

Abstract
Toll-like receptor 5 (TLR5) binding to bacterial flagellin activates NF-κB signaling and triggers an innate immune response to the invading pathogen. To elucidate the structural basis and mechanistic implications of TLR5-flagellin recognition, we determined the crystal structure of zebrafish TLR5, as a VLR-hybrid protein, in complex with the D1/D2 fragment of Salmonella flagellin, FliC, at 2.47 Å resolution. TLR5 interacts primarily with the three helices of the FliC D1 domain using its lateral side. Two TLR5-FliC 1:1 heterodimers assemble into a 2:2 tail-to-tail signaling complex that is stabilized by quaternary contacts of the FliC D1 domain with the convex surface of the opposing TLR5. The proposed signaling mechanism is supported by structure-guided mutagenesis and deletion analysis on CBLB502, a therapeutic protein derived from FliC.
Toll-like receptors (TLRs) play a key role in innate immunity by eliciting the first line of defense against invading pathogens (1). TLRs are membrane-bound antigen-recognition receptors (2) that directly recognize highly conserved molecular structures in pathogens using their horseshoe-shaped, leucine-rich repeat (LRR) ectodomains. Each TLR recognizes a different set or subset of antigens with a distinct ligand-binding mechanism (fig. S1). Nonetheless, all known agonist-activated TLR structures form a similar dimer organization, which brings their C-terminal regions into juxtaposition so that their intracellular TIR domains can initiate the signaling cascades (3-6). To date, structural studies on TLR-ligand recognition have been limited to non-protein antigens, and the structural basis for protein-antigen recognition by a TLR has not been addressed.
TLR5 is the only protein-binding TLR that is conserved in vertebrates from fish to mammals (7-9). TLR5 responds to a monomeric form of flagellin from β- and γ-proteobacteria that constitutes the whip-like flagellar filament responsible for locomotion (8, 10-11). Flagellin (hereafter, termed ‘FliC’ corresponding to the main flagellin gene product in Salmonella) binding to TLR5 induces MyD88-dependent signaling and activates the proinflammatory transcription factor, NF-κB, in epithelial cells, monocytes, and dendritic cells, which results in activation of innate immune responses against flagellated bacteria (8, 12-16).
The key roadblock against TLR5-FliC interaction studies has been the formidable technical challenge in expression of the mammalian TLR5 ectodomain (ECD) in a functionally active, soluble form. We overcame this challenge by testing expression of a number of TLR5 orthologs (17-18). Only the zebrafish, Danio rerio ortholog (drTLR5-ECD) could be successfully expressed in a secreted soluble form in the baculovirus system, but with a low purification yield (< 50 μg from 1 liter culture) (19). To improve the yield, we engineered a series of drTLR5-ECD C-terminal deletion variants as chimeric proteins with the C-terminal fragment of hagfish variable lymphocyte receptor (VLR) B.61 using a strategy successfully used for crystallization of other TLRs (4, 20). Three chimeric proteins containing 6, 12 and 14 N-terminal LRR modules (TLR5-N6VLR, TLR5-N12VLR, and TLR5-N14VLR, respectively) (fig. S2A) were expressed, purified to homogeneity, and tested for their interaction with CBLB502 (21) (fig. S2B), a pharmacologically optimized derivative of FliC from Salmonella enterica subspecies enterica serovar Dublin (sdFliC), by size-exclusion chromatography and native electromobility shift assay (EMSA) (Fig. 1A). Stable 1:1 complexes with CBLB502 were obtained for TLR5-N12VLR and TLR5-N14VLR (but not TLR5-N6VLR) and used for structural studies.
Fig. 1.
