

Abstract
Background
Upon stimulation with different cytokines, macrophages can undergo classical or alternative activation to become M1 or M2 macrophages. Alternatively activated (or M2) macrophages are defined by their expression of specific gene products and play an important role in containing inflammation, removing apoptotic cells and repairing tissue damage. Whereas it is well-established that IL-4 can drive alternative activation, if lack of TGFβ signaling at physiological levels affects M2 polarization has not been addressed.
Results
Vav1-Cre x TβRIIfx/fx mice, lacking TβRII function in hematopoietic cells, exhibited uncontrolled pulmonary inflammation and developed a lethal autoimmune syndrome at young age. This was accompanied by significantly increased numbers of splenic neutrophils and T cells as well as elevated hepatic macrophage infiltration and bone marrow monocyte counts. TβRII-/- CD4+ and CD8+ T-cells in the lymph nodes and spleen expressed increased cell surface CD44, and CD69 was also higher on CD4+ lymph node T-cells. Loss of TβRII in bone marrow-derived macrophages (BMDMs) did not affect the ability of these cells to perform efferocytosis. However, these cells were defective in basal and IL-4-induced arg1 mRNA and Arginase-1 protein production. Moreover, the transcription of genes that are typically upregulated in M2-polarized macrophages, such as ym1, mcr2 and mgl2, was also decreased in peritoneal macrophages and IL-4-stimulated TβRII-/- BMDMs. We found that cell surface and mRNA expression of Galectin-3, which also regulates M2 macrophage polarization, was lower in TβRII-/- BMDMs. Very interestingly, the impaired ability of these null mutant BMDMs to differentiate into IL-4 polarized macrophages was Stat6- and Smad3-independent, but correlated with reduced levels of phospho-Akt and β-catenin.
Conclusions
Our results establish a novel biological role for TGFβ signaling in controlling expression of genes characteristic for alternatively activated macrophages. We speculate that lack of TβRII signaling reduces the anti-inflammatory M2 phenotype of macrophages because of reduced expression of these products. This would cause defects in the ability of the M2 macrophages to negatively regulate other immune cells such as T-cells in the lung, possibly explaining the systemic inflammation observed in Vav1-Cre x TβRIIfx/fx mice.
Keywords: TGFβ, Macrophage polarization, Lung, Alveolar macrophage, M1, M2, Hematopoietic, Inflammation, TGFBR2, LGALS3
Background
Transforming growth factor beta (TGFβ) is a multi-functional cytokine that regulates cell proliferation, differentiation, migration and survival. It plays a critical role in development, wound healing and immune responses through its regulatory effects on many cell types including epithelial and hematopoietic cells [1]. Three isoforms (TGFβ1, β2 and β3), which are encoded by different genes and share high homology, have been identified in mammals. TGF-β1 is the predominant isoform expressed in immune cells, but all three isoforms have similar properties in vitro. However, in vivo, they have both redundant and distinctive functions because of spatial and temporal expression during development [2-4]. TGFβ signals are transmitted via a cell surface receptor complex consisting of the TGFβ type I receptor (TβRI) and TGFβ type II receptor (TβRII). To initiate signal transduction, TGFβ binds to TβRII, which in turn recruits TβRI, leading to the formation of a tetrameric receptor complex. The constitutively active serine/threonine kinase of TβRII phosphorylates TβRI, which activates Smad2 and Smad3 via phosphorylation. Activated Smad2/3 binds Smad4. Subsequently, the Smad2/3/4 complex translocates to nucleus to regulate gene expression [5]. Besides the Smad-dependent pathways, TGFβ also activates the Erk [6], PI3K [7], p38 and JNK [8] pathways, each of which results in a unique pattern of gene expression and thus physiological function.
The importance of TGFβ signaling in the immune system is highlighted by the finding that mice lacking TGFβ1 develop a severe lethal wasting syndrome within 3 weeks of birth, associated with a mixed inflammatory cell infiltration and lesions in different organs including the heart and lungs [9,10]. Since TβRII is the primary receptor for TGFβ, Leveen et al. [11] generated a conditional ablation model for TGFβ signaling using TβRIIfx/fx x Mx1-Cre mice. Upon deletion of the TβRII in the bone marrow of adult mice, a lethal inflammatory disorder that is similar to the phenotype of the TGFβ1-null mutants was observed. Such lethal disorder was transferable through bone marrow transplantation, indicating that a deficiency of TGFβ signaling in cells of bone marrow origin is sufficient to cause a lethal inflammatory disease [11]. Further studies on TGFβ1-null mutants or mice with bone marrow-specific deletion of TβRII showed, that TGFβ signaling is critical for T-cell immunity [12-14], whereas possible functional defects of macrophages lacking TGFβ signaling were largely overlooked.
