

INTRODUCTION
Some patients with central nervous system (CNS) lesions suffer chronic pain that appears at variable times following the onset of the CNS lesion. Various estimates indicate that 8 - 18% of patients with ischemic stroke, 30% of patients with multiple sclerosis, and 34% of patients with spinal cord injury experience pain attributable to the CNS lesion and therefore referred to as central pain (CP). CP is also recognized as a complication of Parkinson’s disease, with an incidence of approximately 10% [ 4 ]. Comprehensive reviews of various aspects of CP have appeared recently [ 95, 8, 101, 113, 19, 45, 55 ].
Nearly all patients with CP experience pain within an area of reduced pain and temperature sensation; this appears to be largely independent of the location or type of lesion although tumor-related CP appears to be rare [ 59, 9, 105, 93, 14, 19 ]. Typically, the pain is constant but modulated by mood and environmental factors. Many patients with CP experience allodynia or hyperalgesia within the area of sensory loss and pain. These observations suggest that an impairment of functions mediated through the spinothalamic tract causes an increase of resting and evoked activity within central nociceptive pathways [ 28 ]. However, a loss of thermal and nociceptive sensory function, although perhaps necessary, is not sufficient for the development of CP because most patients with this type of sensory loss do not develop CP [ 49 ]. Direct recordings and stimulation within the somatosensory thalamus suggest that long-term changes in excitability have occurred [ 61, 42, 46, 60 ] and functional imaging studies have shown exaggerated thalamic and cortical neurovascular responses in patients with CP [ 29, 80, 54 ]. A sample of CP patients with focal cerebral infarctions showed a widely distributed loss of brain opioid receptor binding capacity that included the thalamus and several pain-activated cortical areas, suggesting either opioid receptor loss or increased receptor occupancy [ 118 ]. Nonetheless, a neurophysiological basis for the pain of CP remains unknown [ 49, 12, 35, 19 ].
The observations cited above suggest that CP develops because of unique changes in the excitability of some central structures and pathways that process nociceptive and thermal information. Therefore, we investigated the magnitude and distribution of activation intensity changes in a sample population of healthy individuals, patients with focal CNS lesions and a painless loss of pain and heat sensation, and patients with CP. We used contact heat stimuli below and above the noxious range and measured a normalized estimate of the change in regional cerebral blood flow (rCBF) from the resting condition. We examined group differences in both resting and stimulus-evoked activity. Because these measurements require estimates of baseline activity that are temporally more stable than functional magnetic resonance imaging [ 98, 119 ], we used positron emission tomography (PET) with intravenous H215O as the tracer.
METHODS
Participants
The demographic and relevant clinical characteristics of all participants are shown in Table 1. Basic eligibility for participation required that each participant be within the age range of 40 to 70 years, able to communicate fluently in English, and sign informed consent documents approved by the Internal Review Boards of the Medical Centers of The University of Michigan and The Veteran’s Affairs Medical Center, Ann Arbor. In summary, we recruited a healthy control (HC) group of 15 males ages 42 to 64 years, 10 patients with central pain (CP) ages 48 to 66 years, and 10 patients, ages 40 to 68 years, with central nervous system (CNS) lesions but no pain (NP). We recruited HC participants through newspaper and poster notifications placed at the Veteran’s Affairs Medical Center (VAMC Ann Arbor) and at the University of Michigan Medical Center. For the HC group, these notifications specified the required age range and that each participant should be pain free, in excellent general health, without a history of neurological disease, and not taking analgesic or psychoactive medication. We also used newspaper and poster notifications to recruit patients in both the CP and NP groups; these notifications were similar to those used for the HC group but specified that a neurological diagnosis of brain or spinal cord damage was required for participation in the study. In addition, patients attending the Neurology Clinic at the Ann Arbor VAMC were informed of the opportunity to participate in the study if their history and examination was consistent with eligibility. Eligibility for participation in the CP and NP groups included clinical evidence for a single, localized CNS lesion resulting in a unilateral impairment of nociceptive sensation detectable on the neurological examination; we excluded volunteers with multifocal, diffuse, or degenerative neurological disease or with pain unrelated to the neurological deficit. We obtained supportive imaging evidence (CT or MRI) on all participants unless contraindicated or complicated by the presence of metal (NP patient 8, Table 1). We required CP patients to have nociceptive sensory impairment within the site of neurologic impairment and pain within this site as their chief complaint; patients selected for the NP group also had lesion-related nociceptive sensory impairment but without pain. One of us (KLC) performed a complete neurological examination on all patients and a screening neurological examination on all HC participants. Each participant received $350 for completing both the psychophysical and PET imaging components of this investigation.
Table 1.
A. Demographic characteristics of CP patients. B. Demographic characteristics of NP patients. Summary of the demographic characteristics of the 15 HC participants is shown below.
| A | |||||||
|---|---|---|---|---|---|---|---|
| CENTRAL PAIN (CP) | |||||||
| ID | GENDER | AGE | ETHNICITY | CLINICAL EVALUATION | PAIN ONSET/DURATION | PAIN Rx | RATING OF ONGOING PAIN |
| 1 | M | 51 | C | Lt. spinothalamic post-op stroke at T8; Rt. heat & tactile allodynia, temp and pinprick loss | <1 month/2 years | NT (-) | 0 (resting)-6 (allodynia) |
| 2 | M | 60 | C | Lt. thalamic stroke, Rt. hemibody electric dysesthesias, deep pain & temp loss, tactile allodynia | immediate/3 years | CB(-) | n/a; attempted suicide for pain |
| 3 | M | 66 | C | Rt. thalamic stroke, Lt. hemibody painful paresthesias, heat & cold allodynia, pinprick hyperalgesia | immediate/5 years | NT (+/-) | 5-7 |
| 4 | M | 54 | C | Lt. C6-T2 syrinx, Lt. arm & hand pain, absent temp, reduced pinprick & tactile | unknown/2 years | OP (+/-); GP(+/-) | 6-10 |
| 5 | M | 52 | C | Rt. hemisphere stroke, Lt. hemiparesis, arm and hand pain; tactile allodynia | immediately post-op/2years | OP (+/-) | 6-10 |
| 6 | M | 56 | C | Rt. thalamic stroke, Lt. hemibody pain; pin, temp. sensory loss | immediate/7 years | none | 4-5 |
| 7 | M | 52 | C | Lt. temporopariet al stroke, Rt. hemibody pain, tactile & pinprick loss; valproic acid for seizures | 7months/5 years | NT (-) | 4-10 |
| 8 | M | 51 | C | Lt. thalamic stroke; pain Rt. trunk, arm and hand (elevated laser pain threshold); cold allodynia | < 1 week/1 year | NT (+/-) | 0-5 |
| 9 | M | 48 | AA | Rt. hemisphere stroke, Lt. hemibody loss tactile & pinprick; pressure allodynia | <1month/1 year | NT (-) | n/a; pain is chief complaint |
| 10 | F | 48 | As | Rt. thalamic hemorrhage, Lt. hemibody global hypalgesia;arm & foot pain | 4months/9 months | GP (-) | 4-10 |
| B | |||||||
| CENTRAL LESION, NO PAIN (NP) | |||||||
| ID | GENDER | AGE | ETHNICITY | CLINICAL EVALUATION | LESION ONSET/DURATION | ||
| 1 | M | 55 | C | Lt. thalamic stroke, Rt. hemibody loss temp & pinprick > touch | <1 month/2 years | ||
| 2 | M | 68 | AA | Lt. hemisphere stroke, Rt. hemibody decreased pinprick & cold | immediate/3 years | ||
| 3 | M | 47 | C | Lt. parieto-temporal stroke; decreased pinprick Rt. arm & leg | immediate/5 years | ||
| 4 | M | 40 | C | Rt. hemisphere perinatal stroke; valproic acid for seizures; pin, temp, & tactile sensory loss | unknown/2 years | ||
| 5 | M | 50 | C | Lt. thalamic stroke, Rt. hemibody decreased pinprick > temp | immediately post-op/2years | ||
| 6 | M | 58 | C | Lt. thalamic stroke, Rt. painless paresthesias arm & leg, reduced pinprick & tactile sensation | immediate/7 years | ||
| 7 | M | 50 | C | Lt. hemisphere trauma, Rt. hemibody decreased pinprick, temp, tactile sensation | 7months/5 years | ||
| 8 | M | 50 | C | Rt. hemisphere shrapnel wound; Lt. temp, pin, tactile hemibody sensory loss; phenytoin for seizures | < 1 week/1 year | ||
| 9 | M | 47 | C | Lt. cervical dorsal cord lesion due to trauma; Lt. Babinski reflex; areflexic, flaccid Lt.arm; global sensory loss | <1month/1 year | ||
| 10 | M | 50 | C | Lt. int capsule/thalamic stroke. Rt. hemibody decreased temp >touch | 4months/9 months | ||
C = Caucasian, AA = African-American, As = Asian
(-) = no effect on pain, (+) = reduces pain, , GP= gabapentin, OP= opioid, NT= nortriptyline, CB = carbamazapine
HEALTHY CONTROL (HC) GROUP (summary)
mean age: 49, median age: 49, age range: 42-64
ethnicity: 8 C, 6 AA, 1 As
Psychophysical testing
We obtained bilateral thresholds for heat detection (HD), heat pain threshold (HPT), and heat tolerance (HPTol) on all participants in separate sessions outside the PET scanner several days before PET scanning. Thermal stimulation was performed by a single examiner using the method of limits with a 16mm2 digitally controlled contact thermode (TSA)-2001 thermal stimulator, Medoc Advanced Medical Systems, Ramat Yishay, Israel) applied to the skin of the clinically involved and uninvolved sites (usually volar forearm). Baseline thermode temperature was 32°C and the rate of temperature increase was 1°C/s for each determination of at least 5 trials. The area of stimulation was slightly altered for each trial to prevent sensitization.