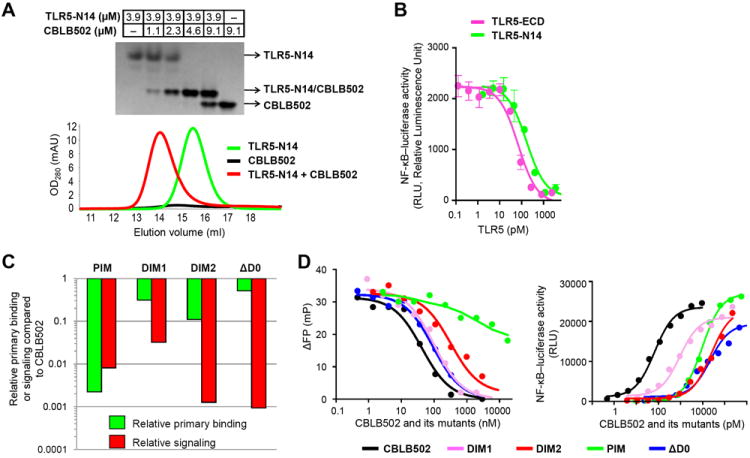
TLR5-FliC interactions and mutational studies. (A) Native EMSA (upper part) and size-exclusion chromatography (lower part) reveal formation of a 1:1 TLR5-N14VLR/CBLB502 heterodimer. (B) TLR5-ECD and TLR5-N14VLR display a comparable binding for CBLB502 as revealed by a competition assay (IC50 values of 67±4 (standard deviation; SD) pM and 139±28 (SD) pM, respectively, in the presence of 90 pM CBLB502) using NF-κB luciferase HEK293 reporter cells that express hsTLR5. Data are expressed as mean ± SD (n = 3). (C) Mutational studies, using dimerization interface-disruption mutants (CBLB502-DIM1 and -DIM2) and D0-deletion mutant (CBLB502-ΔD0), demonstrate that both the dimerization interface of the complex structure and FliC D0 domain contribute to formation of an active signaling complex. TLR5 primary binding efficiency was analyzed by a competitive FP assay where TLR5-N14VLR interaction with fluorescein-labeled CBLB502 was inhibited by unlabeled CBLB502 or its mutants. Relative primary binding efficiency was derived from IC50 ratio of CBLB502 to mutants. TLR5 signaling was assessed in an NF-κB-dependent luciferase reporter assay and relative signaling efficiency was presented using EC50 ratio of CBLB502 to mutants. The smaller ratio corresponds to weaker (or less efficient) binding or signaling. (D) A competitive FP assay (left) and an NF-κB-dependent luciferase reporter cell assay (right) of CBLB502 and its mutants shown in Fig. 1C. FP assay results are representative of two independent experiments (left) and cell assay data are expressed as mean (n = 2) with SD below 2000 RLU (right).
The largest chimeric protein TLR5-N14VLR inhibited CBLB502-induced signaling as efficiently as full-length drTLR5-ECD in a competitive assay using NF-κB-luciferase reporter HEK293 cells stably expressing human TLR5 (hsTLR5) (Fig. 1B). Importantly, the observed IC50 values for both derivatives of drTLR5 (67 and 139 pM; Fig. 1B) are in the same range as the EC50 of CBLB502-hsTLR5 signaling efficiency (77 pM) measured in the same reporter system (Fig. 1, C and D). Although none of these values can be interpreted as a true dissociation constant, this observation supports the relevance of drTLR5 as a model for hsTLR5 interactions with FliC.
To provide the structural basis for TLR5-FliC interaction, crystal structures of TLR5-N12VLR and a complex of TLR5-N14VLR with a D0 deletion variant of sdFliC (sdFliC-ΔD0; fig. S2B) were determined at 2.83 Å and 2.47 Å resolutions, respectively (22) (Fig. 2A and table S1). In the TLR5-FliC complex, the FliC D1 domain is located on the lateral side of TLR5 forming an extensive primary binding interface that defines a 1:1 heterodimer. Two heterodimers further oligomerize to a symmetric 2:2 complex where TLR5 from the first heterodimer makes additional, weaker, interactions with both FliC’ and TLR5’ from the second heterodimer, forming a secondary dimerization interface. This TLR5-FliC interaction mode is completely different from other TLR interactions with non-protein ligands including the stoichiometry, ligand arrangement, and binding interfaces (23). Nonetheless, flagellin binding results in assembly of two TLR5s in a tail-to-tail organization, which juxtaposes the C-terminal tail regions of TLR5-ECD for signaling by analogy with other agonist-bound TLRs (fig. S1). Thus, the TLR5-N14VLR/FliC-ΔD0 structure illustrates distinct, as well as conserved, features compared to other TLRs for receptor activation.
Fig. 2.