Macrophages are important immune cells that have diverse biological functions, which are, to a large extent, determined by their activation states. Exposure to LPS and IFNγ induces macrophages to undergo classical activation and differentiate into M1 macrophages. M1 macrophages are characterized by the production of pro-inflammatory cytokines (TNFα and IL-6) and the induction of iNOS (NOS2), which is essential for generating reactive oxygen species such as NO [15,16]. Because of their pro-inflammatory and cytotoxic activities, M1 macrophages play a critical role in elimination of pathogens and initiation of inflammation. In contrast, when macrophages are exposed to IL-4 (or IL-13), they undergo alternative activation and polarize into M2 macrophages, which are anti-inflammatory. M2 macrophages produce anti-inflammatory cytokines (IL-10) and have increased expression of Arginase-1, which competes with iNOS for a common limiting substrate L-arginine [17]. M2 macrophages are primarily involved in phagocytosis of apoptotic cells, resolution of inflammation, tissue repair, and wound healing [18-21].
Although TGFβ plays a critical role in T-cell immunity, surprisingly, few studies have evaluated the effect of TGFβ signaling on macrophages. To begin to answer this question, we generated mice lacking TβRII and thus TGFβ signaling in hematopoietic cells using TβRIIfx/fx and Vav1-Cre mice. Vav1-Cre has been shown to direct Cre-mediated recombination in cells of the hematopoietic lineage [22]. Interestingly, our studies reveal that macrophages lacking TβRII have defects in expression of a set of genes that form the hallmark of the M2 polarizing program, suggesting that TGFβ signaling is needed for the alternative activation of macrophages.
Results
Mice lacking TβRII in hematopoietic cells develop a lethal inflammatory and autoimmune syndrome
Vav1 is a hematopoietic-specific activator for Rac GTPases, and the Vav1 promoter has been widely used to direct expression of genes selectively in the hematopoietic lineage [22]. Thus, in Vav1-Cre x TβRIIfx/fx(TβRII-/-) mice, the Vav1-Cre transgene only ablates TβRII in hematopoietic cells. We found that mice lacking TβRII function in such cell types were ~50% lighter than wild type (WT) littermates (Figure 1A) and usually died at the age of 24-28 days. These TβRII-/- mice showed classical hallmarks of acute lung inflammation. Total cell numbers in the BALF of TβRII-/- mice were significantly higher than those of WT mice (Figure 1B). Whereas BALF cell populations of age-matched WT mice consisted, as expected, almost entirely of alveolar macrophages, those of TβRII-/- mice instead contained many neutrophils, lymphocytes and monocytes (Figure 1 C and D).
Figure 1.
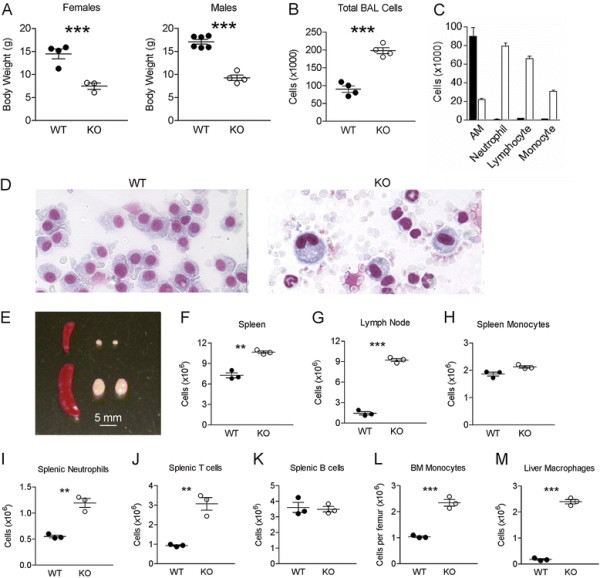
Mice lacking TβRII in hematopoietic cells develop a lethal inflammatory and autoimmune syndrome. (A) Body weight of male and female Vav-Cre x TβRIIfx/fx(TβRII-/-) mice and littermate controls at postnatal day 18-21. (B) Total cell counts and (C) differential cell counts in BALF of WT and TβRII-/- mice at postnatal day 18-21. Solid bar, WT; Open bar, TβRII-/- samples. Differences are significant (p < 0.01) for all cell populations in panel C. (D) Wright-Giemsa-stained BALF cells from the indicated genotypes (18-day old littermates). Note the presence of a single cell type - the alveolar macrophage - in the control lung, whereas the TβRII-/- sample contains neutrophils, monocytes and lymphocytes. (E) Photographic images of spleens and lymph nodes; total cell counts in (F) spleens and (G) lymph nodes; total numbers of (H) monocytes, (I) neutrophils, (J) T-cells and (K) B-cells in spleen; total counts of bone marrow monocytes (L) and liver macrophages (M) in WT (filled circles)and TβRII-/- (KO, open circles) mice (n = 3, **p < 0.01; ***p < 0.001).
