space group | C2221 | C2221 | ||
| a (Å) | 58.0 | 57.7 | ||
| b (Å) | 96.0 | 96.7 | ||
| c (Å) | 166.7 | 166.8 | ||
| No. of molecules in ASU | 1 | 1 | ||
| Data collection | ||||
| Resolution (Å) | 2.64 | 2.39 | 2.39 | 2.39 |
| Wavelength (Å) | 0.9500 | 0.9797 | 0.9794 | 0.9500 |
| Total observations | 230,354 | 94,780 | 91,174 | 96,420 |
| Unique reflections | 14,209 | 17,556 | 17,609 | 19,441 |
| Completeness (%) | 97.8 (99.8) | 98.1 (93.3) | 98.2 (90.9) | 98.6 (91.6) |
| Redundancy | 6.2 (4.5) | 6.3 (4.2) | 6.2 (4.1) | 6.4 (4.3) |
| Rsyma | 0.069 (0.29) | 0.059 (0.30) | 0.065 (0.25) | 0.067 (0.23) |
| 〈I/σ(I)〉 | 21.4 (5.4) | 20.2 (5.3) | 20.7 (6.5) | 19.2 (5.1) |
| Refinement (SeMet crystal) | ||||
| Rcryst/Rfreeb | 0.248/0.266, with |F| > 3.0σ(|F|) | |||
| 0.260/0.280, with |F| > 0σ(|F|) | ||||
| Resolution range (Å) | 20–2.39 | |||
| Reflections (working/test) | 13244/669 |F| > 3.0σ(|F|) | |||
| 15614/797 for |F| > 0σ(|F|) | ||||
| Final model | ||||
| Nonhydrogen atoms | 2,765 | |||
| Water | 114 | |||
| Average B-factors (Å2) | ||||
| Protein (all atoms) | 66.65 | |||
| Protein (with two loops removed) | 54.86 (2186 atoms) | |||
| Waters | 49.48 | |||
| RMSD | ||||
| In bond lengths (Å) | 0.013 | |||
| In bond angles (°) | 1.605 | |||
| Ramachandran plot values (%) | ||||
| Most favored | 77.3 | |||
| Additional allowed | 15.7 | |||
| Generously allowed | 4.7 | |||
| Disallowed | 2.4 |
Values in parentheses are for the highest resolution shell (native, 2.73–2.64 Å; selenomethionine [SeMet], 2.55–2.39 Å). For clarity of presentation, single values for Se-Met crystal and Final model have been categorized under the Inflection column head, leaving other columns without data.
a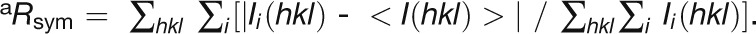 .
.
b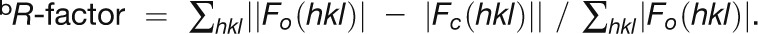 . Rfree was calculated with 5% of the data that were not used for refinement.
. Rfree was calculated with 5% of the data that were not used for refinement.
ASU, asymmetric unit; RMSD, root mean square deviation.
Yeast Two-Hybrid Analysis
The genes encoding various lengths of truncated CYP38 protein were cloned into a binding domain (BD) vector (pGBT9BS) and an activation domain (AD) vector (pGADGH) between the EcoRI and SalI sites. (Primers are listed in Supplemental Table 1 online.) The resulting BD and AD constructs were selectively introduced into the yeast strain AH109 by a lithium acetate-mediated transformation method. The appropriate pairs of BD and AD vectors that were used in the analysis are described in figure legends. Transformants carrying both BD and AD vectors were initially selected on synthetic complete agar-Leu-Trp medium. To screen protein–protein interaction, exponentially grown yeast cells were harvested and adjusted to OD600 = 0.5 with sterilized double-distilled water and diluted 1/10, 1/100, and 1/1000. Five-microliter yeast cells were spotted onto the interaction selection media (synthetic complete agar-Leu-Trp-His-Ala with 1.5 mM 3-amino-1,2,4-aminotriazole) to score growth as an indicator of protein–protein interaction (Lee et al., 2007).