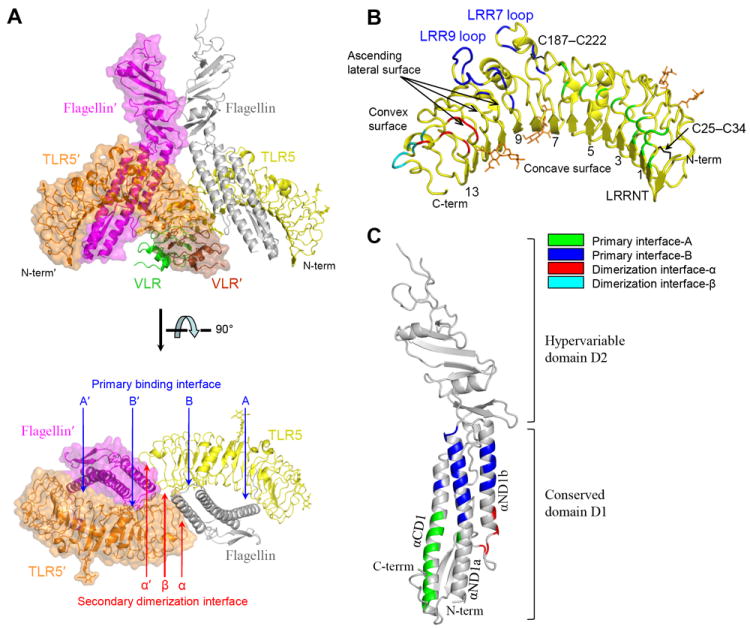
Overall structure of the 2:2 TLR5-N14VLR/FliC-ΔD0 complex. (A) TLR5-N14 interacts with FliC-ΔD0 into a 2:2 quaternary complex structure that organizes two TLR5 molecules in a tail-to-tail orientation where their C-terminal regions are disposed in the center of the complex. The 2:2 complex consists of two copies of 1:1 complex, 1:1 TLR5-N14VLR/FliC-ΔD0 (ribbons; yellow TLR5-N14, green VLR, and gray FliC D1-2) and 1:1 TLR5-N14VLR′/FliC-ΔD0′ (ribbons with translucent surface; orange TLR5-N14′, brown VLR′, magenta FliC′ D1-2). The prime denotes that the molecule or residue comes from the second 1:1 complex in the 2:2 assembly. The 1:1 complex formation is mediated by the primary binding interface (A, B, A′, and B′) and its homodimerization to the 2:2 complex is mediated by the secondary dimerization interface (α, α′, and β). For clarity, only TLR5-N14 and FliC D1 domain are shown in the lower panel. (B) The TLR5-N14 structure of the complex. Interface residues are color-coded according to the color scheme indicated in the figure. Two disulfide bonds and four N-linked glycans are represented by black and orange sticks, respectively. Four N-linked glycans are scattered over the TLR5 surface, but not involved in TLR5-FliC interfaces. LRR7 and LRR9 are atypically long with 32 and 36 residues, compared to other LRRs that contain 23-27 residues, and protrude as long loops (fig. S3). The ascending lateral surface of the LRR domain refers to the region connecting the C-terminal end of the concave surface to the N-terminal end of the convex surface in each LRR module (39). (C) The FliC D1-2 structure observed in the complex. Each α-helix is labeled according to previous nomenclature (40).
TLR5 adopts a single domain LRR structure that consists of an N-terminal β-hairpin capping motif (LRRNT), 13 complete LRR modules (LRR1-13), and two residues from LRR14 (24) (Fig. 2B and fig. S3). The concave surface displays a smooth, curved β-sheet structure that is formed from 2 antiparallel β-strands of LRRNT and 13 parallel β-strands of LRR modules. However, the convex surface is less regular with an assortment of helices and extended structures. Comparison of TLR5 with other known TLR structures indicates that TLR5 can be categorized more as a TLR3-like LRR structure than TLR4- or TLR2-like, consistent with the evolutionary closeness of TLR3 and TLR5 (25) (fig. S4).
Two (D1 and D2) of the three FliC-ΔD0 domains could be modeled in the complex structure (26) (fig. S5), but only D1 is involved in direct interaction with TLR5 (Fig. 2C). The D1 domain consists of four elongated segments that vertically assemble into a long, largely helical rod. The four segments correspond to two N-terminal α-helices (αND1a and αND1b), one C-terminal α-helix (αCD1), and a segment of β-hairpin/extended structure that connects αND1b to the D2 domain (Fig. 2C and fig S6).
The extensive primary binding interface responsible for the specific recognition of FliC by TLR5 is formed between the ascending lateral surface of TLR5 LRRNT-LRR10 and one side of the three long α-helices of FliC D1 with ~1320 Å2 of buried accessible surface area (b.s.a.) on each side (Fig. 3A). This interface can be described as two adjacent, but spatially separated surfaces, interfaces-A and -B, and includes residues previously proposed as TLR5 binding sites by a mutational study (11) (fig. S7). Moreover, the FliC interface coincides with residues involved in FliC oligomerization in the flagellar filament structure and, thus, is well conserved across species of β- and γ-proteobacteria (figs. S6 and S8). These observations support the hypothesis that TLR5 evolved to target a common molecular pattern of FliC that is functionally crucial for flagellar assembly and, hence, evolutionarily constrained (11).