During scanning, we applied heat to the skin of each volar forearm using a thermal probe (Cygnus/GLC, Inc., Paterson, N.J.) with preset temperatures as previously described [ 26 ]. We used the previously obtained threshold information to guide the determination of these thresholds by the method of limits while each participant was in the scanner. In addition, we instructed all participants in the use of a visual analog scale (ruler with sliding marker) to rate the intensity of a series of 5s duration heat stimuli using the following anchors: 0 = no heat sensation, 5 = just painful heat, and 10 = intolerable heat. Participants were instructed to identify HPTol as that heat pain sensation that could not be tolerated if applied for longer than 5s; however, to avoid tissue damage, a limit of 50°C was established for an HPTol stimulus. After receiving instructions on distinguishing sensory intensity from sensory unpleasantness [ 86 ], all participants were asked to rate the unpleasantness of the stimulation using the following anchors: 0 = not unpleasant, 10 = the most unpleasant imaginable. We used the left forearm for threshold determinations in the HC group; for the patients, we determined thresholds separately for the clinically affected and unaffected limbs.
PET Procedures
To detect group or individual differences in regional neuronal excitability, it is necessary to have a temporally stable measure of rCBF in the resting (no stimulus) condition so that the change in activation intensity during heat stimulation can be estimated accurately; this is especially true when comparing healthy individuals and patients with variable types of CNS pathology. Because of the problem of baseline drift in fMRI [ 98, 119 ], we used PET H215O activation methods to provide long duration (~60s) estimates of baseline regional perfusion (rCBF).
We performed all scans on a Siemens/CTI ECAT EXACT PET scanner operated in 3D data acquisition mode to maximize signal intensity, thus permitting multiple scans of individual subjects with intravenous H215O doses at relatively low levels of radioactivity (10 mCi/scan). We monitored the blood pressure and oxygenation (pulse oximeter) of all participants during scanning. The scanner provides 47 tomographic slices covering an axial field of view of approximately 16 cm. We used a soft restraint and laser beam positioning on facial fiducial marks to help maintain head position; additionally, a computer coregistration algorithm corrected small head motion for each subject before the beginning of analysis [ 68 ]. We used a transmission scan to correct attenuation on emission scans. To allow for radioactive decay, at least 10 min elapsed between each scan. Data acquisition began 5s after the estimated arrival of radioactivity in the brain and continued for approximately 60 sec. We analyzed only those voxels with normalized CBF values greater than 60% of the global value (gray matter voxels) using stereotactic anatomical standardization techniques [ 67, 70, 69 ]. After normalizing each image set to whole brain counts [ 40 ], we created mean radioactivity concentration images estimating regional cerebral blood flow (rCBF) for each experimental condition for each subject. Subtraction images were made for each subject by subtracting the images acquired during baseline (rest) from those acquired during heat stimulation. A voxel-by-voxel statistical subtraction analysis (Z-score) with adjustment for multiple comparisons was performed as described previously [ 27, 102 ] by estimating the smoothness of subtraction images following 3 dimensional Gaussian filtering (FWHM = 9mm) to enhance signal-to-noise ratio and compensate for anatomical variance. Voxels showing a significantly increased CBF compared to the average noise variance computed across all voxels (pooled variance) were identified. The critical level of significance (Z = 2.0) was determined by adjusting p=0.05 using this information. Only those voxels with normalized CBF values larger than 60% of the global value were analyzed in this study. We used these estimates of rCBF to compute the stimulation-induced percentage change of rCBF from the resting (no stimulus) condition.
The structures of interest for this study were selected based on the results of previous PET studies of heat pain performed in this and other facilities [ 22, 1 ]. To reduce residual anatomical variance in the stereotactic coordinate system, we first used a standard atlas [ 104 ] to identify the stereotactic coordinates of peak activation within each structure of interest within the group of normal subjects; we used these coordinates as a center for the development of volumes of interest (VOI) within those structures. We developed standard three-dimensional VOI (Table 2) from the rCBF responses (activation threshold: Z > 2.5) of normal subjects to the highest intensity stimulus (HPTol) as described previously [ 27 ]. We determined the final coordinate peaks (Table 2) and the size and shape of each VOI separately for each of the structures by methods used and described previously [ 15, 26, 27 ]. With the exception of the dorsal and ventral caudate, one or more of the peak activation coordinates of adjacent or nearby structures is separated by at least 5mm, well within the spatial resolution reported in previous studies using this functional imaging technique. We applied these VOI to each hemisphere of each subject at rest and in each stimulus condition within the structures of interest. These standard VOI were applied throughout the study to compare the levels of activation in each stimulus condition for each individual and between groups.
Table 2.
Identification, abbreviation, and Talairach coordinates [100] of the peak activation coordinates of volumes of interest (VOI) based on the activation within structures of interest in the group of HC participants.
| STRUCTURE (ABBREVIATION) | VOI COORDINATE PEAKS (X,Y,Z) |
|---|---|
| MEDIAL THALAMUS (MT) | (6,-15, 9/11) |
| INTRALAMINAR THALAMUS (IT) | (8, -10, 9) |
| VENTROLATERAL THALAMUS (VT) | (17, -13, 16) |
| PUTAMEN (P) | (24, 5, 7) |
| DORSAL CAUDATE (DC) | (17, 14, 11) |
| VENTRAL CAUDATE (VC) | (15, 14, 4) |
| ANTERIOR INSULA (AI) | (39, 12, 9) |
| MID-INSULA (MI) | (44, 3, 9) |
| INFERIOR FRONTAL CORTEX (B45) | (46, 23, 7) |
| INFERIOR FRONTAL CORTEX (B47) | (37, 41, -4) |
| MEDIAL FRONTAL CORTEX (B10) | (26, 46, -4) |
| ORBITOFRONTAL CORTEX (B11) | (24, 44,-18/20) |
| DORSAL PREMOTOR CORTEX (B6) | (12, -1, 63) |
| LATERAL PREMOTOR CORTEX (LPM) | (53, 3, 11) |
| SECONDARY SOMATOSENSORY CORTEX (S2) | (44, -15, 16) |
| SENSORIMOTOR CORTEX (M1/S1) | (26, -22, 58) |
| SENSORIMOTOR CORTEX (S1/M1) | (33, -24, 65) |
| PREGENUAL CINGULATE CORTEX (PGC) | (21, 35, 4) |
Experimental procedure during scanning
After participants were positioned in the scanner and before scanning began, we again applied 5s contact heat temperatures at the previously determined HD, HPT, and HPTol levels to determine their reliability, making 1-2 °C adjustments as necessary to accommodate any perceptual changes associated with the scanning environment. During scanning, these 3 levels of heat were applied pseudorandomly to the volar surfaces of each forearm (or lower legs of CP patient #1) of the clinically affected and unaffected sides of the body. Heat stimuli were preset at one of the three threshold temperatures and applied repetitively for 5s, moving each contact site between stimuli, throughout the duration of each scan (about 60s). We applied 5s duration contact heat stimuli repetitively to different sites After each scan, we showed the VAS ruler to each participant and moved the sliding indicator to the number chosen for rating the intensity and unpleasantness of that series of heat stimuli. Inter-scan intervals were approximately 10 minutes. Table 3 shows the stimulation protocol and sequence for the 3 stimulus intensities used during each PET data acquisition session.
Table 3.
Protocol sheet for the acquisition of PET scanning data for each participant.
| CENTRAL PAIN PROTOCOL: 3D PET | |||||||
|---|---|---|---|---|---|---|---|
| Thermal Stimulation | Detection | Threshold | Tolerance | ||||
| Temp1 | Temp2 | Temp3 | |||||
| VAS scores | |||||||
| Side | Stimulation | Time | Counts | Pain | Unpleasantness | COMMENTS | |
| SCAN1 | RIGHT | Temp1 | |||||
| SCAN2 | RIGHT | Temp3 | |||||
| SCAN3 | RIGHT | Temp2 | |||||
| SCAN4 | LEFT | Temp1 | |||||
| SCAN5 | LEFT | Temp3 | |||||
| SCAN6 | LEFT | Temp2 | |||||
| SCAN7 | BASELINE | None | |||||
| SCAN8 | LEFT | Temp3 | |||||
| SCAN9 | LEFT | Temp1 | |||||
| SCAN10 | LEFT | Temp2 | |||||
| SCAN11 | RIGHT | Temp3 | |||||
| SCAN12 | RIGHT | Temp1 | |||||
| SCAN13 | RIGHT | Temp2 | |||||
| SCAN14 | BASELINE | NONE | |||||
| SCAN15 | RIGHT | Temp2 | |||||
| SCAN16 | RIGHT | Temp3 | |||||
| SCAN17 | RIGHT | Temp1 | |||||
| SCAN18 | LEFT | Temp2 | |||||
| SCAN19 | LEFT | Temp3 | |||||
| SCAN20 | LEFT | Temp1 | |||||
| SCAN21 | BASELINE | NONE | |||||
Data Analysis
We used SPSS (Chicago, IL, U.S.A.) version 17 for all statistical analysis. For the psychophysical studies, we used univariate ANOVA with Tukey post-hoc comparisons to analyze group differences in the temperatures and VAS numbers chosen for each heat stimulus category (HD, HPT, and HPTol). We used repeated measures ANOVA to examine group and stimulus intensity differences and interactions in the normalized VAS rating differences between the clinically involved and uninvolved body sides. In addition, we used non-parametric statistics (Kruskal-Wallis) to analyze group differences, by number of participants and number of stimuli, in the lateralization (side difference) of ratings of heat intensity. In the event of a group effect, we used the Mann-Whitney U test to examine group differences between CP and NP patient groups.