To identify potential interaction partners for CYP38 in thylakoids, we constructed a chloroplast lumenal protein/domain yeast two-hybrid library. DNA fragments encoding experimentally identified lumenal proteins, predicted lumenal proteins (Peltier et al., 2002; Schubert et al., 2002), lumenal domains of PSII major proteins, low molecular mass subunits (Shi and Schröder, 2004; Loll et al., 2005), lumenal domains of photosystem I subunits (Amunts et al., 2007), and Cytb(6)f complex (Stroebel et al., 2003) were cloned into the AD vector pGADGH to form the mini-library. This library was screened against BD vectors harboring DNA fragments encoding full-length CYP38, CYP38 N-terminal stretch 92 to 232, or the CYP domain (residues 236 to 437), respectively, using the same protocol mentioned above.
PPIase Assay
PPIase activity was measured in coupled reactions with chymotrypsin as described before (Kofron et al., 1991; Lima et al., 2006; Edvardsson et al., 2007). The peptide substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Sigma-Aldrich) was dissolved in 2,2,2-trifluorethanol containing 400 mM LiCl to increase the proportion of the cis isomer (Kofron et al., 1991). The reactions were started by adding the peptide substrate (at a final concentration of 71 µM) to a mixture containing 43 mM HEPES, pH 7.8, 86 mM NaCl, 0.64 µM chymotrypsin, and 20 µg/mL protein sample at 10°C. The increase in absorbance at 390 nm due to the release of p-nitroaniline by the cleavage of the trans isomer of N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide was monitored using a Cary 3E UV-Visible spectrophotometer (Varian).
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Genome Initiative or GenBank/EMBL databases under accession numbers At3g01480 (At-CYP38), ArthCp049 (At-CP47), and At5g45680 (At-FKBP13). The coordinates and structure factors for At-CYP38 have been deposited at the Protein Data Bank under the entry code 3RFY.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Structural Overlap of CYP38 Helical Bundles.
Supplemental Figure 2. Structure-Based Sequence Alignment of Cyclophilins.
Supplemental Figure 3. Kinetic Traces for PPIase Activity Measurement of GST-FKBP13, GST-CYP38, and Different CYP38 Truncated Forms.
Supplemental Figure 4. Protein Status before and after PPIase Assay.
Supplemental Figure 5. Yeast Two-Hybrid Analysis of Interaction of CP47E with CYP38 and CYP38 Truncations.
Supplemental Figure 6. Sequence Alignment of CYP38 and Spinach TLP40.
Supplemental Table 1. Primers Used in This Study.
Acknowledgments
This project was supported by funding from the Faculty Research Council to K.S. and National University of Singapore research scholarship to D.V. The work was also supported by a grant from the U.S. Department of Energy and the World Class University Program through the National Research Foundation of Korea to S.L.
AUTHOR CONTRIBUTIONS
D.V. and A.F. are equal contributors to this work and performed all experiments. All authors were involved in the experimental design, data analysis, and writing of the article.
Glossary
- PPIase
peptidyl-prolyl cis/trans isomerase
- PSII
photosystem II
- GST
glutathione S-transferase
- MAD
multi-wavelength anomalous dispersion
- BD
binding domain
- AD
activation domain
References
- Ahn J.C., Kim D.W., You Y.N., Seok M.S., Park J.M., Hwang H., Kim B.G., Luan S., Park H.S., Cho H.S. (2010). Classification of rice (Oryza sativa L. Japonica nipponbare) immunophilins (FKBPs, CYPs) and expression patterns under water stress. BMC Plant Biol. 10: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A., Drory O., Nelson N. (2007). The structure of a plant photosystem I supercomplex at 3.4 A resolution. Nature 447: 58–63 [DOI] [PubMed] [Google Scholar]
- Andreeva L., Heads R., Green C.J. (1999). Cyclophilins and their possible role in the stress response. Int. J. Exp. Pathol. 80: 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman A., Fawcett T.W., Ghirardi M.L., Mattoo A.K. (1992). Plant organelles contain distinct peptidylprolyl cis,trans-isomerases. J. Biol. Chem. 267: 21293–21296 [PubMed] [Google Scholar]