Mice lacking TβRII in hematopoietic cells exhibited a normal-sized thymus but enlarged lymph nodes and spleen (Figure 1E), which correlated with a greatly increased cellularity of these tissues (Figure 1 F and G). TβRII-/- spleens contained increased total neutrophil and T-cell numbers, while total monocyte and B-cell numbers were comparable between genotypes (Figure 1H-K). Strikingly, monocyte numbers in bone marrow, and macrophage infiltration in liver were significantly increased in TβRII-/- mice (Figure 1 L and M). Since macrophage infiltration in liver is a sign of autoimmune disease, the increased infiltration of macrophages in the livers of TβRII-/- mice is consistent with the observed inflammatory autoimmune syndrome. We next examined spleen, lymph nodes and thymus for the major T-cell subsets using FACS. Analysis of the thymus showed WT and TβRII-/- mice had a similar distribution of CD4+, CD8+ and double positive (DP) or double negative (DN) subsets (Additional file 1: Figure S1 A and B). This indicates there were no abnormalities in T-cell development, which is consistent with a previous report that TGFβ signaling is not required for thymocyte differentiation [12]. We also found that CD25 expression on CD4+ T cells was comparable in thymus, spleen and lymph nodes of WT and TβRII-/- mice (Additional file 1: Figure S1C), suggesting TβRII signaling does not regulate the development of Treg cells and consistent with previous studies [23,24].
Analysis of the spleen and lymph nodes showed similar percentages of CD4+ and CD8+ T-cells, although their absolute numbers were increased in TβRII-/- mice (not shown). Interestingly, however, there was a dramatic increase in expression of CD44 on T-cells. Around 20% of splenic CD4+ T-cells and 40% of CD8+ T-cells expressed CD44 in WT mice, while 90-95% of T-cells lacking TβRII had CD44 expression. This was also found in lymph nodes (Additional file 1: Figure S1 E and F). In addition, a significantly higher percentage of CD4+ T-cells in lymph nodes of TβRII-/- mice expressed CD69 (Additional file: 1 Figure S1G). Since CD44 and CD69 are T cell activation markers, our data indicate that one physiological role of TβRII signaling is to suppress T cell activation.
Expression of M2 markers on BMDMs is impaired in the absence of TβRII signaling
The lack of normal alveolar macrophages in the BALF of TβRII-/- mice could be associated with intrinsic defects in those macrophages, or could be caused by the environment, since it is well-known that monocytes from the blood can differentiate into different types of macrophages depending upon environmental factors. Treatment with LPS and IFNγ will generate classically activated M1 macrophages, whereas treatment with IL-4 and dexamethasone generates M2a and M2c polarized macrophages, respectively. To examine macrophages lacking TβRII without the presence of other, possibly confounding, cell types, we isolated bone marrow-derived macrophage/monocytes (BMDMs) from the TβRII-/- mice.
FACS analysis of WT and TβRII-/- BMDMs after 11 days in culture with M-CSF, a macrophage growth factor, showed that cells of both genotypes were uniformly Ly6GlowCD11bhi and expressed F4/80, a pan-macrophage surface marker (Additional file 1: Figure S2A), indicating that the purity of the isolated BMDMs is very high (~99%). We then treated these BMDMs with stimuli that are typically used to generate different subsets of polarized macrophages. Additional file 1: Figure S2B illustrates that, as reported previously [15,20,25], these stimuli generate morphologically distinct populations. However, BMDMs with or without TβRII were morphologically similar. We also investigated the efficiency of Vav1-Cre-mediated ablation of the TGFβRII in these cells. Quantitative real-time PCR on DNA from BMDMs showed that, although the deletion was efficient, there was a residual signal of about 6% for TβRII present in Vav1-Cre xTβRIIfx/fx BMDMs (Additional file 1: Figure S2E).
We next assayed WT and TβRII-/- BMDM for their ability to phagocytose apoptotic cells. As shown in Additional file 1: Figure S2C, there were no significant differences in the ability of M0 BMDMs with or without TβRII to perform efferocytosis. Next, we polarized them using different agents including LPS/IFNγ (M1), IL-4 (M2) and hTGFβ1. BMDMs polarized towards the M1 phenotype had significantly decreased ability to phagocytose dying cells (Additional file 1: Figure S2D), but no significant differences were observed between WT and TβRII-/- BMDMs. We also tested migration. In contrast to bone-marrow derived neutrophils, which showed significant migration towards 0.1 ng/ml TGFβ1, bone marrow-derived macrophages did not migrate towards 5 ng/ml TGFβ1 (not shown).
A classical readout for macrophage polarization states is the induction of either iNOS (M1) or Arginase-1 (M2), two enzymes that compete for the common substrate L-arginine. As illustrated in Figure 2A, iNOS was not expressed in M0 or M2 macrophages. Lack of TβRII allowed the normal production of iNOS upon stimulation with LPS/IFNγ. As reported previously [26], there was a baseline level of Arginase-1 (Arg-1) expression in non-polarized macrophages, which was enhanced by treatment with LPS. Remarkably, TβRII-/- BMDMs had very low baseline levels of Arg-1, showed severely impaired induction of Arg-1 by LPS and had profoundly reduced induction of Arg-1 by IL-4 treatment. Similar defects in Arg-1 expression were obtained when M1-polarized (LPS/IFNγ-treated) TβRII-/- macrophages were subsequently treated with IL-4 to re-polarize them to an M2 phenotype. Interestingly, macrophages lacking Smad3 had normal induction of Arg-1, indicating that the mechanism is Smad3-independent (Additional file 1: Figure S3).
Figure 2.