We chose non-parametric analytical methods to examine group differences in regional excitability (percentage change from baseline) among VOI because of the small sample size in this study and the individual pathophysiological differences, such as the nature and location of the central lesion, among participants in the patient groups. After examining the frequency distribution of the percentage change from the resting baseline of the rCBF during stimulation at HPT of all VOI in all HC participants, we used the Kruskal-Wallis and median tests to examine group differences in the number of VOI with out of range Z-scores (≥ 2 or < -2 ; Z = [individual %change from rest – HC group mean %change from rest]/HC group standard deviation %change from rest). Outliers were identified as values outside 1.5 times the 25th or 75th percentiles as provided in SPSS v 17. We considered VOI with Z scores ≥ 2 as hyperactive at rest or hyper-responsive during stimulation; VOI with Z scores < -2 were considered hypoactive at rest or hypo-responsive during stimulation. We used the Kruskal-Wallis and median tests also to examine group differences in the regional (VOI-wise) distribution of these Z score measurements. In the event of a group effect, we used the Mann-Whitney U test to examine group differences between CP and NP patient groups. For group comparisons, we corrected for the larger number of participants in the HC group by applying the following formula: (# (or sum) of Z scores /N) × 100.
RESULTS
Psychophysics
The average threshold temperatures selected for the stimulus categories in the noxious range is not different among groups; however, both the CP and NP patients have average heat detection (HD) thresholds that are slightly higher than the HC group (F2,32 = 7.15; p < 0.003; Table 4). Only 2 patients have HD thresholds below the mean or median of the HC group. In addition, the CP group gave higher average VAS heat intensity ratings for each stimulus category (F2,101 = 133.2, p < 0.008; Table 4). We detected no group rating differences of unpleasantness. Note that these threshold and rating differences do not reflect abnormalities on the side of clinical involvement because, for this pre-scanning analysis in the patient groups, we used data obtained from stimulating the clinically uninvolved side.
Table 4.
Average (s.e.m.) temperatures and VAS ratings chosen by HC, NP, and CP participants for perceived heat detection (HD), heat pain threshold (HPT), and heat pain tolerance (HPTol) during stimulation of the right (HC group) or clinically uninvolved volar forearm (patients) immediately before scanning.
| mean (s.e.m.) | ||||
|---|---|---|---|---|
| HC | NP | CP | ||
| HD | °C | 38.6 (0.37) | 41.2 (0.71)* | 40.6 (0.72)* |
| VAS | 2.35 (0.24) | 1.89 (0.21) | 2.98 (0.50)** | |
| HPT | °C | 46.4 (0.97) | 46.9 (0.66) | 47.2 (0.45) |
| VAS | 4.64 (0.35) | 4.70 (0.47) | 5.43 (0.41)** | |
| HPTol | °C | 49.4 (0.91) | 49.2 (0.55) | 50.0 (0.36) |
| VAS | 7.82 (0.23) | 7.55 (0.37) | 8.46 (0.38)** | |
higher than HC group (post hoc comparisons: p< 0.031 for NP and p< 0.004 for CP groups)
greater than HC and NP groups (post hoc comparisons: p<0.003 for NP and <0.015 for HC groups)
To examine lateralized (side-to-side) differences in the heat intensity and unpleasantness ratings during scanning, we first normalized the lateralized rating differences for each patient ([VAS clinically normal side – VAS clinically affected side]/VAS clinically normal side) so that a positive number reflects a decreased rating on the clinically affected side; this normalization is necessary because of individual differences in the numbers chosen by each participant for the VAS rating. For the HC group, we applied the same formula but substituted the right side for “normal” and the left side for “affected”. The average normalized differences in lateralized VAS ratings across all stimulus intensities are positive for both CP and NP groups (0.182 +/- 0.065se; 0.084 +/-0.066se respectively), but nearly zero (- 0.007) for the HC group, consistent with reduced heat intensity perception on the clinically involved side among patients and with a lack of side preference among healthy participants. A repeated measures ANOVA on these average normalized differences reveals a trend of group effect (p = 0.079; F2,91 =2.605), no effect of stimulus intensity (p = 0.562), and no group by stimulus intensity interaction (p = 0.532). However, this analysis is sensitive to the degree of lateralized perceived difference, which is of less interest in this study than the distribution of differences (independent of degree) among the 3 groups. Thus, in a non-parametric analysis, we find a significant group effect on the number of participants who show a lateralized average normalized difference in the VAS rating for heat intensity (but not for unpleasantness) for 2 or more of the 3 stimulus intensities (Kruskal – Wallis χ2df 2= 7.92, p < 0.019). As shown in Figure 1, the entire NP group and 7 (70%) of the CP group have lower intensity ratings of the same stimulus applied to the clinically affected side. Only 7 (47%) of the HC group shows a lateralized preference for 2 or more stimulus intensities. We also find a group effect on the number of stimuli for which there is a lateralized difference for intensity ratings at all applied intensities (Kruskal-Wallis χ2df2= 15.8, p < 0.010) and a similar group effect for perceived unpleasantness (Kruskal-Wallis χ2df 2= 9.16, p < 0.010). Thus, 22 (73%) of the stimuli applied to the CP group and 25 (83%) of the stimuli applied to the NP group receive lower intensity ratings when the same stimulus is applied to the clinically affected side; in contrast, only 23 (51%) of the stimuli applied to the HC group show a lateralized intensity preference.
Figure 1.

Number of healthy control participants (HC; N = 15), central pain patients (CP; N = 10), and patients with sensory loss but no pain (NP; N = 10), who have a lower average normalized VAS rating of an identical heat stimulus on the clinically affected (patients) or left (HC) side for 3 (black bars), 2 (gray bars), or 1-0 (white bars) applied stimulus intensity levels.
The results of the psychophysical studies thus confirm the clinical impression that, in comparison with the HC group of males in the same age range, both patient groups have a reduced perception of heat intensity and, by at least one measure, heat unpleasantness on the clinically affected side. In addition, there is an impairment of heat intensity perception (heat detection thresholds) on the clinically unaffected side among both NP and CP patients.
Functional imaging
The activation images for the HC group are shown in Figure 2 (see also Table 2). At rest, there is no lateralized (side-to-side) difference in estimated resting rCBF among VOI among healthy subjects (F1,504 = 0.260, p = 0.610), nor is there a significant interaction of side and location of resting activity (F17,504 = 0.945, p = 0.521). Because we use the percentage change from rest as an estimate of excitability during stimulation, and Z-scores of ≥ 2 or < -2 from the HC group mean as evidence of hyper- or hypo-excitability, we examined the distribution of excitability among all VOI in the HC group during stimulation of either side. The distribution of this metric is not normal according to the Kolmogorov-Smirnov test (K-S statistics: 0.062 L stimulation, 0.072 R stimulation; p<0.001) presumably because of 4 outlying frontal lobe activations in one participant. However, the frequency distribution of VOI excitability is unimodal and, by inspection, fits closely a superimposed normal curve as shown in Figure 3 during stimulation at HPT, for example. Therefore, we proceeded with the use of Z scores as estimates of deviation from the mean of VOI excitability within the HC group.
Figure 2.

Statistical parametric maps (Z score color code in center) of the regional cerebral blood flow (rCBF) responses from baseline (H215O PET) to repetitive 5s contact heat stimulation of the left or right volar forearm in a group of healthy participants, ages 42-54. Transverse brain images shown at selected levels above the anterior-posterior commissure plane; right hemisphere is to the reader’s left. Stimulus intensities were psychophysically determined for each individual. HD: heat detection. HPT: heat pain threshold. HPTol: heat pain tolerance.
Figure 3.
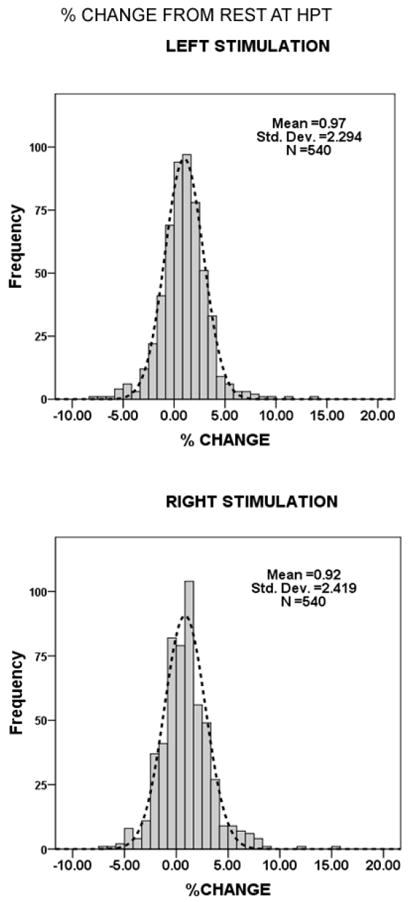
Frequency histograms of the percentage change from baseline rCBF among HC participants during heat stimulation of either side at HPT. A normal distribution curve is superimposed on the histogram.
There is a significant group difference among healthy participants (HC), patients without pain (NP), and patients with central pain (CP) in the total percentage of hyperactive (Z =/> 2) and hypoactive (Z < -2) VOIs at rest (Figure 4; Kruskal Wallis test: hyperactive, χ2df 2 = 7.53, p < 0.023; hypoactive, χ2df 2 = 14.08, p < 0.001). This difference persists after excluding three outlying values. The Mann-Whitney U test shows no difference in these measures between NP and CP groups at rest (U = 49, p = 0.94 and U = 40, p = 0.45 for hyper-active and hypo-active VOIs, respectively).
Figure 4.

Group percentages of total number of hyperactive (Z ≥ 2) and hypoactive (Z < -2) VOI among HC (white bars; N = 15), NP (gray bars; N = 10), and CP (black bars; N = 10) groups at rest (baseline rCBF).
At rest, there is also a significant group difference in the anatomical distribution of hyper-and hypo-active VOIs (Kruskal Wallis test: hyper-active, χ2df 2 = 21.16, p < 0.001; hypo-active, χ2df 2 = 23.49, p < 0.001). This difference persists after excluding three outlying values. Chi-square tests of each VOI show no right-left (lateralized) effect (p > 0.05). Inspection of the data (Figure 5) shows that both NP and CP groups have more hyper- and hypoactive VOI at rest than the HC group; however, the Mann-Whitney U test shows no difference in these measures between NP and CP groups at rest (U = 572, p = 0.36 and U = 637, p = 0.90 for hyper-active and hypo-active VOIs, respectively).
Figure 5.