Fig. 3.
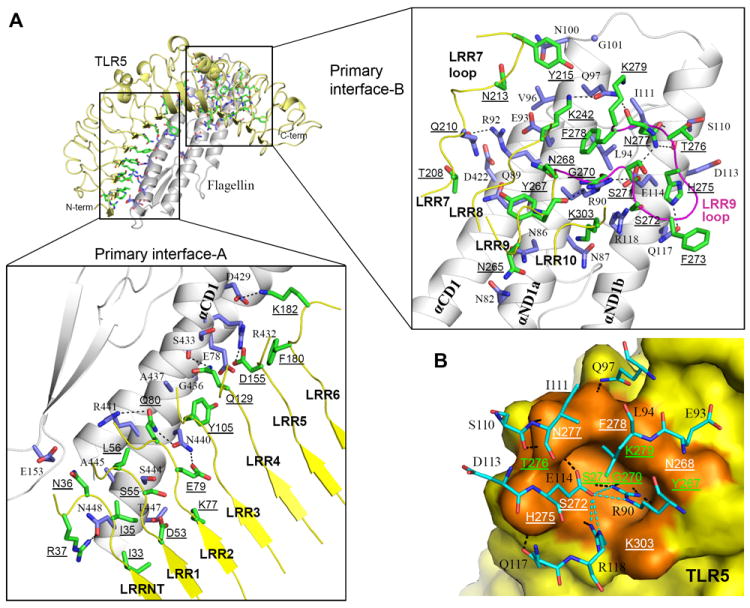
The primary binding interface of the TLR5-FliC 1:1 complex. (A) Residues in primary interfaces-A (left) and -B (right) are shown in sticks (TLR5, green; FliC, light blue) on the 1:1 TLR5/FliC complex (TLR5, yellow ribbon; FliC, gray ribbon). The protruding loop of TLR5 LRR9 is highlighted in magenta. H-bonds and salt bridges are represented by dashed lines. To discriminate from FliC residues, TLR5 residues are underlined throughout the rest of figures. (B) The LRR9 loop forms a groove that provides the major FliC binding site. FliC Arg90 is deeply inserted into the groove and makes four H-bonds with carbonyl oxygens of TLR5 Tyr267, Gly270, and Ser271 (41). Glu114 buttresses and orients the Arg90 side chain toward the groove via H-bonds, and also H-bonds with TLR5 Asn277. The bottom of the groove is constructed from the main chains of Gly270 and Ser271, and its surrounding wall is decorated by 8 LRR9 residues (Tyr267, Asn268, Ser272, His275, Thr276, Asn277, Phe278, and Lys279) and an LRR10 residue (Lys303). The LRR9 loop groove is shown in the orange surface and interacting residues are labeled (green labels for residues that engage only main chains in the interaction with FliC; white labels for residues that engage side chains in the interaction). FliC residues that interact with the LRR9 loop groove are shown in cyan sticks. Inter-molecular and intra-molecular H-bonds are represented by black and cyan dashed lines, respectively.
Interface-A is formed between the concave and ascending sides of TLR5 LRRNT-LRR6 and the C-terminal half of FliC αCD1 with ~530 Å2 b.s.a. (Fig. 3A, bottom, and fig. S9). Interface-A narrows from LRRNT toward LRR6 with LRRNT-LRR2 providing 3-4 residues from both concave and ascending sides, and LRR3-LRR6 contributing 1-2 residues only from the ascending side. Interaction in interface-A is mainly hydrophilic with 5 H-bonds and 3 salt bridges (table S2).
Interface-B is located on the ascending lateral surface of the central LRR modules (LRR7-LRR10) of TLR5 and on the upper part of FliC αND1a and αND1b (Fig. 3A, right), and constitutes ~60% (~790 Å2) of the primary interface (fig. S9, A and B). Interface-B is also mainly hydrophilic with 12 H-bonds and primarily located at a protruding loop of LRR9 that undergoes structural changes upon FliC binding (fig. S10, A and B). In the TLR5-N12VLR structure, the LRR9 loop is highly flexible as indicated by high B-values and poor electron density, but upon FliC binding, the loop undergoes structural rearrangement to form a well-defined groove (Fig. 3B and fig. S10A) that accommodates absolutely conserved FliC residues, Arg90 and Glu114 (fig. S6). The groove side and rim reinforce FliC binding through diverse H-bonds and van der Waals interactions with 8 FliC residues at the C-terminal end of αND1a and N-terminal half of αND1b. This groove is responsible for the half of primary interface H-bonds and ~35% of b.s.a. The functional importance of the LRR9 loop was further demonstrated by an LRR9-loop deletion that severely impaired FliC binding in size-exclusion chromatography, a competitive FP assay (at least ~1000-fold), and competitive cell-based signaling assays (130~490-fold) (fig. S10, C-F). This loop is highly conserved in all species and likely forms the hot-spot in the TLR5-FliC interaction (27).
