

Abstract
A cooperative catalysis approach for the enantioselective formal [3+2] addition of α,β-unsaturated aldehydes to isatins has been developed. The N-heterocyclic carbene (NHC)-catalyzed homoenolate annulations of β-aryl enals require the addition of lithium chloride for high levels of enantioselectivity. This NHC-catalyzed annulation provides efficient access to the 3-hydroxy indole skeleton and has been applied to the first eantioselective total synthesis of maremycin B.
Keywords: catalysis, asymmetric synthesis, N-heterocyclic carbene, homoenolate, Lewis acid, total synthesis
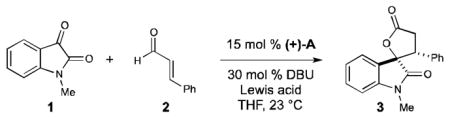
The development of efficient strategies for the stereoselective construction of privileged heterocyclic systems is an ongoing objective in chemical synthesis. Over the last decade, the development of powerful NHC-catalyzed homoenolate equivalents has allowed access to a wide range of hetero- and carbocyclic structural motifs.[1] Despite significant advancements in this NHC-homoenolate field with additions to activated C=X systems by Glorius, Bode, Nair, our group, and others,[2] the combination of these interesting and unconventional nucleophilic species with less active electrophiles, such as ketones, remains challenging.[3] In addition, rendering these carbonyl addition processes enantioselective remains an important goal since these annulation reactions provide an efficient method for accessing bioactive γ-butyrolactones. We have been investigating cooperative carbene catalysis strategies to explore new opportunities with NHCs as Lewis base catalysts.[4] In select cases, the use of a Lewis acid in conjunction with an NHC has: a) enhanced yield and enantioselectivity, b) reversed diastereoselectivity, and c) enabled new reactions between enals and carbonyl reaction partners.[5,6] While strong Lewis acids presumably inhibit carbene catalysis through NHC-Lewis acid complexation, milder Lewis acids such as Mg(OtBu)2 and Ti(OiPr)4 can be essential additives for new NHC-homoenolate reactions.[7] However, the use of alkali metal salts in NHC reactions is underexplored and represents a new combination given that these salts play critical roles in a variety of carbon–carbon bond forming reactions.[8] The impact of alkali metal salt effects on carbene catalyzed reactions has been observed by us,[9a] Lupton,[9b] and You,[9c] and we sought to explore for the first time the potential of these metal salts in the context of NHC-homoenolate additions to ketones. We report herein the highly enantioselective, NHC-catalyzed addition of αβ-unsaturated aldehydes to isatins activated by lithium cations (Figure 1). In this formal [3+2] annulation, the alkali salt significantly enhances the level of enantioselectivity in the resultant spirooxindole products.
Figure 1.

In 2006, Nair reported an interesting NHC-catalyzed annulation of 1,2-diketones with enals to generate racemic spirocyclic lactone products as a 1:1 mixture of diastereomers.[3b, 10] Recently, You reported a related enantioselective annulation which relies on a hydrogen-bonding NHC catalyst.[11] Our group has been interested in the preparation of spirooxindoles[12] due to the prevalence of this structural motif in a number of architecturally complex and biologically relevant natural products (Figure 1).[13] Given our strong interest in the synthesis of these compounds and carbene catalysis, we became interested in developing an NHC-catalyzed method for accessing these privileged structures in a diastereo- and enantioselective manner.
We began our studies by combining N-methyl isatin (1) with cinnamaldehyde (2) in the presence of triazolium precatalyst A (15 mol %) and DBU (30 mol %). Under these conditions, lactone 3 was obtained as a 1:1 mixture of diastereomers in 35% ee (entry 1). With this baseline established for comparison, we turned our attention toward the use of Lewis acid additives to improve both the diastereo- and enantioselectivity. Unfortunately, in the presence of 50 mol % Mg(OtBu)2 or Ti(OiPr)4, lactone 3 was obtained with only a modest increase in diastereoselectivity or enantioselectivity, respectively (entry 2–3). No significant improvement in stereoselectivity was observed upon extensive variation of the reaction components, including stoichiometry, temperature, solvent, and nature of the Lewis acid. We next turned our attention toward the use of lithium chloride as a potential mild Lewis acid additive.9 Gratifyingly, the addition of 50 mol % lithium chloride provided lactone 3 as a ~2:1 mixture of diastereomers, but more importantly, with an increase in enantioselectivity to 70% ee (entry 4). A significant difference in both diastereo- and enantioselectivity was observed by increasing the amount of lithium chloride (entry 4–6). Upon determination that two equivalents was optimal, lactone 3 was obtained in 2.5:1 dr and 90% ee (entry 6).