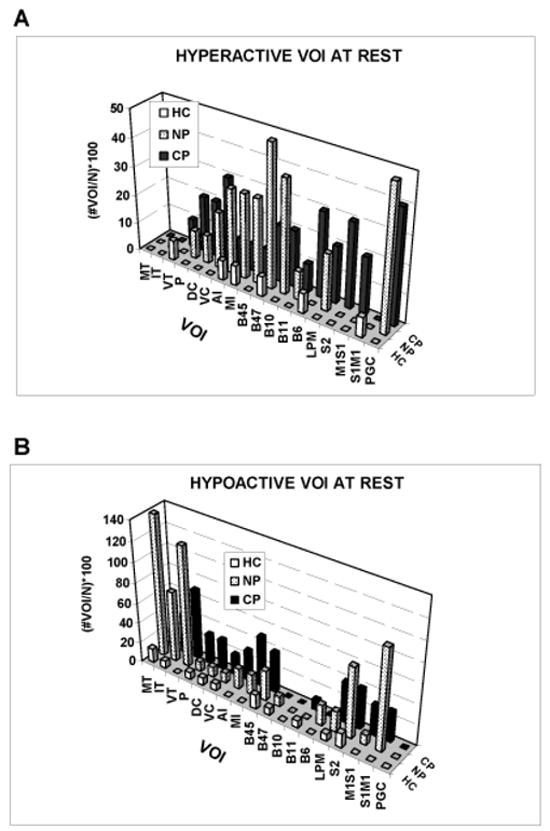
Anatomical distribution of group percentages of the total number of hyperactive and hypoactive VOI among HC, NP, and CP groups. See Table 2 for abbreviations.
During stimulation, there is a group difference in the number of out of range Z scores among subjects (Kruskal Wallis test: hyper-responsive, χ2df 2 = 14.40, p < 0.002; hypo-responsive, χ2df 2 = 23.65, p < 0.001); we did not detect an effect of the three levels stimulus intensity (K-W test: hyper-responsive, χ2df 2 = 1.46, p = 0.48; hypo-responsive, χ2df 2 = 0.045, p = 0.98). Inspection of the data (Figure 6) again shows that both NP and CP groups have more hyper- and hypo-responsive VOI during stimulation than the HC group; however, the Mann-Whitney U test shows no difference in these measures between NP and CP groups at rest (U = 438, p > 0.86 and U = 386, p = 0.34 for hyper-active and hypo-active VOIs, respectively).
Figure 6.
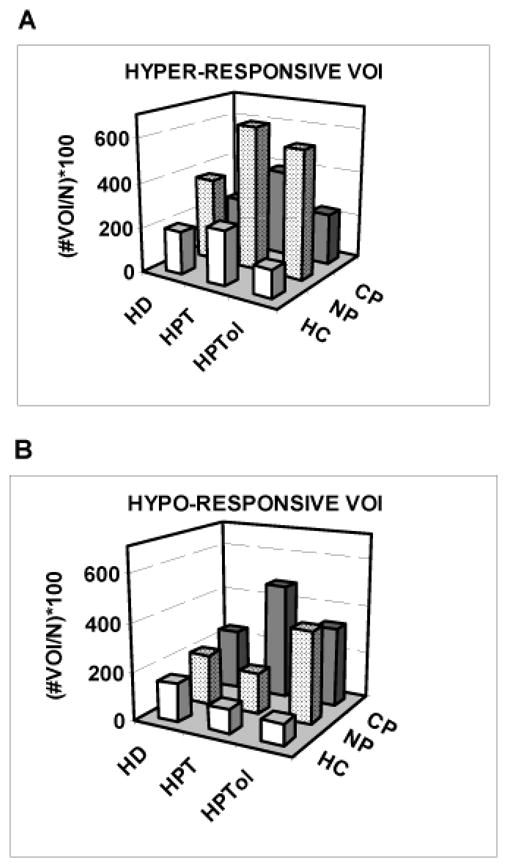
Group percentages of the total number of hyper-responsive and hypo-responsive VOI among HC, NP, and CP groups during heat stimulation at HD, HPT, and HPTol perceived intensity levels.
During stimulation, and across all stimulus intensity levels, there is also a group difference in the anatomical distribution of both hyper- and hypo-responsive VOI (Figure 7; Kruskal Wallis test, hyper-active, χ2df 2 = 7.80, p < 0.020; hypo-active, χ2df 2 = 9.52, p < 0.009). This difference persists, and in fact increases, after excluding two outliers. As noted above, there is no group effect of stimulus intensity. Chi-square tests of each VOI show no right-left (lateralized) effect of stimulation (p > 0.05). Inspection of the data shows that both NP and CP groups have more hyper- and hypo-responsive VOI during stimulation than the HC group. Of particular relevance for the purpose of this study, however, a post-hoc Mann-Whitney U test shows a difference between NP and CP groups in the number of hyper-responsive (U = 452, p = 0.02), but not hypo-responsive (U = 494, p = 0.08), VOI during stimulation.
Figure 7.
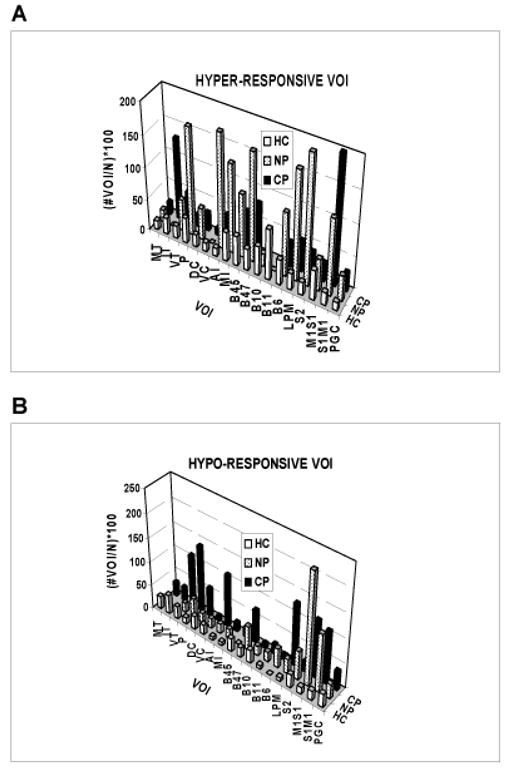
Anatomical distribution of group percentages of the total number of hyper-responsive and hypo-responsive VOI among HC, NP, and CP groups at all levels of heat stimulation.
The frequency distribution of the differences among hyper-responsive VOI (CP group – NP group) is not uniform (Figure 8; Kolmogorov-Smirnov one-sample test, Z = 2.17, p (2-tailed) < 0.001). Inspection of these differences shows that only the intralaminar thalamus (IT), ventral caudate (VC) and the somatosensory-somatomotor (S1M1) cerebral cortex are more frequently hyper-responsive among CP than among NP patients. All other hyper-responsive VOI appear with either less or equal frequency in the CP group. Seven CP patients and 5 NP patients have hyper-responsive VOI within the S1M1 cortex during one or more stimulus trials; 4 CP patients, but no NP patients, have a hyper-responsive VOI in the intralaminar thalamus (IT). There are more hyper-responsive (N = 11 vs 5) and fewer hypo-responsive (N = 1 vs 5) intralaminar thalamic or S1M1 cortical VOI among CP than among NP patients (χ2 df1 =4.54, p < 0.033). The hyper-responsive thalamus or cortex in CP patients is contralateral to the pain in 5 patients, ipsilateral in 2, and bilateral in the remainder.
Figure 8.
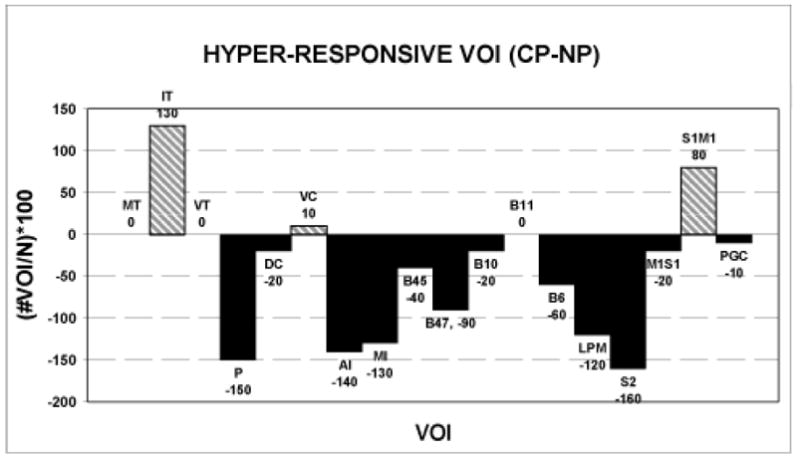
Anatomical distribution of the differences between CP and NP patients (CP-NP) in the group percentage of the total number of VOI that are hyper-responsive during heat stimulation at all levels of perceived intensity.
Figure 9 shows an example of a CP patient (patient # 1, Table 1) with a clinically determined left spinothalamic tract lesion (~T8 level), heat allodynia, and a hyper-responsive intralaminar thalamus. Note that both the left (partially denervated) and right thalamus is hyper-responsive at stimulus levels below and at the heat pain threshold. This patient also has a hyper-responsive left S1M1 cortex during stimulation of either leg but at HPTol levels only. This example demonstrates the remote effects of a single lesion on central nociceptive mechanisms.
Figure 9.