The secondary dimerization interface consists of three surfaces: interface-α (TLR5’-FliC), its 2-fold symmetry related interface-α’ (TLR5-FliC’), and interface-β (TLR5-TLR5’); no contacts are made between FliC molecules (28) (Fig. 4). Dimerization interface-α is located on the convex side of TLR5’ LRR12/13 and on the FliC C-terminal αND1b region and following loop at the bottom of the D1 domain (Fig. 4, left). Dimerization interface-β is formed by TLR5 residues mainly on the ascending lateral side of LRR12/13 (Fig. 4, right). The dimerization interface is relatively limited with a total of ~550 Å2 b.s.a. on each 1:1 complex (α, ~130Å 2; α’, ~130 A2; β, ~290 Å2), but may be greater in the full-length TLR5-FliC interaction (29). This dimerization interface exhibits high sequence conservation among both vertebrate TLR5 orthologs and FliC orthologs from diverse bacteria (figs. S3, S6 and S11), and high shape complementarity (Sc, 0.64-0.76), underscoring its functional significance in the secondary dimerization of TLR5-FliC (30).
Fig. 4.
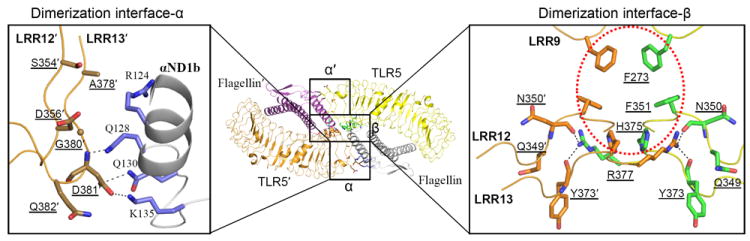
Secondary dimerization interface in the 2:2 TLR5-FliC assembly. Two of the 1:1 TLR5-FliC complex homodimerize to the 2:2 complex using dimerization interfaces-α, α′, and β. The overall 2:2 complex is shown in the middle, and enlarged dimerization interfaces-α and β are shown in the left and right panels, respectively (TLR5 residues, brown sticks; TLR5 residues, green sticks; FliC residues, lilac sticks). In interface-α, TLR5′ Asp381′, which is conserved as an acidic residue (Asp or Glu) in all TLR5 orthologs, forms three H-bonds with FliC Gln128, Gln130, and Lys135. Interface-β is created by van der Waals interactions among two sets of three equivalent aromatic residues (Phe273, Phe351, and His375 of TLR5 and TLR5′), which creates a largely hydrophobic core (red dotted circle) that is conserved in all other TLR5 sequences (Phe at residue 273, Leu at residue 351, and His at residue 375; see fig. S3), and by four H-bonds (Arg377-Asn350′, Arg377-Tyr373′, Arg377′-Asn350, and Arg377′-Tyr373).
We used structure-guided mutagenesis to assess whether the observed dimerization interface contributes to TLR5 activation for downstream NF-κB signaling. A series of CBLB502 derivatives with the proposed dimerization surface disrupted by point mutations and/or deletions were tested in two assays: (i) competitive FP assay to assess primary binding for drTLR5 and (ii) NF-κB-luciferase reporter assay to assess signaling activity that results from both primary binding and secondary dimerization. The latter assay with hsTLR5 in HEK293 cells also provided an opportunity to further validate the drTLR5 structure as a surrogate for hsTLR5. The binding and signaling data from the CBLB502 mutants (Fig. 1, C and D, and table S3) support the proposed contribution of the dimerization interface to signaling. Dimerization interface mutant 1 (DIM1; R124D/Q128A/Q130A/K135A in αND1b at the dimerization interface-α, Fig. 4) shows a ~30-fold decrease in signaling, but essentially no decrease (~3-fold) in binding. For the second mutant (DIM2; deletion of residues 126-128 and T129G/Q130G/K135E mutations to disrupt αND1b and connecting loop structures and introduce charge repulsion), the disruption in signaling efficiency is even more pronounced (~800-fold decrease versus only 9-fold decrease in binding). In the control mutant CBLB502-PIM (with Q89A/R90A/Q97A mutations in the primary interface), comparable decreases in primary binding (450-fold) and signaling efficiency (~120-fold) were observed (31). Decoupling of antigen binding and receptor signaling was also observed for CBLB502-ΔD0, used as a control, where a minor (2-3 fold) change in binding affinity both for TLR5-N14VLR and TLR5-ECD contrasts with an ~1,000-fold drop in signaling efficiency (Fig 1, C and D, and fig. S12). Thus, both dimerization interface mutants (CBLB502-DIM1 and -DIM2) clearly correlate in activity with the signaling-impaired CBLB502-ΔD0 mutant than with the primary binding-impaired CBLB502-PIM mutant.