Expression of M2 markers on BMDMs is impaired in the absence of TβRII signaling. (A) Western blot analysis of WT and TβRII-/- BMDMs cultured for 24 hrs with medium alone (M0), with LPS/IFNγ (M1), IL-4 (M2), or hTGFβ1. β-actin serves as a loading control. Increased Arg-1 in KO BMDM upon hTGFβ1stimulation due to residual WT cells, see Additional file 1: Figure S2E. (B) Real-time RT-PCR for the indicated genes after incubation for 24 hrs with medium alone (M0) or IL-4 (M2). (C) Real-time RT-PCR for the indicated genes in naïve peritoneal macrophages. Fold change is with respect to the expression level of M0 WT. Solid bar, WT; Open bar, TβRII-/- peritoneal macrophages. The results shown are representative of one of two independently derived sets of BMDMs from different mice.
TGFβ1 is generally regarded as an anti-inflammatory cytokine and it is frequently listed in the same category as IL-10 and glucocorticoids such as dexamethasone to generate M2c-polarized macrophages. However, we found that WT BMDMs reacted to hTGFβ1 stimulation with an increased expression of Arg-1 (Figure 2A), similar to the classical M2a stimulus IL-4, whereas dexamethasone (M2c) failed to induce Arg1 (not shown).
To examine if TβRII signaling only regulates Arg-1 expression, or that there are defects in the expression of other M2 markers, we next performed real-time RT-PCR for a number of molecules that are typically up-regulated in IL-4-polarized macrophages such as ym1mrc2 and mgl1/2[27-30]. As shown in Figure 2B, lack of TβRII was associated with decreased basal and IL-4-induced transcription of arg1 and correlated with decreased transcription of mcr2, mgl2, and ym1, which are also induced by IL-4. The transcription of mgl1 was not affected. A similar pattern of reduced basal levels of M2 markers was also observed in naïve peritoneal macrophages (Figure 2C). Collectively, our data indicate that TβRII signaling is critical for the optimal expression of genes characteristic for M2 polarization.
Lack of TβRII signaling is correlated with decreased galectin-3, β-catenin and phospho-Akt levels
The molecular pathways that underlie macrophage polarization are of intense interest due to the importance of this cell type in a wide array of pathologies, ranging from autoimmune disorders to cancer, and the desire to be able to modulate their polarization state. However, there are very few reports of null mutants in which macrophage polarization defects have been reported. One of these is Galectin-3 (lgals3). Macrophages lacking Galectin-3 show impaired IL-4-stimulated M2 polarization and lgals3 null mutants have reduced TGFβ-associated liver fibrosis [31,32].
We therefore performed real-time RT-PCR for lgals3. Interestingly, TβRII-/- BMDMs stimulated with IL-4 had reduced mRNA levels of lgals3 (Figure 3A). To examine this in more detail, we stimulated WT and TβRII-/- BMDMs with different polarizing agents and measured cell surface Galectin-3 expression using FACS. Whereas the expression of cell surface CD11b was comparable in these different treatment conditions (not shown), BMDMs lacking TβRII showed clearly decreased Galectin-3 expression, especially when exposed to LPS/IFNγ (Figure 3B).
Figure 3.

Lack ofTβRIIsignaling is correlated with decreased Galectin-3, β-catenin and phospho-Akt levels. (A) Real-time RT-PCR for lgals3 in BMDMs. Fold change is with respect to the expression of M0 WT. Solid bar, WT; Open bar, TβRII-/-. (B) Cell surface expression of Galectin-3 on WT and TβRII-/- BMDMs measured using FACS. MFI, mean fluorescent intensity. The result shown is representative of one of two independently derived sets of BMDMs from different mice for both A and B. (C) Western blot analysis of β-catenin expression. BMDM were treated with medium alone (M0), LPS/IFNγ (M1), or IL-4 (M2). Results are representative of 3 pairs of matched WT and TβRII-/- mice. Western blot analysis of phosphorylated-Stat6 (D) and phosphorylated-Akt (E) in WT and TβRII-/- BMDMs that had been treated with IL-4 for 1 hr. Results are shown for 2 independent mice per genotype. The numbers at the bottom represent the band intensity ratio of phosphorylated protein versus total protein, normalized to lane 1 on each blot.
The E-cadherin/β-catenin complex was recently shown to be a selective marker for alternatively activated macrophages [33]. Interestingly, when WT and TβRII-/- BMDMs were stimulated by IL-4, β-catenin levels were ~35% lower in TβRII-/- than in WT BMDMs (Figure 3C), further indicating that TβRII signaling promotes M2 macrophage polarization.
To explore the mechanism by which TGFβ contributes to IL-4-induced M2 polarization, we examined the downstream targets of IL-4. STAT6 phophorylation is a key signaling event downstream of the IL-4 receptor complex, and Akt also becomes activated in this pathway [34-36]. Thus we investigated if STAT6 and/or Akt are also involved in the IL-4 stimulated polarization that is co-regulated by TGFβ signaling. As shown in Figure 3D, p-Stat6 levels were comparable in WT and TβRII-/- BMDMs stimulated with IL-4. However, p-Akt was decreased ~50% in the absence of TGFβ signaling (Figure 3E). This indicates that TGFβ contributes to IL-4-induced M2 polarization through co-signaling to Akt, which is one of the TGFβ1 non-Smad-associated signal transduction pathways reported in other cell types [37,38].