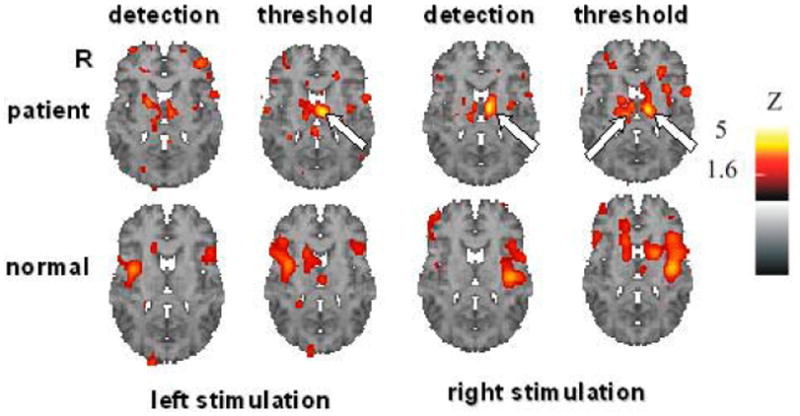
Statistical parametric maps (Z score color code at right) of the regional cerebral blood flow (rCBF) responses from baseline (H215O PET) to repetitive 5s contact heat stimulation at heat detection (HD) and heat pain threshold levels (HPT). Imaging orientation conventions as in Figure 2. Top row of images are from CP patient # 1, who had heat allodynia of the right body below the T8 dermatome due to a lesion clinically identified as a stroke involving the right spinothalamic tract. Bottom row of images show the average responses at the same levels from the (normal) HC group. Arrows indicate the excessive responses primarily, but not exclusively, in the thalamus ipsilateral to the spinal cord lesion.
DISCUSSION
Psychophysics
Both NP and CP patients had reduced heat intensity sensation on the clinically affected side, a finding in accord with those of other investigators [ 10, 9, 108, 25, 13, 38 ]. In addition, both NP and CP patients had a higher HD on the clinically unaffected side compared with the HC group. This finding contrasts with the generalized increased pain sensitivity found during experimental acute or chronic nociceptive pain [ 57 ]; moreover, it has not been reported in previous similar investigations possibly because the clinically unaffected side was used as a within-patient comparison. The HD finding suggests that the focal CNS lesions in both patient groups caused a bilateral impairment of heat sensory function that was clinically detected only contralateral to the lesion. It is possible that undetected CNS lesions caused sensory losses on the unaffected side but this is unlikely because such lesions would have to be present in 80% (16/20) of patients. This result may also reflect the presence of the few ventrolateral thalamic and S1 cortical neurons that are bilaterally responsive to heat stimuli in non-human primates [ 52, 32 ]. Clinical observations have long provided evidence for bilateral nociceptive transmission that is normally suppressed when both spinothalamic pathways are intact [ 74, 11, 73, 72 ]. Finally, we note that CP patients chose higher VAS ratings for each stimulus category compared to both NP and HC groups but we do not have a neurophysiological hypothesis for this observation. Overall, the psychophysical results reveal a bilateral effect of a CNS lesion that produces a clinically contralateral effect.
Imaging at rest
At rest, NP and CP patients had more VOI that were hyperactive or hypoactive than HC participants. We did not detect a laterality difference in this measurement nor did we find a group difference between NP and CP patients. The anatomical distribution of hyperactive and hypoactive VOI was also different among the three groups but not between NP and CP patients. The group difference in the anatomical distribution of hyperactive and hypoactive VOI cannot be attributed to any particular VOI or group of VOI; it probably reflects both the wide distribution and the larger number of out of range Z scores among VOI within the patient groups. Thus, the results of the study during the resting condition do not suggest a neurophysiological basis for the resting pain experienced by CP patients. However, the large number and wide distribution of VOI with abnormal activity among NP and CP patients shows that the CNS lesion causing the focal sensory loss has divergent and widely distributed effects.
Imaging during stimulation
During stimulation, NP and CP patients had more hyper-responsive and hypo-responsive VOI than HC participants and there was no group effect of stimulus intensity or laterality. The group difference in the anatomical distribution of these excitability changes again reflects primarily the wide distribution and much larger number of hyper- and hypo-responsive VOI within the patient groups. However, we detected a difference in the anatomical distribution of hyper-responsive, but not hypo-responsive, VOI between NP and CP patients and these differences were not uniformly distributed. As shown in Figures 7 and 8, several structures had fewer hyper-responsive VOI in CP than in NP patients; those with the greatest differences relative to both CP and HC groups include the putamen and the anterior insular, mid-insular, inferior prefrontal (B47), lateral premotor, and secondary somatosensory (S2) cortices. It is possible that the relatively reduced number of these hyper-responsive VOI contributes to the pain of our CP patients because they could participate in endogenous analgesic mechanisms [ 90, 56, 63, 5, 6, 48 ]. If this hypothesis is correct, however, we would expect to find a relatively increased number of hypo-responsive VOI within these structures among CP patients and thus a significant difference between NP and CP patients in the distribution of hypo-responsive VOI. However, only hyper-responsive VOI were distributed differently between the NP and CP groups. It is therefore unlikely that a relatively reduced hyper-responsiveness of some structures contributes to the pain of CP patients. Rather, the pain of our CP patients is more likely attributable to the uniquely increased number of hyper-responsive VOI within the intralaminar thalamus (IT) and primary sensory-motor cortex (S1/M1) as shown in Figures7 and 8. The ventral caudate (VC) also has slightly more hyper-responsive VOI among CP patients, but the total number of VOI is small compared to the other two structures. All CP patients but one (90%) showed hyper-responsiveness in either the intralaminar thalamus, S1/M1 cortex, or in both structures; in contrast, 5 NP patients (50%) had hyper-responsive VOI in these structures.
Physiological significance of IL thalamic and S1M1 hyper-responsiveness in CP patients
Our findings may not apply to CP patients generally because our sample size is small and the pathophysiology of CP is not likely to be identical or even similar across a larger sample of patients. Nonetheless, the intralaminar thalamus and primary sensory-motor cortex are likely candidates for participation in mediating the pain of CP. The primate intralaminar thalamus receives direct input from the spinothalamic tract [ 65, 7, 50, 99, 2, 36 ], shows cellular responses to noxious stimuli [ 21, 16, 58, 51, 91 ], and is among the thalamic structures regularly activated in functional imaging studies of pain in humans [ 22, 81, 85, 1 ]. Furthermore, thalamo-cortical intralaminar thalamic cells send axons directly to the anterior cingulate cortex [ 111, 110 ], which also shows differential cellular and evoked potential responses to noxious stimuli [ 62, 96, 47, 97 ] and is among the structures most commonly activated in human pain imaging studies [ 22, 81, 1 ]. Focal lesions within the anterior cingulate cortex have been used to alleviate the affective component of chronic pain [ 39, 33, 117 ]. Based on clinical and neurophysiological evidence, the intralaminar thalamic-anterior cingulate pathway has long been associated with mediating the affective component of pain [ 39, 112, 109 ]. Similarly, the sensory-motor cortex receives nociceptive input via the ventrolateral thalamic nuclei, shows differential or selective cellular and evoked potential responses to noxious stimuli in human and non-human primates [ 52, 84, 53, 83, 77, 116 ], and is among the structures activated in pain imaging studies of humans [ 17, 81, 1 ]. Lesions within the territory of the primary sensory-motor cortex may attenuate pain in restricted body areas according to a few reports [ 92, 64 ]. Notably, electrical stimulation of the motor cortex has been reported to be effective in the treatment of chronic pain, including intractable CP [ 106, 79, 76, 20, 89, 63 ]. Overall, the weight of clinical and electrophysiological evidence supports the hypothesis that the sensory-motor cortex participates in mediating the sensory-discriminative, but not affective, components of pain [ 82, 94, 83 ]. Thus, a unique nociceptive excitability increase in the intralaminar thalamus and somatosensory cortex may contribute to the pain in some patients with central pain syndrome.
Widespread excitability changes in NP and CP patients
Our results provide additional evidence for the anatomically distributed effects of focal CNS lesions, a phenomenon clearly recognized in the 19th and early 20th centuries [ 71 ] and further documented in contemporary brain imaging studies [ 78, 103, 3 ]. Focal lesions are often followed by an extensive anatomical and functional reorganization of sensory and motor pathways [ 31, 114, 115, 75, 30 ]. The changes following focal injury include widespread altered excitability [ 66 ] and changes in the amount and activity of several neurotransmitters and neurotrophic factors [ 100, 41, 66, 87 ], gamma-amino-butyric acid (GABA) being among the most frequently implicated in the pathophysiology of post-stroke or post-traumatic central pain [ 88, 107, 18, 24, 43 ]. The recent study of Willoch and colleagues [ 118 ] reveals further the widespread effect of focal subcortical lesions on cerebral opioid receptor binding and its presumed effect on endogenous analgesia mechanisms [ 14, 120, 23 ]. Given the results presented here, some aspects of a thalamic disinhibition hypothesis deserve consideration also [44, 37, 34 ].
Conclusion
NP and CP patients have bilaterally reduced heat detection, unilaterally reduced heat sensory ratings, and more brain areas with altered resting activity and heat-evoked excitability than an aged-matched group of healthy individuals. Compared to HC participants and NP patients, CP patients have more hyper-responsive areas in the intralaminar thalamus and sensory-motor cortex. Our results thus confirm the widely distributed effects of focal CNS lesions and suggest that a unique pattern of nociceptive excitability change may underlie the pain in some patients with central pain syndrome.
Acknowledgments
Supported by NIH grant P01-HD33986 and the Department of Veteran’s Affairs. The authors thank Susan King and Gail VanDusen for administrative and technical support; we also thank Jill Rothley, Todd Hauser, Paul Kison, Edward McKenna, and Andrew Weeden for technical support with PET scanning.