In conclusion, the D1 domain makes substantial contributions to both high affinity binding and TLR5 signaling (32), whereas D0 contributes to TLR5 activation, but has no or little effect on binding. Furthermore, the TLR5-FliC structure and unique features of ligand recognition and activation provide a template to enhance the therapeutic activity of the radioprotective drug (CBLB502) or flu vaccine (VAX102) in clinical trials, as well as to design new vaccine adjuvants or antagonistic therapeutics for hyper-inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Y S Choo (Sanford-Burnham Medical Research Institute) for critical comments on the manuscript, H Tien and D Marciano (The Joint Center for Structural Genomics) for automated crystal screening, and X Dai and M-A Elsliger (The Scripps Research Institute) for expert technical assistance. We also thank K Namba (Osaka University, Japan) and K Yonekura (Harima Institute, Japan) for generously sharing coordinates of flagellar filament. X-ray diffraction datasets were collected at the SSRL beamline 11-1 and the APS beamlines 23ID-B and 23ID-D. The work was funded in part by NIH grants (AI042266, I.A.W.; R01 AI080446, A.V.G.; RC2 AI087616, A.V.G.), the Skaggs Institute for Chemical Biology (I.A.W.), and research grants from Cleveland BioLabs, Inc. (CBLI) to Roswell Park Cancer Institute (A.V.G.) and Sanford-Burnham Medical Research Institute (A.L.O.). A.L.O. and A.V.G. are paid consultants of Cleveland BioLabs, Inc. (CBLI), which have developed the flagellin derivative, CBLB502, into a radiation countermeasure. A.V.G. is also a shareholder of CBLI. This is manuscript no. 21369 from The Scripps Research Institute. The data presented in this paper are tabulated in the main paper and the supporting online material. Structure factors and coordinates for TLR5-N12VLR and TLR5-N14VLR/FliC-ΔD0 are deposited in the Protein Data Bank under accession codes, 3V44 and 3V47, respectively.
Footnotes
Materials and Methods
SOM Text
References (42-57)
References and Notes
- 1.Takeda K, Kaisho T, Akira S. Annu Rev Immunol. 2003;21:335. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.There are 10 functional TLRs (TLR1-10) in human and 12 functional TLRs (TLR1-9 and TLR11-13) in mouse.
- 3.Liu L, et al. Science. 2008;320:379. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin MS, et al. Cell. 2007;130:1071. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Park BS, et al. Nature. 2009;458:1191. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 6.This tail-to-tail TLR dimerization is believed to be prerequisite for signal transduction, consistent with lack of signaling by antagonist-bound TLR 1:1 complexes and a head-to-head TLR dimer of a TLR4 regulator, RP105 (20, 33-35).
- 7.Stockhammer OW, Zakrzewska A, Hegedus Z, Spaink HP, Meijer AH. J Immunol. 2009;182:5641. doi: 10.4049/jimmunol.0900082. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi F, et al. Nature. 2001;410:1099. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 9.Iqbal M, et al. Infect Immun. 2005;73:2344. doi: 10.1128/IAI.73.4.2344-2350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen-Nissen E, et al. Proc Natl Acad Sci U S A. 2005;102:9247. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith KD, et al. Nat Immunol. 2003;4:1247. doi: 10.1038/ni1011. [DOI] [PubMed] [Google Scholar]
- 12.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. J Immunol. 2001;167:1882. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 13.Means TK, Hayashi F, Smith KD, Aderem A, Luster AD. J Immunol. 2003;170:5165. doi: 10.4049/jimmunol.170.10.5165. [DOI] [PubMed] [Google Scholar]
- 14.McDermott PF, Ciacci-Woolwine F, Snipes JA, Mizel SB. Infect Immun. 2000;68:5525. doi: 10.1128/iai.68.10.5525-5529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eaves-Pyles T, et al. J Immunol. 2001;166:1248. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- 16.The significance of the current study is discussed in SOM text as supporting material on Science Online.