Discussion
In the current study, we found that the phenotype of mice born without TβRII function in hematopoietic cells overlaps with that of mice born without systemic TGFβ1 production. In both cases, the null mutants develop a severe wasting syndrome at around the same time, at 3 weeks of age [9,10]. However, our mice exhibited splenomegaly and lymph node enlargement with a corresponding large increase in cellularity, whereas Shull et al. reported reduced size of spleen and Peyer’s patches [10].
We measured dramatically increased expression (15-40% in WT, 80-95% in KO) of the activation marker CD44, a receptor for the extracellular matrix (ECM) protein hyaluronic acid, on both CD4+ and CD8+ T cells in the spleen and lymph nodes. Similarly increased CD44 expression was also observed in mice transplanted with bone marrow of mice transgenic for a dominant negative TβRII construct [39]. CD69 is a C-type lectin that is expressed on the surface of all leukocytes during activation, and engagement of CD69 maintains high expression of membrane-bound TGFβ1 on T-cells [40]. We found that the numbers of CD4+ and CD8+ T-cells in the lymph nodes that expressed CD69 were around twice that of control cells. This result is consistent with the increased numbers of CD69+ T-cells found in the lymph nodes of mice transplanted with bone marrow from TβRIIfx/fx x Mx1-Cre mice and with the signs of T-cell activation in lymph nodes and spleen in those mice [12].
Alveolar macrophages are a specialized subclass of differentiated tissue macrophages that are generally regarded as regulatory and anti-inflammatory [41]. At three weeks of age, Vav1-Cre x TβRIIfx/fx mice developed spontaneous pulmonary inflammation in which few normal alveolar macrophages were found. The lack of normal alveolar macrophages could be explained by the pro-inflammatory environment in the lung, which would polarize peripheral blood monocytes that migrate into the lung to an M1 phenotype and/or by re-polarization of alveolar macrophages into proinflammatory M1 macrophages. Also, reduced production of monocytes in the bone marrow could result in lower levels of alveolar macrophages. However, we did not find increased levels of IFNγ, a classic proinflammatory cytokine, in the peripheral blood of the null mutant mice (Additional file 1: Figure S4) and there was no evidence for impaired generation of monocytes in the bone marrow of mice lacking hematopoietic TβRII.
To examine possible macrophage-intrinsic defects, we studied naïve, bone-marrow derived monocyte/macrophages for possible cell-intrinsic defects. This analysis showed an overall decreased ability of macrophages lacking TβRII signaling to produce the normal amounts of arg1 mRNA and protein, either at basal levels or, more markedly, when BMDMs were polarized towards an M2 phenotype with IL-4. In agreement with the regulation of Arg-1 by TGFβ1, we found increased expression of Arg-1 in WT macrophages stimulated with this cytokine. Moreover, transcription of other M2 markers including mgl2, ym1 and mcr2, but not mgl1, was also deceased in TβRII-/- BMDMs, showing that signals through TβRII modulate the M2 transcription program.
Ym1 is a secretory lectin that promotes Th2 cytokine expression [42], whereas Mrc2 is a C-type receptor for mannose that promotes remodeling of the ECM and the uptake and degradation of collagen [29]. Both Mgl1 (clec10a) and Mgl2 as markers for alternatively activated macrophages are type C galactose-binding lectins with a different binding specificities to carbohydrate structures [43]. These results show that TGFβ signaling is responsible for the transcriptional upregulation of several M2-polarization genes in macrophages that are important for binding to carbohydrate structures present in the ECM and on other cells and are consistent with the role of TGFβ as a master regulator of ECM reorganization [44,45].
We also found that macrophages lacking TβRII produce less mRNA for lgals3, which encodes a β-galactoside-binding lectin, and have markedly reduced Galectin-3 cell surface expression on M1 (LPS/IFNγ-polarized) macrophages. Interestingly, Galectin-3 negatively regulates LPS-induced inflammation [46]. Moreover, BMDMs lacking lgals3 have reduced Arg-1 production and are defective in M2 polarization [32]. These results suggest that some of the defects in TβRII-/- BMDMs could be caused by the reduced Galectin-3 in those cells. Importantly, Akt phosphorylation was decreased in the IL-4-polarized M2 TβRII-/- BMDMs. This suggests that TGFβ and IL-4 co-signal to maximally activate the Akt pathway. Although further experiments will be needed to determine the detailed molecular mechanisms through which TGFβ regulates M2 polarization, our findings suggest that modulation of the TGFβ signaling pathway may be a method to medically regulate macrophage polarization.