With these initial results, we next investigated this ion effect on enantioselectivity.[14] The sequestration of the lithium cation from the reaction mixture by addition of an excess of 12-crown-4 delivered lactone 3 with diminished diastereo- and enantioselectivity, similar to when the reaction was run without Lewis acid (entry 7). Other alkali metal chloride salts, such as NaCl and KCl, afforded lactone 3 with lower enantioselectivity than with LiCl (entry 8–9), providing further evidence that this effect was particular to lithium. However, lithium tetrafluoroborate and lithium triflate did not produce lactone 3 with high levels of stereoinduction as were achieved with lithium chloride (entry 10–11).[15] The data currently supports that both ions are necessary for high levels of enantioselectivity and there is not an observable increase in overall rate vs. reaction without Li+ (e.g., Table 1, entry 1 vs. entry 6). Further investigation of this phenomenon is underway. Having established that lithium chloride was the optimal Lewis acid additive, various chiral NHCs were examined to enhance the diastereo- and enantioselectivity of the reaction. Employing chiral azolium catalysts B-E resulted in a wide range of diastereo- and enantioselectivities, with the 2,6-diethylphenyl substituted triazolium precatalyst B being the catalyst of choice (entry 12–15). We discovered that with only 5 mol % catalyst B similar levels of conversion and selectivity were obtained (entry 16).
Table 1.
Optimization of the reaction conditions.
| ||||
|---|---|---|---|---|
| entry | variation of the standard conditions | conv[a] | dr[a] | ee[b] |
| 1 | no Lewis acid | 99 | 1.1:1 | 34 |
| 2 | Mg(OtBu)2 (50 mol %) | 99 | 1.5:1 | 35 |
| 3 | Ti(OiPr)4 (50 mol %) | 99 | 1:1 | 52 |
| 4 | LiCl (50 mol %) | 99 | 1.8:1 | 70 |
| 5 | LiCl (1 equiv) | 99 | 2:1 | 77 |
| 6 | LiCl (2 equiv) | 99 | 2.5:1 | 90 |
| 7 | LiCl (2 equiv), 12-crown-4 (4 equiv) | 99 | 1.2:1 | 35 |
| 8 | NaCl (2 equiv) | 99 | 1.2:1 | 53 |
| 9 | KCl (2 equiv) | 99 | 1:1 | 47 |
| 10 | LiBF4 (2 equiv) | 99 | 1:1 | 62 |
| 11 | LiOTf (2 equiv) | 99 | 3.7:1 | 39 |
| 12 | (+)-B, LiCl (2 equiv) | 99 | 2.8:1 | 92 |
| 13 | (−)-C, LiCl (2 equiv) | 66 | 1.1:1 | 38 |
| 14 | (−)-D, LiCl (2 equiv) | 99 | 1.7:1 | 32 |
| 15 | (−)-E, LiCl (2 equiv) | 99 | 1.4:1 | 50 |
| 16 | (+)-B (5 mol %), DBU (10 mol %) LiCl (2 equiv) | 99 | 2.4:1 | 96 |
|
| ||||
|
| ||||
Determined by 1H NMR spectroscopy (500 MHz) of the unpurified reaction.
Enantiomeric excess determined by HPLC.