- Brünger A.T., et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921 [DOI] [PubMed] [Google Scholar]
- DeLano W.L. (2002). The PyMOL Molecular Graphics System. (San Carlos, CA: DeLano Scientific). http://www.pymol.org [Google Scholar]
- Doublié S. (1997). Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276: 523–530 [PubMed] [Google Scholar]
- Edvardsson A., Shapiguzov A., Petersson U.A., Schröder W.P., Vener A.V. (2007). Immunophilin AtFKBP13 sustains all peptidyl-prolyl isomerase activity in the thylakoid lumen from Arabidopsis thaliana deficient in AtCYP20-2. Biochemistry 46: 9432–9442 [DOI] [PubMed] [Google Scholar]
- Fu A., He Z., Cho H.S., Lima A., Buchanan B.B., Luan S. (2007). A chloroplast cyclophilin functions in the assembly and maintenance of photosystem II in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 15947–15952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulgosi H., Vener A.V., Altschmied L., Herrmann R.G., Andersson B. (1998). A novel multi-functional chloroplast protein: Identification of a 40 kDa immunophilin-like protein located in the thylakoid lumen. EMBO J. 17: 1577–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galat A. (2003). Peptidylprolyl cis/trans isomerases (immunophilins): Biological diversity-targets-functions. Curr. Top. Med. Chem. 55: 423–436 [DOI] [PubMed] [Google Scholar]
- Gopalan G., He Z., Balmer Y., Romano P., Gupta R., Héroux A., Buchanan B.B., Swaminathan K., Luan S. (2004). Structural analysis uncovers a role for redox in regulating FKBP13, an immunophilin of the chloroplast thylakoid lumen. Proc. Natl. Acad. Sci. USA 101: 13945–13950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan G., He Z., Battaile K.P., Luan S., Swaminathan K. (2006). Structural comparison of oxidized and reduced FKBP13 from Arabidopsis thaliana. Proteins 65: 789–795 [DOI] [PubMed] [Google Scholar]
- Harper J.F., Breton G., Harmon A. (2004). Decoding Ca(2+) signals through plant protein kinases. Annu. Rev. Plant Biol. 55: 263–288 [DOI] [PubMed] [Google Scholar]
- He Z., Li L., Luan S. (2004). Immunophilins and parvulins. Superfamily of peptidyl prolyl isomerases in Arabidopsis. Plant Physiol. 134: 1248–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. (1993). Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Holt N.E., Fleming G.R., Niyogi K.K. (2004). Toward an understanding of the mechanism of nonphotochemical quenching in green plants. Biochemistry 43: 8281–8289 [DOI] [PubMed] [Google Scholar]
- Ingelsson B., Shapiguzov A., Kieselbach T., Vener A.V. (2009). Peptidyl-prolyl isomerase activity in chloroplast thylakoid lumen is a dispensable function of immunophilins in Arabidopsis thaliana. Plant Cell Physiol. 50: 1801–1814 [DOI] [PubMed] [Google Scholar]
- Ivery M.T. (2000). Immunophilins: Switched on protein binding domains? Med. Res. Rev. 20: 452–484 [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kleffmann T., Russenberger D., von Zychlinski A., Christopher W., Sjölander K., Gruissem W., Baginsky S. (2004). The Arabidopsis thaliana chloroplast proteome reveals pathway abundance and novel protein functions. Curr. Biol. 14: 354–362 [DOI] [PubMed] [Google Scholar]
- Kofron J.L., Kuzmic P., Kishore V., Colón-Bonilla E., Rich D.H. (1991). Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30: 6127–6134 [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. (1993). PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283–291 [Google Scholar]
- Lee S.C., Lan W.Z., Kim B.G., Li L., Cheong Y.H., Pandey G.K., Lu G., Buchanan B.B., Luan S. (2007). A protein phosphorylation/dephosphorylation network regulates a plant potassium channel. Proc. Natl. Acad. Sci. USA 104: 15959–15964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima A., Lima S., Wong J.H., Phillips R.S., Buchanan B.B., Luan S. (2006). A redox-active FKBP-type immunophilin functions in accumulation of the photosystem II supercomplex in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 103: 12631–12636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll B., Kern J., Saenger W., Zouni A., Biesiadka J. (2005). Towards complete cofactor arrangement in the 3.0 A resolution structure of photosystem II. Nature 438: 1040–1044 [DOI] [PubMed] [Google Scholar]