Conclusions
Taken together, our results establish a critical biological role for TGFβ signaling in promoting the alternative activation of macrophages. Moreover, our results suggest a distinct contribution of monocytes/macrophages to the systemic inflammation observed in mice with defective TβRII function, because lack of TβRII signaling in macrophages inhibits the polarization of macrophages to an anti-inflammatory M2 phenotype, this could lead to defective down-regulatory interactions with other immune cells such as T-cells in the lung. These findings further advance our knowledge of the physiological role of TGFβ signaling and enhance our understanding of the regulation of macrophage activation states.
Methods
Animals
The TβRIIfx/fx mice [47] on a C57BL/6 background were obtained from the NCI Mouse Repository (Strain number: 01XN5) and were bred to Vav1-Cre mice, which were generously provided by Dr. Dimitris Kioussis (National Institute for Medical Research, UK) [22], to generate Vav1-Cre x TβRIIfx/fx mice and littermate controls. TβRII-/- mice develop a wasting syndrome 24-28 days after birth. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Children’s Hospital Los Angeles.
Bronchoalveolar lavage
To prepare BALF, tracheas were exposed and cannulated with an 18-gauge angiocath. Lungs were lavaged five times with 0.8 ml of cold sterile PBS. Analysis of resident and recruited cells was performed on cells pooled from the five washes.
Cytospin and differential staining
50-80,000 cells were spun onto glass slides at 800 rpm for 5 min in a Shandon Cytospin II Cytocentrifuge. Slides were stained using a Kwik-Diff stain kit (Thermo Fisher Scientific). Cells were differentially (morphologically) counted using light microscopy.
Tissue culture and isolation of bone marrow-derived macrophages and peritoneal macrophages
Tissue culture reagents were from Invitrogen. LPS (cat no. L2630) was from Sigma-Aldrich. IL-4, hTGFβ1, IL-10 and IFNγ were purchased from PeproTech. The L929 cell line was from the American Type Culture Collection. The supernatant collected from L929 cultures that had grown for 7 days was used to prepare L929 conditioned media, as a source of M-CSF, for macrophage maturation and expansion. Bone marrow-derived macrophages (BMDMs) were prepared by maturing bone marrow cells, which were isolated from femur and tibiae of mice, in DMEM containing 15% FBS and 20% L929 conditioned media for 11 days. BMDMs were expanded and treated with IFNγ (10 ng/ml) and LPS (100 ng/ml) for M1 polarization and IL-4 (10 ng/ml) for M2 polarization. BMDMs were also treated with hTGFβ1 at 5 ng/ml or dexamethasone at 40 ng/ml. Peritoneal cells were harvested from the peritoneal cavity of mice by lavage. Cells from 3-4 mice of the same genotype were pooled and stained for FITC-Ly6G, PE-CD11b, PerCP-CD45 and APC-F4/80. Peritoneal macrophages (CD45+CD11b+Ly6G-F4/80+) were sorted out using a BD FACSAria Sorter. Sorted cells were lyzed in Trizol reagent (Invitrogen) for total RNA extraction.
Real-time RT-PCR
Total RNA was prepared using RNeasy kits (Qiagen). cDNA was synthesized using a First Strand cDNA Synthesis kit (Invitrogen). The cDNA was amplified by 40 two-step cycles (15 sec at 95°C for denaturation of the DNA, 1 min at 60°C for primer annealing and extension). cDNA was analyzed using a SYBR green-based quantitative fluorescence method (Applied Biosystems) in duplicate. The PCR primers were as follows:
GAPDH: forward 5'-AGA GGG AAA TCG TGC GTG AC-3', reverse 5'-CAA TAG TGA TGA CCT GGC CGT-3'; ym1: forward 5'-AGA AGG GAG TTT CAA ACC TGG G-3', reverse 5'-GTC TTG CTC ATG TGT GTA AGT GA-3'; arginase 1: forward 5'-CTC CAA GCC AAA GTC CTT AGA G-3', reverse 5'-AGG AGC TGT CAT TAG GGA CAT C-3'; marc2: forward 5'-TAC AGC TCC ACG CTA TGG ATT-3', reverse 5'-CAC TCT CCC AGT GTA GGT ACT-3'; mgl1: forward 5'-TGA GAA AGG CTT TAA GAA CTG GG-3', reverse 5'-GAC CAC CTG TAG TGA TGT GGG-3'; mgl2: forward 5'-TTA GCC AAT GTG CTT AGC TGG-3', reverse 5'-GGC CTC CAA TTC TTG AAA CCT'-3’; lgals3: forward 5'-TTG AAG CTG ACC ACT TCA AGG TT-3', reverse 5'- AGG TTC TTC ATC CGA TGG TTG T-3’; TβRII: forward 5’-GGG ATT GCC ATA GCT GTC AT-3’; reverse 5’-TGA TGG CAC AAT TGT CAC TG-3’.
Flow cytometry and quantification of different cell populations in tissues
Data on fluorochrome-labeled monoclonal antibody-stained cells were acquired on an Accuri cytometer (Accuri Cytometers Inc). Data were analyzed using the Accuri software provided by the manufacturer. FITC-Ly6G, FITC-CD4, PE-CD8, PE-CD25, APC-CD69, APC-CD44, PE-Gal 3, PerCP-CD45, and APC-CD11b, were from BioLegend.