Footnotes
The authors have no conflicts of interest related to any aspect of the contents of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Apkarian AV, Hodge CJ. Primate spinothalamic pathways: III. Thalamic terminations of the dorsolateral and ventral spinothalamic pathways. J Comp Neurol. 1989;288:493–511. doi: 10.1002/cne.902880309. [DOI] [PubMed] [Google Scholar]
- 3.Baron JC, Levasseur M, Mazoyer B, Legault-Demare F, Mauguiere F, Pappata S, Jedynak P, Derome P, Cambier J, Tran-Dinh S. Thalamocortical diaschisis: positron emission tomography in humans. J Neurol Neurosurg Psychiat. 1992;55:935–942. doi: 10.1136/jnnp.55.10.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beiske AG, Loge JH, Ronningen A, Svensson E. Pain in Parkinson’s disease: Prevalence and characteristics. Pain. 2009;141:173–177. doi: 10.1016/j.pain.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Benarroch EE. Descending monoaminergic pain modulation: Bidirectional control and clinical relevance. Neurol. 2008;71:217–221. doi: 10.1212/01.wnl.0000318225.51122.63. [DOI] [PubMed] [Google Scholar]
- 6.Bingel U, Tracey I. Imaging CNS Modulation of Pain in Humans. Physiology. 2008;23:371–380. doi: 10.1152/physiol.00024.2008. [DOI] [PubMed] [Google Scholar]
- 7.Boivie J. The termination of the spinothalamic tract in the cat. An experimental study with silver impregnation methods. Exp Brain Res. 1971;12:331–353. doi: 10.1007/BF00234489. [DOI] [PubMed] [Google Scholar]
- 8.Boivie J. Central post-stoke pain. In: Cervero F, Jensen TS, editors. Pain. Vol. 81. Edinburgh: Elsevier; 2006. pp. 715–730. [Google Scholar]
- 9.Boivie J, Leijon G, Johansson I. Central post-stroke pain---a study of the mechanisms through analyses of the sensory abnormalities. Pain. 1989;37:173–185. doi: 10.1016/0304-3959(89)90128-0. [DOI] [PubMed] [Google Scholar]
- 10.Bowsher D. Central pain: Clinical and physiological characteristics. J Neurol Neurosurg Psychiat. 1996;61:62–69. doi: 10.1136/jnnp.61.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowsher D. Contralateral mirror-image pain following anterolateral cordotomy. Pain. 1988;33:63–65. doi: 10.1016/0304-3959(88)90204-7. [DOI] [PubMed] [Google Scholar]
- 12.Bowsher D. Central pain: clinical and physiological characteristics. J Neurol Neurosurg Psychiat. 1996;61:62. doi: 10.1136/jnnp.61.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowsher D, Leijon G, Thoumas KA. Central poststroke pain - Correlation of MRI with clinical pain characteristics and sensory abnormalities. Neurol. 1998;51:1352–1358. doi: 10.1212/wnl.51.5.1352. [DOI] [PubMed] [Google Scholar]
- 14.Bowsher D, Leijon G, Thuomas KA. Central poststroke pain: correlation of MRI with clinical pain characteristics and sensory abnormalities. Neurol. 1998;51:1352. doi: 10.1212/wnl.51.5.1352. [DOI] [PubMed] [Google Scholar]
- 15.Burton H, Videen TO, Raichle ME. Tactile-vibration-activated foci in insular and parietal-opercular cortex studied with positron emission tomography: Mapping the second somatosensory area in humans. Somatosens Mot Res. 1993;10:297–308. doi: 10.3109/08990229309028839. [DOI] [PubMed] [Google Scholar]
- 16.Bushnell MC, Duncan GH. Sensory and affective aspects off pain perception: is medial thalamus restricted to emotional issues? Exp Brain Res. 1989;78:415–418. doi: 10.1007/BF00228914. [DOI] [PubMed] [Google Scholar]
- 17.Bushnell MC, Duncan GH, Hofbauer RK, Ha B, Chen JI, Carrier B. Pain perception: Is there a role for primary somatosensory cortex? Proc Natl Acad Sci USA. 1999;96:7705–7709. doi: 10.1073/pnas.96.14.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canavero S, Bonicalzi V. The neurochemistry of central pain: evidence from clinical studies, hypothesis and therapeutic implications. Pain. 1998;74:109–114. doi: 10.1016/s0304-3959(97)00089-4. [DOI] [PubMed] [Google Scholar]
- 19.Canavero S, Bonicalzi V. Central Pain Syndrome: Pathophysiology, Diagnosis, and Management. Cambridge: Cambridge University Press; 2006. pp. 1–382. [Google Scholar]
- 20.Carroll D, Joint C, Maartens N, Shlugman D, Stein J, Aziz TZ. Motor cortex stimulation for chronic neuropathic pain: a preliminary study of 10 cases. Pain. 2000;84:431–437. doi: 10.1016/s0304-3959(99)00245-6. [DOI] [PubMed] [Google Scholar]
- 21.Casey KL. Unit analysis of nociceptive mechanisms in the thalamus of the awake squirrel monkey. J Neurophysiol. 1966;29:727–750. doi: 10.1152/jn.1966.29.4.727. [DOI] [PubMed] [Google Scholar]
- 22.Casey KL. Forebrain mechanisms of nociception and pain: Analysis through imaging. Proc Natl Acad Sci USA. 1999;96:7668–7674. doi: 10.1073/pnas.96.14.7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casey KL. Central pain: distributed effects of focal lesions. Pain. 2004;108:205–206. doi: 10.1016/j.pain.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Casey KL. Pathophysiology of Central Poststroke Pain: The Contribution of Functional Imaging and a Hypothesis. In: Henry JL, Panju A, Yashpal K, editors. Central Neuropathic Pain: Focus on Poststroke Pain. Seattle: IASP Press; 2007. pp. 115–131. [Google Scholar]
- 25.Casey KL, Beydoun A, Boivie J, Sjolund B, Holmgren H, Leijon G, Morrow TJ, Rosen I. Laser-evoked cerebral potentials and sensory function in patients with central pain. Pain. 1996;64:485–491. doi: 10.1016/0304-3959(95)00143-3. [DOI] [PubMed] [Google Scholar]
- 26.Casey KL, Minoshima S, Berger KL, Koeppe RA, Morrow TJ, Frey KA. Positron emission tomographic analysis of cerebral structures activated specifically by repetitive noxious heat stimuli. J Neurophysiol. 1994;71:802–807. doi: 10.1152/jn.1994.71.2.802. [DOI] [PubMed] [Google Scholar]
- 27.Casey KL, Minoshima S, Morrow TJ, Koeppe RA. Comparison of human cerebral activation patterns during cutaneous warmth, heat pain, and deep cold pain. J Neurophysiol. 1996;76:571–581. doi: 10.1152/jn.1996.76.1.571. [DOI] [PubMed] [Google Scholar]
- 28.Cassinari V, Pagni CA. Central Pain: A Neurosurgical Survey. Cambridge: Harvard University Press; 1969. [Google Scholar]
- 29.Cesaro P, Mann MW, Moretti JL, Defer G, Roualdes B, Nguyen JP, Degos JD. Central pain and thalamic hyperactivity: a single photon emission computerized tomographic study. Pain. 1991;47:329–336. doi: 10.1016/0304-3959(91)90224-L. [DOI] [PubMed] [Google Scholar]
- 30.Chen R, Cohen LG, Hallett M. Nervous system reorganization following injury. Neurosci. 2002;111:761–773. doi: 10.1016/s0306-4522(02)00025-8. [DOI] [PubMed] [Google Scholar]
- 31.Chollet F, DiPiero V, Wise RJS, Brooks DJ, Dolan RJ, Frackowiak RSJ. The functional anatomy of motor recovery after stroke in humans: a study with positron emission tomography. Ann Neurol. 1991;29:63–71. doi: 10.1002/ana.410290112. [DOI] [PubMed] [Google Scholar]
- 32.Chung JM, Lee KH, Surmeier DJ, Sorkin LS, Kim J, Willis WD. Response characteristics of neurons in the ventral posterior lateral nucleus of the monkey thalamus. J Neurophysiol. 1986;56:370–390. doi: 10.1152/jn.1986.56.2.370. [DOI] [PubMed] [Google Scholar]
- 33.Cohen RA, Kaplan RF, Moser DJ, Jenkins MA, Wilkinson H. Impairments of attention after cingulotomy. Neurol. 1999;53:819–824. doi: 10.1212/wnl.53.4.819. [DOI] [PubMed] [Google Scholar]
- 34.Craig AD. A new version of the thalamic disinhibition hypothesis of central pain. Pain Forum. 1998;7:1–14. [Google Scholar]
- 35.Craig AD. The functional anatomy of lamina 1 and its role in post-stroke central pain. In: Sandkuhler J, Bromm B, Gebhart GF, editors. Nervous System Plasticity and Chronic Pain. Vol. 129. Amsterdam: Elsevier; 2000. pp. 137–151. [DOI] [PubMed] [Google Scholar]
- 36.Craig AD. Distribution of trigeminothalamic and spinothalamic lamina I terminations in the macaque monkey. J Comp Neurol. 2004;477:119–148. doi: 10.1002/cne.20240. [DOI] [PubMed] [Google Scholar]
- 37.Craig AD, Bushnell MC. The thermal grill illusion: Unmasking the burn of cold pain. Science. 1994;265:252–255. doi: 10.1126/science.8023144. [DOI] [PubMed] [Google Scholar]
- 38.Finnerup NB, Johannesen IL, Fuglsang-Frederiksen A, Bach FW, Jensen TS. Sensory function in spinal cord injury patients with and without central pain. Brain. 2003;126:57. doi: 10.1093/brain/awg007. [DOI] [PubMed] [Google Scholar]
- 39.Foltz EL, White LE. Pain “relief” by frontal cingulumotomy. J Neurosurg. 1962;19:89–100. doi: 10.3171/jns.1962.19.2.0089. [DOI] [PubMed] [Google Scholar]
- 40.Fox PT, Raichle ME. Stimulus rate dependence of regional cerebral blood flow in human striate cortex, demonstrated by positron emission tomography. J Neurophysiol. 1984;51:1109–1120. doi: 10.1152/jn.1984.51.5.1109. [DOI] [PubMed] [Google Scholar]