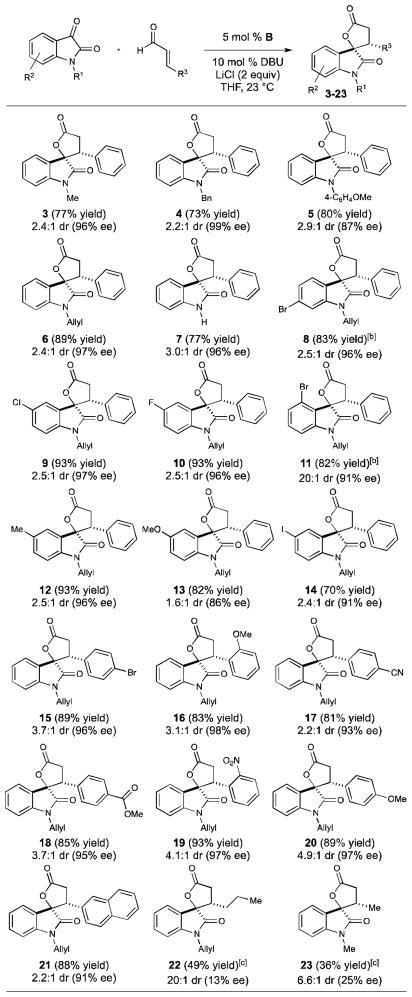
With the optimized reaction conditions, we surveyed isatins with varying nitrogen protecting groups and substitution around the aromatic ring (Table 2, 3-14). Electron withdrawing and donating substituents on the aromatic ring were well accommodated, providing the lactone products in 1.6–3.0:1 dr and 77–99% ee. Notably, the free N–H isatin provides the desired product 7 in high ee and yield. A slight decrease in enantioselectivity was observed in the case of the isatin bearing a p-methoxyphenyl protecting group (5) and with 5-methoxy isatin (13) (87% and 86% ee, respectively). Interestingly, the 6-bromo isatin delivered lactone 11 with exquisite diastereoselectivity (20:1) and high enantioselectivity (90%). Structural modification of the cinnamaldehyde component was also explored (Table 2, 15-21). Both electron-withdrawing and donating groups were tolerated, furnishing the desired lactones in good yields (81–93%) and high enantioselectivity (91–98%). With enals bearing a β-alkyl substituent, both the yields and level of enantioinduction decreased significantly, as is typical with many NHC-homoenolate reactions.[2] With 2-hexenal and crotonaldehyde, lactones 22 and 23 could be obtained with good diastereoselectivity, but in modest yield (49% and 39%, respectively), and low enantioselectivity (13% and 25% ee, respectively). Since a number of 3-hydroxy indole natural products contain alkyl substituents, for example the maremycins (Figure 1), it was necessary to re-examine conditions for the β-alkyl enals.
Table 2.
Isatin and enal reaction scope.a
|
All reactions performed on 0.3 mmol scale. Isolated yield after chromatography. Diastereomeric ratios determined by 1H NMR spectroscopy (500 MHz). Enantiomeric excess determined by HPLC.
Structure confirmed by X-ray crystallography, see Supporting Information.
20 mol % B and 40 mol % DBU.
Table 3.
Optimization of conditions with crotonaldeyde.
| ||||||
|---|---|---|---|---|---|---|
| entry | NHC | additive | conv[a] (yield)[b] | 23/24[a] | dr[a] | ee[a] |
| 1 | (+)-A | LiCl (2 equiv) | 99 (36) | 4:1 | 6.6:1 | 25 |
| 2 | (+)-A | none | 99 (76) | >20:1 | 8:1 | <10 |
| 3 | (+)-B | none | 99 | >20:1 | 6.2:1 | <10 |
| 4 | (−)-C | none | 99 | >20:1 | 7.4:1 | 19 |
| 5 | (−)-D | none | 99 | >20:1 | 6.8:1 | 30 |
| 6[d] | (+)-E | none | 99 (76) | >20:1 | 5:1 | −78 (−99)[e] |
Determined by 1H NMR spectroscopy (500 MHz).
Isolated yield after chromatography.
Enantiomeric excess determined by HPLC.
5 mol % catalyst (+)-E and 10 mol % DBU.
Enantiomeric excess after recrystallization in hexanes/EtOAc (44% overall yield of 99% ee lactone 23).