For quantification of different cell populations in tissues (spleen, lymph node, thymus, bone marrow and liver), tissues were harvested and processed for total live cell counts using Trypan blue and a hemacytometer. Cells were then stained with appropriate antibodies. The percentages of cell populations were calculated using Accuri software. The total cell number for each leukocyte subset was calculated by multiplying total cell counts with the percentage of that specific cell population within the total cell population.
Efferocytosis assay
The ability of BMDM to perform phagocytosis of apoptotic cells (efferocytosis) was tested on apoptotic primary thymocytes isolated from 2-4-week old WT mice. After red blood cell lysis, thymocytes were cultured overnight in RPMI 1640 supplemented with 10% FBS, L-glutamine, sodium pyruvate, penicillin/streptomycin and 50 μM β-mercaptoethanol, with 6 μM CFDA. Thymocytes were induced to undergo apoptosis by treatment of 5x106 cells/ml with 1 μM dexamethasone for 6 hrs, which was monitored by annexin V-FITC/propidium iodide staining and FACS. BMDM were starved for 24 hr without CSF-1 in DMEM + 15% FBS; in some experiments, polarizing cytokines were added. After a wash with cold DMEM, BMDM and thymocytes were preincubated at a ratio of 1:40 on ice. Efferocytosis was initiated by addition of prewarmed DMEM/15% FBS, followed by incubation at 37°C for different times. Controls kept on ice showed no engulfment of apoptotic thymocytes. After co-incubation, cells were washed three times in ice-cold PBS-/- and background fluorescence was quenched on ice for 15 min. with 0.25 mg/ml Trypan blue in 0.02 M ammonium acetate, 150 mM NaCl, pH 4.0. Cells were washed 3 times, fixed and stained with TRITC-phalloidin. Phagocytosis was evaluated by counting 200–300 macrophages per slide from triplicate experiments. Results for the phagocytosis index are expressed as the percentage of macrophages that had engulfed one or more thymocyte.
Western blots
Cell lysates were resolved by SDS-PAGE and immunoblotted with anti-Arginase-1 (BD Bioscience), anti-phospho-Stat6 (Tyr641, Millipore), anti-phospho-Akt (Ser473), anti-β-catenin, and anti-iNOS antibodies (Cell Signaling). Blots were stripped and re-blotted with anti–β-actin (Sigma), anti-GAPDH (Millipore), anti-Stat6 or anti-Akt (Cell Signaling) antibodies. Band intensities were analyzed using Un-Scan-It software (Silk Scientific, Orem, UT) on the scanned images of the blots.
ELISA
Blood was collected from WT and TβRII-/- mice at postnatal day 18-21. After clotting of blood, serum was harvested by centrifugation at 800 g for 10 min. Serum IFNγ levels were measured using a mouse IFNγ ELISA kit (Biolegend) according to the manufacturer’s protocol.
Statistical analysis
Data are expressed as mean ± SEM and analyzed by the unpaired Student's t test using Prism (GraphPad) software, unless indicated otherwise in the figure legend. p < 0.05 was considered to be statistically significant.
Abbreviations
BAL, Bronchoalveolar lavage; BALF, Bronchoalveolar lavage fluid; BMDMs, Bone marrow-derived macrophages; ECM, Extracellular matrix; TGFβ, Transforming growth factor beta; TβRII, TGFβ type II receptor.
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
DG designed and performed experiments, collected and analyzed data, and drafted the manuscript. SY designed and performed the efferocytosis and motility assays. WS and HC provided Vav1-Cre and TGFβRIIfx/fx mice and revised the manuscript. NH and JG designed and coordinated experiments, evaluated and interpreted data, and prepared the manuscript. All authors read and approved the manuscript.
Supplementary Material
Figure S1. Analysis of T-cells in thymus, spleen and lymph nodes of WT and TβRII-/- mice. (A) FACS of WT and TβRII-/- thymus for CD4- and CD8-positive cells. (B) Cell counts of double negative (DN), double positive (DP), CD4 single positive and CD8 single positive cells in thymus. (C) Percentage of CD4 + CD25+ T-cells in thymus, lymph node and spleen of WT and TβRII-/- mice. Solid bar, WT; Open bar, TβRII-/- mice. (D) Left, single-cell suspensions of lymph nodes were gated for lymphocytes (P1) and right, CD4+ and CD8+ T cells. (E) Representative histogram of CD44 expression on CD4+ T cells of TβRII-/- (red line) and control (black line) mice. (F) Percentage of CD44+ cells in spleen and lymph nodes using the strategy in (D). (G) Percentage of CD69+ cells in lymph nodes. **, p < 0.05; ***p < 0.005. Figure S2 BMDMs with or without TβRII function. (A) Gating strategy for FACS on BMDMs of the indicated genotypes using CD11b and Ly6G. Right panel, the expression of F4/80, a pan-macrophage surface marker. (B) BMDMs were treated with the stimuli as indicated to the right for 24 hrs. Representative phase contrast images are shown. (C) Percentage BMDMs that had engulfed at least one apoptotic thymocyte (efferocytosis) in standard medium. (D) Efferocytosis of BMDMs with or without TβRII function treated for 24 hrs with medium alone (M0); 100 ng/ml LPS and 10 ng/ml IFNγ (M1), 10 ng/ml IL-4 (M2a), or 5 ng/ml hTGFβ1. M0 compared to M1, **p = 0.003. (E) Genomic DNA was extracted from WT and TβRII-/- BMDMs (n = 4). Real-time PCR was used to quantify the levels of TβRII. The level of TβRII in one WT sample was set to 1 and the levels of TβRII in other samples were normalized to that of the chosen WT sample. Figure S3 Expression of iNOS and Arg1 in WT and smad3-/- BMDMs. Western blot analysis of Arg1 and iNOS in BMDMs of WT and smad3-/- mice. Figure S4 IFNγ levels in the blood of WT and TβRII-/- mice. ELISA was used to measure IFNγ levels in the serum of WT and TβRII-/- mice on postnatal day 18-21 (n = 4).