- Luan S., Albers M.W., Schreiber S.L. (1994). Light-regulated, tissue-specific immunophilins in a higher plant. Proc. Natl. Acad. Sci. USA 91: 984–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortuza G.B., Haire L.F., Stevens A., Smerdon S.J., Stoye J.P., Taylor I.A. (2004). High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431: 481–485 [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp K.A., Honig B. (1991). Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology: Macromolecular Crystallography, Vol. 276, C.W. Carter, Jr. and R.M. Sweet, eds (New York: Academic Press), pp.307–326. [DOI] [PubMed] [Google Scholar]
- Peltier J.B., Emanuelsson O., Kalume D.E., Ytterberg J., Friso G., Rudella A., Liberles D.A., Söderberg L., Roepstorff P., von Heijne G., van Wijk K.J. (2002). Central functions of the lumenal and peripheral thylakoid proteome of Arabidopsis determined by experimentation and genome-wide prediction. Plant Cell 14: 211–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A., Morris R.M., Lamzin V.S. (1999). Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Quintero F.J., Martinez-Atienza J., Villalta I., Jiang X., Kim W.Y., Ali Z., Fujii H., Mendoza I., Yun D.J., Zhu J.K., Pardo J.M. (2011). Activation of the plasma membrane Na/H antiporter Salt-Overly-Sensitive 1 (SOS1) by phosphorylation of an auto-inhibitory C-terminal domain. Proc. Natl. Acad. Sci. USA 108: 2611–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak T., Carrello A. (1996). Cyclophilin 40 (CyP-40), mapping of its hsp90 binding domain and evidence that FKBP52 competes with CyP-40 for hsp90 binding. J. Biol. Chem. 271: 2961–2965 [DOI] [PubMed] [Google Scholar]
- Romano P.G.N., Horton P., Gray J.E. (2004). The Arabidopsis cyclophilin gene family. Plant Physiol. 134: 1268–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger R., Hentz M., Fait J., Mumby M., Walter G. (1994). Molecular model of the A subunit of protein phosphatase 2A: Interaction with other subunits and tumor antigens. J. Virol. 68: 123–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M., Petersson U.A., Haas B.J., Funk C., Schröder W.P., Kieselbach T. (2002). Proteome map of the chloroplast lumen of Arabidopsis thaliana. J. Biol. Chem. 277: 8354–8365 [DOI] [PubMed] [Google Scholar]
- Shapiguzov A., Edvardsson A., Vener A.V. (2006). Profound redox sensitivity of peptidyl-prolyl isomerase activity in Arabidopsis thylakoid lumen. FEBS Lett. 580: 3671–3676 [DOI] [PubMed] [Google Scholar]
- Shi L.X., Schröder W.P. (2004). The low molecular mass subunits of the photosynthetic supracomplex, photosystem II. Biochim. Biophys. Acta 1608: 75–96 [DOI] [PubMed] [Google Scholar]
- Sirpiö S., Khrouchtchova A., Allahverdiyeva Y., Hansson M., Fristedt R., Vener A.V., Scheller H.V., Jensen P.E., Haldrup A., Aro E.M. (2008). AtCYP38 ensures early biogenesis, correct assembly and sustenance of photosystem II. Plant J. 55: 639–651 [DOI] [PubMed] [Google Scholar]
- Stroebel D., Choquet Y., Popot J.L., Picot D. (2003). An atypical haem in the cytochrome b(6)f complex. Nature 426: 413–418 [DOI] [PubMed] [Google Scholar]
- Vallon O. (2005). Chlamydomonas immunophilins and parvulins: Survey and critical assessment of gene models. Eukaryot. Cell 4: 230–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D., Gopalan G., He Z., Luan S., Swaminathan K. (2005). Expression, purification, crystallization and preliminary X-ray diffraction analysis of Arabidopsis thaliana cyclophilin 38 (AtCyp38). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61: 1087–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks C.M., Blessing R.H., Miller R., Mungee R., Potter S.A., Rappleye J., Smith G.D., Xu H., Furey W. (2002). Towards automated protein structure determination: BnP, the SnB-PHASES interface. Z. Kristallogr. 217: 686–693 [Google Scholar]