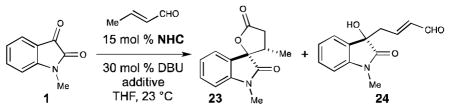
With the previously optimized conditions, lactone 23 was obtained in 39% yield and 25% ee (Table 3, entry 1). The low yield could be partially attributed to the unexpected formation of enal 24 through a γ-alkylation pathway. An extensive screen of solvents and Lewis base additives yielded no improvement in efficiency, enantioselectivity, or suppression of enal 24 (results not shown). Interestingly, when the annulation reaction was carried out with catalyst A in the absence of lithium chloride, lactone 23 was obtained in 76% yield, with no observable amounts of 24, albeit with diminished levels of enantioselectivity. The increase in yield prompted us to re-examine azolium catalysts B-E with crotonaldehyde in the absence of lithium chloride. While most of the catalysts provided unsatisfactory results, with catalyst E lactone 23 was obtained in 76% yield, 5:1 dr, and 78% ee (entry 6). The target lactone 23 could then be accessed in 99% ee after recrystallization. The modified reaction conditions produced a similar outcome for the reaction with 2-hexenal, as now lactone 22 could be isolated in 91% yield, 7:1 dr, and 79% ee.
For β-aryl-substituted enals, we propose that the high levels of enantioselectivity from lithium cations generating an organized transition state through coordination of the enol oxygen atom of the NHC-bound homoenolate and the 1,2-dicarbonyl of the isatin (Figure 2). The indene subunit on the NHC controls the facial bias of the homoenolate equivalent and the major diastereomer results from avoiding a destabilizing interaction between the β-aryl group and isatin ring. The trends in enantioselectivity observed upon variation of the alkali metal and counterion are presumably due to the oxophilicity and coordination states of lithium compared to sodium or potassium and fine-tuning of the Lewis acidity upon variation of the lithium counterion. For β-alkyl-substituted enals, initial modeling suggests that the NHC-homoenolate may adopt a conformation leading to opposite facial accessibility since NHC E has the bis-phenyl directing groups in a stacked orientation (vs. flat for NHC A).
Figure 2.

Model for stereoinduction with β-aryl enals
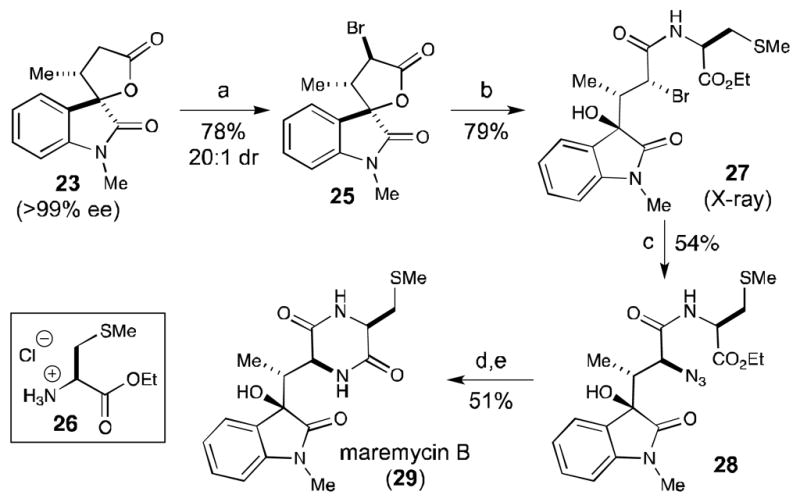
With the ability to engage a wide range of αβ-unsaturated aldehydes in this enantioselective annulation reaction, we sought to apply this methodology toward the synthesis maremycin B (29) given its interesting 3-hydroxy indole core architecture and anticancer activity.[16] We envisioned accessing maremycin B through functionalization of previously prepared β-alkyl substituted lactone 23. To begin, the treatment of lactone 23 (99% ee after recrystallization) with LiHMDS followed by the addition of bromine generated α-bromo lactone 25 in 78% yield with 20:1 diastereoselectivity (Scheme 1). Attempts to displace the bromide with a variety of nitrogen nucleophiles were unsuccessful at this stage. Instead, the introduction of the cysteine side chain was pursued prior to displacement of the bromide. Toward this end, α-bromo lactone 25 was converted to α-bromo amide 27 in the presence of S-methyl cysteine ethyl ester hydrochloride (26) and trimethylaluminum in 79% yield, without compromising the sensitive α-bromo carbonyl functionality.[ 17 ] The subsequent displacement of the halogen was achieved by modifying the Deshong-Smith protocol with TBAT and trimethylsilylazide.[18] A Staudinger reduction of 28 with trimethylphosphine to the amine,[19] followed by treatment with imidazole in methanol furnished maremycin B (29) in 51% yield over the two steps.[20] The spectral data (1H, 13C NMR) for maremycin B matched the literature with confirmation of the absolute configuration by optical rotation ([α]D 20 = + 9.0 (c 0.11, MeOH) [Lit [α]D 20 = + 2.9 (c 0.21, MeOH)]).[15] This cooperative carbene catalysis approach to the maremycins highlights the efficiency of this formal [3+2] reaction compared to recent synthetic studies on these compounds: this is the first enantioselective total synthesis of maremycin B in five steps from lactone 23 (only six steps from commercial materials) without protecting groups and in 17% overall yield.