- 41.Franke H, Krugel U, Illes P. P2 receptors and neuronal injury. Pflüg Arch Eur J Physiol. 2006;452:622–644. doi: 10.1007/s00424-006-0071-8. [DOI] [PubMed] [Google Scholar]
- 42.Gorecki J, Hirayama T, Dostrovsky JO, Tasker RR, Lenz FA. Thalamic stimulation and recording in patients with deafferentation and central pain. Stereotact Funct Neurosurg. 1989;52:219–226. doi: 10.1159/000099504. [DOI] [PubMed] [Google Scholar]
- 43.Goudet C, Magnaghi V, Landry M, Nagy F, Gereau RW, Pin JP. Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev. 2009;60:43–56. doi: 10.1016/j.brainresrev.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Head H, Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254. [Google Scholar]
- 45.Henry JL, Panju AA, Yashpal K. Central Neuropathic Pain: Focus on Poststroke Pain. Seattle: IASP Press; 2007. pp. 3–281. [Google Scholar]
- 46.Hirayama T, Dostrovsky JO, Gorecki J, Tasker RR, Lenz FA. Recordings of abnormal activity in patients with deafferentation and central pain. Stereotact Funct Neurosurg. 1989;52:120–126. doi: 10.1159/000099492. [DOI] [PubMed] [Google Scholar]
- 47.Hutchison WD, Davis KD, Lozano AM, Tasker RR, Dostrovsky JO. Pain-related neurons in the human cingulate cortex. Nat Neurosci. 1999;2:403–405. doi: 10.1038/8065. [DOI] [PubMed] [Google Scholar]
- 48.Jensen MP. A Neuropsychological Model of Pain: Research and Clinical Implications. J Pain. 2010;11:2–12. doi: 10.1016/j.jpain.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 49.Jensen TS, Lenz FA. Central post-stroke pain: A challenge for the scientist and the clinician. Pain. 1995;61:161–164. doi: 10.1016/0304-3959(94)00227-6. [DOI] [PubMed] [Google Scholar]
- 50.Jones EG, Burton H. Cytoarchitecture and somatic sensory connectivity of thalamic nuclei other than the ventrobasal complex. J Comp Neurol. 1974;154:395–432. doi: 10.1002/cne.901540404. [DOI] [PubMed] [Google Scholar]
- 51.Kawakita K, Dostrovsky JO, Tang JS, Chiang CY. Responses of neurons in the rat thalamic nucleus submedius to cutaneous, muscle and visceral nociceptive stimuli. Pain. 1993;55:327–338. doi: 10.1016/0304-3959(93)90008-D. [DOI] [PubMed] [Google Scholar]
- 52.Kenshalo DR, Jr, Isensee O. Responses of primate S1 cortical neurons to noxious stimuli. J Neurophysiol. 1983;50:1479–1496. doi: 10.1152/jn.1983.50.6.1479. [DOI] [PubMed] [Google Scholar]
- 53.Kenshalo DR, Iwata K, Sholas M, Thomas DA. Response properties and organization of nociceptive neurons in Area 1 of monkey primary somatosensory cortex. J Neurophysiol. 2000;84:719–729. doi: 10.1152/jn.2000.84.2.719. [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Greenspan JD, Coghill RC, Ohara S, Lenz FA. Lesions Limited to the Human Thalamic Principal Somatosensory Nucleus (Ventral Caudal) Are Associated with Loss of Cold Sensations and Central Pain. J Neurosci. 2007;27:4995–5004. doi: 10.1523/JNEUROSCI.0716-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klit H, Finnerup NB, Andersen G, Jensen TS. Central poststroke pain: A population-based study. Pain. 2011;152:818–824. doi: 10.1016/j.pain.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 56.Koechlin E, Ody C, Kouneiher F. The architecture of cognitive control in the human prefrontal cortex. Science. 2003;302:1181–1185. doi: 10.1126/science.1088545. [DOI] [PubMed] [Google Scholar]
- 57.Kosek E, Ordeberg G. Abnormalities of somatosensory perception in patients with painful osteoarthritis normalize following successful treatment. Eur J Pain. 2000;4:229–238. doi: 10.1053/eujp.2000.0175. [DOI] [PubMed] [Google Scholar]
- 58.Krauthamer GM, Krol JG, Grunwerg BS. Effect of superior colliculus lesions on sensory unit responses in the intralaminar thalamus of the rat. Brain Res. 1992;576:277–286. doi: 10.1016/0006-8993(92)90691-2. [DOI] [PubMed] [Google Scholar]
- 59.Leijon G, Boivie J, Johansson I. Central post-stroke pain--Neurological symptoms and pain characteristics. Pain. 1989;36:13–25. doi: 10.1016/0304-3959(89)90107-3. [DOI] [PubMed] [Google Scholar]
- 60.Lenz FA, Gracely RH, Baker FH, Richardson RT, Dougherty PM. Reorganization of sensory modalities evoked by microstimulation in region of the thalamic principal sensory nucleus in patients with pain due to nervous system injury. J Comp Neurol. 1998;399:125–138. [PubMed] [Google Scholar]
- 61.Lenz FA, Kwan HC, Dostrovsky JO, Tasker RR. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res. 1989;496:357–360. doi: 10.1016/0006-8993(89)91088-3. [DOI] [PubMed] [Google Scholar]
- 62.Lenz FA, Rios M, Zirh A, Chau D, Krauss G, Lesser RP. Painful stimuli evoke potentials recorded over the human anterior cingulate gyrus. J Neurophysiol. 1998;79:2231–2234. doi: 10.1152/jn.1998.79.4.2231. [DOI] [PubMed] [Google Scholar]
- 63.Lima MC, Fregni F. Motor cortex stimulation for chronic pain: Systematic review and meta-analysis of the literature. Neurol. 2008;70:2329–2337. doi: 10.1212/01.wnl.0000314649.38527.93. [DOI] [PubMed] [Google Scholar]
- 64.Marshall J. Sensory disturbances in cortical wounds with special reference to pain. J Neurol Neurosurg Psychiat. 1951;14:187–204. doi: 10.1136/jnnp.14.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehler WR, Feferman ME, Nauta WJH. Ascending axon degeneration following anterolateral cordotomy. An experimental study in the monkey. Brain. 1960;83:718–750. doi: 10.1093/brain/83.4.718. [DOI] [PubMed] [Google Scholar]
- 66.Menon B, Shorvon SD. Ischaemic stroke in adults and epilepsy. Epilepsy Res. 2009;87:1–11. doi: 10.1016/j.eplepsyres.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 67.Minoshima S, Berger KL, Lee KS, Mintun MA. An automated method for rotational correction and centering of three-dimensional functional brain images. J Nucl Med. 1992;33:1579–1585. [PubMed] [Google Scholar]
- 68.Minoshima S, Koeppe RA, Fessler JA, Mintun MA, Berger KL, Taylor SF, Kuhl DE. Integrated and automated data analysis method for neuronal activation studies using 15O-water PET. In: Uemura K, Jones T, Lassen NA, Kanno I, editors. Quantification of Brain Function. Tracer Kinetics and Image Analysis in Brain PET. Amsterdam: Excerpta Medica; 1993. pp. 409–415. [Google Scholar]
- 69.Minoshima S, Koeppe RA, Frey KA, Kuhl DE. Anatomic standardization: linear scaling and nonlinear warping of functional brain images. J Nucl Med. 1994;35:1528–1536. [PubMed] [Google Scholar]
- 70.Minoshima S, Koeppe RA, Mintun MA, Berger KL, Taylor SF, Frey KA, Kuhl DE. Automated detection of the intercommissural line for stereotactic localization of functional brain images. J Nucl Med. 1993;34:322–329. [PubMed] [Google Scholar]
- 71.Monakow Cv. Die Localisation im grosshirn und der abbau der funktion durch kortikale herde. Wiesbaden: J.F Bergmann; 1914. [Google Scholar]
- 72.Nagaro T, Amakawa K, Arai T, Ochi G. Ipsilateral referral of pain following cordotomy. Pain. 1993;55:275–276. doi: 10.1016/0304-3959(93)90157-K. [DOI] [PubMed] [Google Scholar]
- 73.Nagaro T, Amakawa K, Kimura S, Arai T. Reference of pain following percutaneous cervical cordotomy. Pain. 1993;53:205–211. doi: 10.1016/0304-3959(93)90082-Z. [DOI] [PubMed] [Google Scholar]
- 74.Nathan PW. Reference of sensation at the spinal level. J Neurol Neurosurg Psychiatry. 1956;19:88–100. doi: 10.1136/jnnp.19.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nelles G, Spiekramann G, Jueptner M, Leonhardt G, Muller S, Gerhard H, Diener HC. Evolution of functional reorganization in hemiplegic stroke: a serial positron emission tomographic activation study. Ann Neurol. 1999;46:901–909. doi: 10.1002/1531-8249(199912)46:6<901::aid-ana13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 76.Nguyen JP, Lefaucheur JP, Decq P, Uchiyama T, Carpentier A, Fontaine D, Brugières P, Pollin B, Fève A, Rostaing S, Cesaro P, Keravel Y. Chronic motor cortex stimulation in the treatment of central and neuropathic pain. Correlations between clinical, electrophysiological and anatomical data. Pain. 1999;82:245–251. doi: 10.1016/S0304-3959(99)00062-7. [DOI] [PubMed] [Google Scholar]
- 77.Ohara S, Crone NE, Weiss N, Treede RD, Lenz FA. Cutaneous Painful Laser Stimuli Evoke Responses Recorded Directly From Primary Somatosensory Cortex in Awake Humans. J Neurophysiol. 2004;91:2734–2746. doi: 10.1152/jn.00912.2003. [DOI] [PubMed] [Google Scholar]
- 78.Pappata S, Mazoyer B, Tran Dinh S, Cambon H, Levasseur M, Baron JC. Effects of capsular or thalamic stroke on metabolism in the cortex and cerebellum: a positron tomography study. Stroke. 1990;21:519–524. doi: 10.1161/01.str.21.4.519. [DOI] [PubMed] [Google Scholar]
- 79.Peyron R, Garcia-Larrea L, Deiber MP, Cinotti L, Convers P, Sindou M, Mauguière F, Laurent B. Electrical stimulation of precentral cortical area in the treatment of central pain: Electrophysiological and PET study. Pain. 1995;62:275–286. doi: 10.1016/0304-3959(94)00211-V. [DOI] [PubMed] [Google Scholar]