- 17.Materials and methods are available as supporting material on Science Online.
- 18.Human, mouse, frog, trout, and zebrafish TLR5 ectodomains were screened for expression in a baculovirus system. In addition, human and mouse TLR5 ectodomains were also tested in an HEK293 cell expression system.
- 19.Zebrafish contains two highly homologous tlr5 genes, tlr5a and tlr5b. Previous tlr5a/b knockdown embryo experiments showed that TLR5a/b are responsible for FliC-specific immune responses (7). TLR5b was used for this study.
- 20.Kim HM, et al. Cell. 2007;130:906. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 21.CBLB502 contains only D0 and D1 domains from sdFliC that consists of four domains including D0, D1, D2 and D3. CBLB502 activates TLR5 as FliC, and protects the hematopoietic system and gastrointestinal tissues from radiation-induced damage, creating opportunities for therapeutic applications (36).
- 22.Despite the tight complex formation, co-crystallization trials of drTLR5 chimeric proteins with sdFliC and CBLB502 failed to yield diffractable crystals, most likely due to the excessive flexibility of the D0 domain, which appeared dispensable for formation of a 1:1 complex with TLR5 as assessed by binding of CBLB502-ΔD0 in a competitive fluorescence polarization (FP) assay (fig. S12). Although CBLB502-ΔD0 also failed to yield diffractable co-crystals, sdFliC-ΔD0 in complex with TLR5-N14VLR was successful in producing diffraction quality crystals.
- 23.Discussion on ‘unique mode of ligand recognition by TLR5 but with some structural similarity to TLR4 binding to MD-2LPS’ is available in SOM text as supporting material on Science Online.
- 24.The TLR5 structures in TLR5-N12VLR and FliC-bound TLR5-N14VLR superimpose well, except for the protruding loop of LRR9 that undergoes substantial conformational changes upon FliC binding (Cα-RMSD without loop, 0.56 Å). Thus, for TLR5 structure analysis, the TLR5-N14VLR structure will be described unless specified.
- 25.Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW. BMC Genomics. 2008;9:62. doi: 10.1186/1471-2164-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The FliC D3 domain could not be built due to poor electron density, although it exhibits an apparent molecular envelope (fig. S5).
- 27.Although protein-to-protein binding interface is generally extensive, most of the binding stability and free energy is provided by a small cluster of hot-spot residues. We propose that the binding hot-spot of the primary drTLR5-FliC binding interface is located in the LRR9 loop, given that deletion of the LRR9 loop almost completely abrogates FliC binding. Moreover, the LRR9 loop sequence is more conserved than other primary interface residues. Accordingly, the LRR9 loop is anticipated to also function as a key determinant in human TLR5 interaction with FliC. ~62% of the total interactions observed at the drTLR5 LRR9 loop would be conserved in human TLR5, with 8 out of 10 H-bonds recapitulated (fig. S13). In contrast, other interface residues from LRRNT-LRR8 show lower sequence conservation, consistent with our observation that a TLR5-VLR hybrid (TLR5-N6VLR) that contains a majority of interface-A residues at LRRNT-LRR6 does not form a tight complex with FliC or CBLB502 in solution. Therefore, the network of hsTLR5/FliC interactions in this region could be somewhat different. Only ~34% of the total interactions observed at the drTLR5 LRRNT-LRR8 would be formed in human TLR5, with 2 out of 10 H-bonds functionally conserved. Importantly, the key interactions in the TLR5-flagellin interface are supported by our mutagenesis data (PIM-mutant of CBLB502) using a reporter system with human TLR5 (Fig. 1, C and D), as well as by highly efficient, competitive inhibition of CBLB502-induced signaling (Fig. 1B).
- 28.The secondary dimerization interaction is very weak in solution for these ectodomain constructs. The 2:2 complex was not detectable by size exclusion chromatography and native EMSA even with full-length TLR5-ECD and FliC proteins. Only a trace amount of the 2:2 complex was observed between the full-length TLR5-ECD and CBLB502 with a Kd of ~0.5 mM in analytical ultracentrifugation, suggesting the instability of the secondary dimerization in solution. Nonetheless, we anticipate that FliC-mediated TLR5 dimerization would be enhanced on the cell surface, e.g. due to the constraint of two-dimensional movement in the membrane (37), engagement of intracellular and transmembrane domains of TLR5, or contribution of other unknown co-receptor/adaptor molecules.
- 29.Discussion on ‘contribution of the FliC D0 domain and the TLR5-ECD C-terminal region to the TLR5-FliC interaction’ is available in SOM text as supporting material on Science Online.