Scheme 1.
Total synthesis of maremycin B (29).[a]
[a] See Supporting Information for details. Reagents and conditions: (a) LiHMDS, −78 °C to 0 °C, THF; then Br2, −78 °C, 78% (20:1 dr). (b) AlMe3, 26, THF, 79%. (c) TMSN3, TBAT, THF, 54% (d) PMe3, THF, then H2O, 60 °C (e) Imidazole, MeOH, 60 °C, 51% (2 steps). TBAT = tetrabutylammonium difluorotriphenylsilicate
In conclusion, an enantioselective NHC/Lewis acid-catalyzed homoenolate annulation of enals with isatins has been developed. This approach is one of the few general methods of enantioselective addition of NHC-generated homoenolate equivalents to ketones and opens up the possibility to include new, less reactive electrophilic partners with NHC-homoenolates using cooperative catalysis. The addition of lithium chloride as a Lewis acid with β-aryl substituted enals generates lactone products with high levels of enantioselectivity (>90%). Interestingly, with enals bearing β-alkyl substituents, lithium chloride was detrimental due to the promotion of a competing pathway. In the absence of lithium chloride, a different triazolium precatalyst was employed to generate the alkyl-substituted lactones in good yield and enantioselectivity. The utility of this highly selective formal [3+2] annulation method was highlighted by a concise total synthesis of maremycin B. Our investigations to define a clear model for stereoinduction in this reaction and integrating NHC catalysis with other modes of activation (including Lewis acid and transition metal catalysis) are continuing and will be reported in due course.
Acknowledgments
Financial support has been generously provided by the NIH (NIGMS RO1-GM073072). T.B.H.S. was supported by a 2011 Northwestern Summer Undergraduate Research Grant. D.T.C thanks the ACS Division of Organic Chemistry for a 2011–12 Graduate Fellowship (sponsored by Organic Synthesis/Organic Reactions). We thank Michael Katz (NU) and John M. Roberts (NU) for assistance with X-ray crystallography.
Footnotes
Supporting information for this article is available on the Web under http://www.angewandte.org or from the author.
Contributor Information
Dr. Julien Dugal-Tessier, Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute Northwestern University 2145 Sheridan Road, Evanston, IL 60208 (USA)
Dr. Elizabeth A. O’Bryan, Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute Northwestern University 2145 Sheridan Road, Evanston, IL 60208 (USA)
Thomas B. H. Schroeder, Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute Northwestern University 2145 Sheridan Road, Evanston, IL 60208 (USA)
Daniel T. Cohen, Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute Northwestern University 2145 Sheridan Road, Evanston, IL 60208 (USA)
Prof. Karl A. Scheidt, Email: scheidt@northwestern.edu, Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute Northwestern University 2145 Sheridan Road, Evanston, IL 60208 (USA),
References
- 1.a) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; b) Nair V, Vellalath S, Babu BP. Chem Soc Rev. 2008;37:2691–2698. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]; c) Phillips EM, Chan A, Scheidt AK. Aldrichim Acta. 2009;42:55–65. [PMC free article] [PubMed] [Google Scholar]; d) Biju AT, Kuhl N, Glorius F. Acc Chem Res. 2011;44:1182–1195. doi: 10.1021/ar2000716. [DOI] [PubMed] [Google Scholar]; e) Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem Soc Rev. 2011;40:5336–5346. doi: 10.1039/c1cs15139h. [DOI] [PubMed] [Google Scholar]