- 80.Peyron R, Garcia-Larrea L, Gregoire MC, Convers P, Lavenne F, Veyre L, Froment JC, Mauguière F, Michel D, Laurent B. Allodynia after lateral-medullary (Wallenberg) infarct. A PET study. Brain. 1998;121:345–356. doi: 10.1093/brain/121.2.345. [DOI] [PubMed] [Google Scholar]
- 81.Peyron R, Laurent B, Garcia-Larrea L. Functional imaging of brain responses to pain. A review and meta-analysis. Neurophysiol Clin. 2000;30:263–288. doi: 10.1016/s0987-7053(00)00227-6. [DOI] [PubMed] [Google Scholar]
- 82.Ploner M, Freund HJ, Schnitzler A. Pain affect without pain sensation in a patient with a postcentral lesion. Pain. 1999;81:211–214. doi: 10.1016/s0304-3959(99)00012-3. [DOI] [PubMed] [Google Scholar]
- 83.Ploner M, Gross J, Timmermann L, Schnitzler A. Cortical representation of first and second pain sensation in humans. Proc Natl Acad Sci U S A. 2002;99:12444–12448. doi: 10.1073/pnas.182272899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ploner M, Schmitz F, Freund HJ, Schnitzler A. Parallel activation of primary and secondary somatosensory cortices in human pain processing. J Neurophysiol. 1999;81:3100–3104. doi: 10.1152/jn.1999.81.6.3100. [DOI] [PubMed] [Google Scholar]
- 85.Porro CA. Functional Imaging and Pain: Behavior, Perception, and Modulation. The Neuroscientist. 2003;9:354–369. doi: 10.1177/1073858403253660. [DOI] [PubMed] [Google Scholar]
- 86.Price DD, McGrath PA, Rafii A, Buckingham B. The validation of visual analogue scales as ratio scale measures for chronic and experimental pain. Pain. 1983;17:45–56. doi: 10.1016/0304-3959(83)90126-4. [DOI] [PubMed] [Google Scholar]
- 87.Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(S2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ralston HJ, Ohara PT, Meng XW, Wells J, Ralston DD. Transneuronal changes of the inhibitory circuitry in the macaque somatosensory thalamus following lesions of the dorsal column nuclei. J Comp Neurol. 1996;371:325–335. doi: 10.1002/(SICI)1096-9861(19960722)371:2<325::AID-CNE11>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 89.Rasche D, Ruppolt M, Stippich C, Unterberg A, Tronnier VM. Motor cortex stimulation for long-term relief of chronic neuropathic pain: A 10 year experience. Pain. 2006;121:43–52. doi: 10.1016/j.pain.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 90.Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- 91.Ren Y, Zhang L, Lu Y, Yang H, Westlund KN. Central Lateral Thalamic Neurons Receive Noxious Visceral Mechanical and Chemical Input in Rats. J Neurophysiol. 2009;102:244–258. doi: 10.1152/jn.90985.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Russell WR. Transient disturbances following gunshot wounds of the head. Brain. 1945;68:79–97. doi: 10.1093/brain/68.2.79. [DOI] [PubMed] [Google Scholar]
- 93.Schmahmann JD, Leifer D. Parietal pseudothalamic pain syndrome: Clinical features and anatomic correlates. Arch Neurol. 1992;49:1032–1037. doi: 10.1001/archneur.1992.00530340048017. [DOI] [PubMed] [Google Scholar]
- 94.Schnitzler A, Ploner M. Neurophysiology and functional neuroanatomy of pain perception. J Clin Neurophysiol. 2000;17:592–603. doi: 10.1097/00004691-200011000-00005. [DOI] [PubMed] [Google Scholar]
- 95.Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- 96.Sikes RW, Vogt BA. Nociceptive neurons in area 24 of rabbit cingulate cortex. J Neurophysiol. 1992;68:1720–1732. doi: 10.1152/jn.1992.68.5.1720. [DOI] [PubMed] [Google Scholar]
- 97.Sikes RW, Vogt LJ, Vogt BA. Distribution and properties of visceral nociceptive neurons in rabbit cingulate cortex. Pain. 2008;135:160–174. doi: 10.1016/j.pain.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith SM. Preparing fMRI data for statistical analysis. In: Jezzard P, Matthews PM, Smith SM, editors. Functional MRI: An Introduction to Methods. Oxford: Oxford University Press; 2001. pp. 229–241. [Google Scholar]
- 99.Stevens RT, Hodge CJ, Jr, Apkarian AV. Medial, intralaminar, and lateral terminations of lumbar spinothalamic tract neurons: A fluorescent double-label study. Somatosens Res. 1989;6:285–308. doi: 10.3109/08990228909144678. [DOI] [PubMed] [Google Scholar]
- 100.Stroemer RP, Kent TA, Hulsebosch CE, Feeney DM. Enhanced Neocortical Neural Sprouting, Synaptogenesis, and Behavioral Recovery With D-Amphetamine Therapy After Neocortical Infarction in Rats. Stroke. 1998;29:2381–2395. doi: 10.1161/01.str.29.11.2381. [DOI] [PubMed] [Google Scholar]
- 101.Svendsen KB, Flemming BW. Pain in multiple sclerosis. In: Cervero F, Jensen TS, editors. Pain. Vol. 81. Edinburgh: Elsevier B.V.; 2006. pp. 731–745. [Google Scholar]
- 102.Svensson P, Minoshima S, Beydoun A, Morrow TJ, Casey KL. Cerebral processing of acute skin and muscle pain in humans. J Neurophysiol. 1997;78:450–460. doi: 10.1152/jn.1997.78.1.450. [DOI] [PubMed] [Google Scholar]
- 103.Szelies B, Herholz K, Pawlik G, Karbe H, Hebold I, Heiss W-D. Widespread functional effects of discrete thalamic infarction. Arch Neurol. 1991;48:178–182. doi: 10.1001/archneur.1991.00530140072019. [DOI] [PubMed] [Google Scholar]
- 104.Talairach J, Tournoux A. A Coplanar Stereotaxic Atlas of the Human Brain. New York: Thieme Medical Publishers, Inc.; 1988. [Google Scholar]
- 105.Tasker RR, de Carvalho G, Dostrovsky JO. The History of Central Pain Syndromes,with Observations Concerning Pathophysiology and Treatment. In: Casey KL, editor. Pain and Central Nervous System Disease. New York: Raven Press; 1991. pp. 31–58. [Google Scholar]
- 106.Tsubokawa T, Katayama Y, Yamamoto T, Hirayama T, Koyama S. Treatment of thalamic pain by chronic motor cortex stimulation. Pacing & Clinical Electrophysiology. 1991;14:131–134. doi: 10.1111/j.1540-8159.1991.tb04058.x. [DOI] [PubMed] [Google Scholar]
- 107.van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vestergaard K, Nielsen J, Andersen G, Ingeman-Nielsen M, Arendt-Nielsen L, Jensen TS. Sensory abnormalities in consecutive, unselected patients with central post-stroke pain. Pain. 1995;61:177–186. doi: 10.1016/0304-3959(94)00140-A. [DOI] [PubMed] [Google Scholar]
- 109.Vogt BA. Pain and emotion inteactions in subregions of the cingulate gyrus. Nat Rev Neurosci. 2005;6:533–544. doi: 10.1038/nrn1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vogt BA, Pandya DN, Rosene DL. Cingulate cortex of the rhesus monkey: I. Cytoarchitecture and thalamic afferents. J Comp Neurol. 1987;262:256–270. doi: 10.1002/cne.902620207. [DOI] [PubMed] [Google Scholar]
- 111.Vogt BA, Rosene DL, Pandya DN. Thalamic and cortical afferents differentiate anterior from posterior cingulate cortex in the monkey. Science. 1979;204:205–207. doi: 10.1126/science.107587. [DOI] [PubMed] [Google Scholar]
- 112.Vogt BA, Sikes RW, Vogt LJ. Anterior Cingulate Cortex and the Medial Pain System. In: Vogt BA, Gabriel M, editors. Neurobiology of Cingulate Cortex and Limbic Thalamus: A Comprehensive Handbook. Boston: Birkhauser; 1993. [Google Scholar]
- 113.Wassner G, Deuschl G. Pain in Parkinson’s disease. In: Cervero F, Jensen TS, editors. Pain. Vol. 81. Edinburgh: Elsevier B.V; 2006. pp. 747–760. [Google Scholar]
- 114.Weiller C, Chollet F, Friston KJ, Wise RJ, Frackowiak RS. Functional reorganization of the brain in recovery from striatocapsular infarction in man. Ann Neurol. 1992;31:463–472. doi: 10.1002/ana.410310502. [DOI] [PubMed] [Google Scholar]
- 115.Weiller C, Ramsay SC, Wise RJ, Friston KJ, Frackowiak RS. Individual patterns of functional reorganization in the human cerebral cortex after capsular infarction. Ann Neurol. 1993;33:181–189. doi: 10.1002/ana.410330208. [DOI] [PubMed] [Google Scholar]
- 116.Whitsel BL, Favorov OV, Li Y, Quibrera M, Tommerdahl M. Area 3a Neuron Response to Skin Nociceptor Afferent Drive. Cereb Cortex. 2009;19:349–366. doi: 10.1093/cercor/bhn086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wilkinson HA, Davidson KM, Davidson RI. Bilateral anterior cingulotomy for chronic noncancer pain. Neurosurg. 1999;45:1129–1134. doi: 10.1097/00006123-199911000-00023. [DOI] [PubMed] [Google Scholar]
- 118.Willoch F, Schindler F, Wester HJ, Empl M, Straube A, Schwaiger M, Conrad B, Tolle TR. Central poststroke pain and reduced opioid receptor binding within pain processing circuitries: a [11C]diprenorphine PET study. Pain. 2004;108:213–220. doi: 10.1016/j.pain.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 119.Worsley KJ. Statistical analysis of activation images. In: Jezzard P, Matthews PM, Smith SM, editors. Functional MRI: An Introduction to Methods. Oxford: Oxford University Press; 2001. pp. 251–270. [Google Scholar]
- 120.Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]