- 30.The secondary dimerization interface of TLR5-N14/FliC extends to interactions between VLR-VLR’ (b.s.a., ~370 Å2), VLR-FliC’ (b.s.a., ~100 Å2) and VLR’-FliC (b.s.a., ~40 Å2) in the TLR5-N14VLR/FliC-ΔD0 structure. However, the VLR interactions exhibit very low shape complementarity (0.42, 0.16, and 0.04 of Sc values, respectively) and do not include any specific H-bonds and salt bridges. These observations suggest that they make minimal contributions to TLR5-N14VLR/FliC-ΔD0 dimerization.
- 31.Individual alanine mutations at FliC Gln89, Arg90, and Gln97 reduced TLR5 signaling by 4.1, 7.4, and 5.6 fold, respectively, in a previous study (11).
- 32.Discussion on ‘primary binding interface observed in the TLR5-N14VLR/FliC-ΔD0 complex structure’ is available in SOM text as supporting material on Science Online.
- 33.Divanovic S, et al. Nat Immunol. 2005;6:571. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon SI, Hong M, Wilson IA. Nat Struct Mol Biol. 2011;18:1028. doi: 10.1038/nsmb.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohto U, Miyake K, Shimizu T. J Mol Biol. 2011;413:815. doi: 10.1016/j.jmb.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Burdelya LG, et al. Science. 2008;320:226. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavutis M, Jaks E, Lamken P, Piehler J. Biophys J. 2006;90:3345. doi: 10.1529/biophysj.105.072546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersen-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A. J Exp Med. 2007;204:393. doi: 10.1084/jem.20061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bella J, Hindle KL, McEwan PA, Lovell SC. Cell Mol Life Sci. 2008;65:2307. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samatey FA, et al. Nature. 2001;410:331. doi: 10.1038/35066504. [DOI] [PubMed] [Google Scholar]
- 41.Amino-acid variations at groove-forming residue 271 (Ala267 in human, Pro268 in mouse, and Ser271 in zebrafish) were implicated in human versus mouse differences in the cellular response to FliC (38). The same study also showed that mutations of two TLR5 aspartate residues (Asp294 of LRR10 and Asp366 of LRR13 in human TLR5) modulated the FliC response. However, these two acidic residues are not in the TLR5-FliC interface, but are located in the middle of the concave β-strands and are involved in intra-molecular interactions with nearby serine residues and the Asn346-linked glycan, suggesting a critical role in TLR5 protein folding or stability. Thus, it is expected that the mutations would indirectly affect FliC response through modulation of TLR5 protein integrity, rather than directly through FliC binding.
- 42.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, et al. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HM, et al. J Biol Chem. 2007;282:6726. doi: 10.1074/jbc.M608471200. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 47.Vijay-Kumar M, et al. J Immunol. 2008;180:8280. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 48.Gewirtz AT, et al. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1157. doi: 10.1152/ajpgi.00544.2005. [DOI] [PubMed] [Google Scholar]
- 49.O’Neill LA, Bryant CE, Doyle SL. Pharmacol Rev. 2009;61:177. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donnelly MA, Steiner TS. J Biol Chem. 2002;277:40456. doi: 10.1074/jbc.M206851200. [DOI] [PubMed] [Google Scholar]
- 51.Eaves-Pyles TD, Wong HR, Odoms K, Pyles RB. J Immunol. 2001;167:7009. doi: 10.4049/jimmunol.167.12.7009. [DOI] [PubMed] [Google Scholar]
- 52.Murthy KG, Deb A, Goonesekera S, Szabo C, Salzman AL. J Biol Chem. 2004;279:5667. doi: 10.1074/jbc.M307759200. [DOI] [PubMed] [Google Scholar]
- 53.Jacchieri SG, Torquato R, Brentani RR. J Bacteriol. 2003;185:4243. doi: 10.1128/JB.185.14.4243-4247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizel SB, West AP, Hantgan RR. J Biol Chem. 2003;278:23624. doi: 10.1074/jbc.M303481200. [DOI] [PubMed] [Google Scholar]
- 55.Maki-Yonekura S, Yonekura K, Namba K. Nat Struct Mol Biol. 2010;17:417. doi: 10.1038/nsmb.1774. [DOI] [PubMed] [Google Scholar]
- 56.Bell JK, et al. Proc Natl Acad Sci U S A. 2005;102:10976. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McQuiston JR, et al. J Clin Microbiol. 2004;42:1923. doi: 10.1128/JCM.42.5.1923-1932.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.