- 2.For selected NHC-homoenolate reactions, see: Burstein C, Glorius F. Angew Chem Int Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572.Sohn SS, Rosen EL, Bode JW. J Am Chem Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b.Chan A, Scheidt KA. Org Lett. 2005;7:905–908. doi: 10.1021/ol050100f.He M, Bode JW. Org Lett. 2005;7:3131–3134. doi: 10.1021/ol051234w.Nair V, Vellalath S, Poonoth M, Suresh E. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677.Zeitler K. Org Lett. 2006;8:637–640. doi: 10.1021/ol052826h.Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167.Phillips EM, Reynolds TE, Scheidt KA. J Am Chem Soc. 2008;130:2416–2417. doi: 10.1021/ja710521m.Rommel M, Fukuzumi T, Bode JW. J Am Chem Soc. 2008;130:17266–17267. doi: 10.1021/ja807937m.Nair V, Babu BP, Vellalath S, Varghese V, Raveendran AE, Suresh E. Org Lett. 2009;11:2507–2510. doi: 10.1021/ol900571x.Nair V, Vellalath S, Babu BP, Varghese V, Paul RR, Suresh E. Org Biomol Chem. 2010;8:4861–4866. doi: 10.1039/c0ob00180e.Chen ZY, Yu XX, Wu J. Chem Commun. 2010;46:6356–6358. doi: 10.1039/c0cc01207f.
- 3.For the limited number of 1,2-additions of NHC-homoenolates to ketones, see: a) ref 2a; Burstein C, Tschan S, Xie X, Glorius F. Synthesis. 2006:2418–2439.Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. Org Lett. 2006;8:507–509. doi: 10.1021/ol052926n.Nair V, Vellalath S, Poonoth M, Suresh E, Viji S. Synthesis. 2007:3195–3200.Hirano K, Piel I, Glorius F. Adv Synth Catal. 2008;350:984–988.Li Y, Zhao ZA, He H, You S. Adv Synth Catal. 2008;350:1885–1890.Matsuoka Y, Ishida Y, Sasaki D, Saigo K. Chem Eur J. 2008;14:9215–9122. doi: 10.1002/chem.200800942.Struble JR, Bode JW. Tetrahedron. 2009;65:4957–4967. doi: 10.1016/j.tet.2009.03.103.
- 4.For recent reviews of carbene cooperative catalysis, see: Patil NT. Angew Chem Int Ed. 2011;50:1759–1761. doi: 10.1002/anie.201006866.Hirano K, Piel I, Glorius F. Chem Lett. 2011;40:786–791.Cohen DT, Scheidt KA. Chem Sci. 2012;3:53–57. doi: 10.1039/C1SC00621E.
- 5.a) Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat Chem. 2010;2:766–771. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cardinal-David B, Raup DEA, Scheidt KA. J Am Chem Soc. 2010;132:5345–5347. doi: 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cohen DT, Cardinal-David B, Scheidt KA. Angew Chem Int Ed. 2011;50:1678–1682. doi: 10.1002/anie.201005908. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cohen DT, Cardinal-David B, Roberts JM, Sarjeant AA, Scheidt KA. Org Lett. 2011;13:1068–1071. doi: 10.1021/ol103112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For a recent report combining NHC-homoenolates with Brønsted acid activation, see: Zhao X, DiRocco DA, Rovis T. J Am Chem Soc. 2011;133:12466–12469. doi: 10.1021/ja205714g.
- 7.For a computational study on NHC-Lewis acid processes, see: Domingo LR, Zaragozá RJ, Arnó M. Org Biomol Chem. 2011;9:6616–6622. doi: 10.1039/c1ob05609c.For a comparative study of Lewis acid strength, see: Kobayashi S, Busujima T, Nagayama S. Chem Eur J. 2000;6:3491–3194. doi: 10.1002/1521-3765(20001002)6:19<3491::aid-chem3491>3.3.co;2-g.
- 8.a) Tomooka K, Ito M. Main Group Metals in Organic Synthesis. Wiley-VCH Verlag GmbH & Co KGaA; 2004. pp. 1–34. [Google Scholar]; b) Hevia E, Mulvey RE. Angew Chem Int Ed. 2011;50:6448–6450. doi: 10.1002/anie.201102054. [DOI] [PubMed] [Google Scholar]
- 9.For recent examples, see: Phillips EM, Riedrich M, Scheidt KA. J Am Chem Soc. 2010;132:13179–13181. doi: 10.1021/ja1061196.Ryan SJ, Candish L, Lupton DW. J Am Chem Soc. 2011;133:4694–4697. doi: 10.1021/ja111067j.Rong ZQ, Jia MQ, You SL. Org Lett. 2011;13:4080–4083. doi: 10.1021/ol201595f.
- 10.For other NHC-catalyzed ketene/enolate reactions with isatins (not NHC-homoenolates), see: He L, Zhang YR, Huang XL, Ye S. Synthesis. 2008:2825–2829.Shen LT, Shao PL, Ye S. Adv Synth Catal. 2011;353:1943–1948.
- 11.Sun LH, Shen LT, Ye S. Chem Commun. 2011;47:10136–10138. doi: 10.1039/c1cc13860j.For this process, β-alkyl enal are poor substrates (low yields).
- 12.a) Galliford CV, Martenson JS, Stern C, Scheidt KA. Chem Commun. 2006:631–633. doi: 10.1039/b609155e. [DOI] [PubMed] [Google Scholar]; b) Wang J, Crane EA, Scheidt KA. Org Lett. 2011;13:3086–3089. doi: 10.1021/ol200987c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748–8758. doi: 10.1002/anie.200701342.Trost BM, Brennan MK. Synthesis. 2009:3003–3025.Badillo JJ, Hanhan NV, Franz AK. Curr Opin Drug Disc Dev. 2010;13:758–776.For a recent enantioselective additions to isatins, see: Itoh J, Han SB, Krische MJ. Angew Chem Int Ed. 2009;48:6313–6316. doi: 10.1002/anie.200902328.Hanhan NV, Sakin AH, Cheng TW, Fettinger JC, Franz AK. Angew Chem Int Ed. 2010;49:744–747. doi: 10.1002/anie.200904393.Hanhan NV, Ball-Jones NR, Tran NT, Franz AK. Angew Chem Int Ed. 2012;51:989–992. doi: 10.1002/anie.201105739.
- 14.For an early observation of counterion effects from the azolium precatalyst on ee in the NHC-catalyzed Stetter reaction, see: Kerr M, de Alaniz JR, Rovis T. J Am Chem Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v.
- 15.The addition of LiPF6, LiSbF6, MgCl2 resulted in no observed product formation. The addition of n-Bu4Cl and LiI delivered the desired product 75% and 57% ee, respectively. Efforts are underway to explore the impact of azolium counter ion and the origin of stereochemistry in this chiral NHC-lithium(I) process.
- 16.Balk-Bindseil W, Helmke E, Weyland H, Laatsch H. Liebigs Ann. 1995:1291–1294.Tang Y-Q, Sattler I, Thiericke R, Grabley S, Feng X-Z. Eur J Org Chem. 2001:261–267.For total syntheses of maremycin A, see: Ueda T, Inada M, Okamoto I, Morita N, Tamura O. Org Lett. 2008;10:2043–2046. doi: 10.1021/ol800515w.Bergonzini G, Melchiorre P. Angew Chem Int Ed. 2012;51:971–974. doi: 10.1002/anie.201107443.A synthesis of maremycin B in 15 steps from L-isoleucine, was recently reported: Liu Y, Zhang L, Jia Y. Tetrahedron Lett. 2012;53:684–687.
- 17.Harwood LM, Mountford SJ, Yan R. J Pept Sci. 2009;15:1–4. doi: 10.1002/psc.1080. [DOI] [PubMed] [Google Scholar]
- 18.Soli ED, Manoso AS, Patterson MC, DeShong P, Favor DA, Hirschmann R, Smith AB. J Org Chem. 1999;64:3171–3177. doi: 10.1021/jo982302d. [DOI] [PubMed] [Google Scholar]
- 19.Vaultier M, Knouzi N, Carrié R. Tetrahedron Lett. 1983;24:763–764. [Google Scholar]
- 20.Dolence EK, Miller MJ. J Org Chem. 1991;56:492–499. [Google Scholar]