Contributor Information
Dapeng Gong, Email: dgong@chla.usc.edu.
Wei Shi, Email: wshi@chla.usc.edu.
Sun-ju Yi, Email: sunzoo2000@hanmail.net.
Hui Chen, Email: huchen@chla.usc.edu.
John Groffen, Email: jgroffen@chla.usc.edu.
Nora Heisterkamp, Email: heisterk@hsc.usc.edu.
Acknowledgements
We thank Dr. Dimitris Kioussis (National Institute for Medical Research, UK) for providing the Vav1-Cre transgenic mice, and Donna Foster for excellent care of the mice. This work was supported by the National Institutes of Health (HL071945 and HL060231 to JG).
References
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Iseki S, Osumi-Yamashita N, Miyazono K, Franzen P, Ichijo H, Ohtani H, Hayashi Y, Eto K. Localization of transforming growth factor-beta type I and type II receptors in mouse development. Exp Cell Res. 1995;219:339–347. doi: 10.1006/excr.1995.1237. [DOI] [PubMed] [Google Scholar]
- Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. Differential expression of the TGF-beta isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol Reprod Dev. 1992;32:91–98. doi: 10.1002/mrd.1080320203. [DOI] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997;403:57–60. doi: 10.1016/S0014-5793(97)00024-0. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568. doi: 10.1182/blood.V100.2.560. [DOI] [PubMed] [Google Scholar]
- Leveen P, Carlsen M, Makowska A, Oddsson S, Larsson J, Goumans MJ, Cilio CM, Karlsson S. TGF-beta type II receptor-deficient thymocytes develop normally but demonstrate increased CD8+ proliferation in vivo. Blood. 2005;106:4234–4240. doi: 10.1182/blood-2005-05-1871. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ. et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- Mamura M, Lee W, Sullivan TJ, Felici A, Sowers AL, Allison JP, Letterio JJ. CD28 disruption exacerbates inflammation in Tgf-beta1-/- mice: in vivo suppression by CD4 + CD25+ regulatory T cells independent of autocrine TGF-beta1. Blood. 2004;103:4594–4601. doi: 10.1182/blood-2003-08-2897. [DOI] [PubMed] [Google Scholar]
- Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasen S, Hammermann R, Fuhrmann M, Lindemann D, Beck KF, Pfeilschifter J, Racke K. Glucocorticoids inhibit lipopolysaccharide-induced up-regulation of arginase in rat alveolar macrophages. Br J Pharmacol. 2001;132:1349–1357. doi: 10.1038/sj.bjp.0703951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denda-Nagai K, Aida S, Saba K, Suzuki K, Moriyama S, Oo-Puthinan S, Tsuiji M, Morikawa A, Kumamoto Y, Sugiura D. et al. Distribution and function of macrophage galactose-type C-type lectin 2 (MGL2/CD301b): efficient uptake and presentation of glycosylated antigens by dendritic cells. J Biol Chem. 2010;285:19193–19204. doi: 10.1074/jbc.M110.113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, Haslett C, Simpson KJ, Sethi T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A. 2006;103:5060–5065. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180:2650–2658. doi: 10.4049/jimmunol.180.4.2650. [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, Bogaert P, van Hengel J, Guerin CJ, Berx G, Movahedi K, Van den Bergh R, Pereira-Fernandes A, Geuns JM, Pircher H. et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL-4 and polyamine-induced E-cadherin/catenin complexes. Blood. 2009;114:4664–4674. doi: 10.1182/blood-2009-05-221598. [DOI] [PubMed] [Google Scholar]
- Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- Giallourakis C, Kashiwada M, Pan PY, Danial N, Jiang H, Cambier J, Coggeshall KM, Rothman P. Positive regulation of interleukin-4-mediated proliferation by the SH2-containing inositol-5'-phosphatase. J Biol Chem. 2000;275:29275–29282. doi: 10.1074/jbc.M002853200. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Derynck R. Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs. 2011;193:8–22. doi: 10.1159/000320172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- Shah AH, Tabayoyong WB, Kimm SY, Kim SJ, Van Parijs L, Lee C. Reconstitution of lethally irradiated adult mice with dominant negative TGF-beta type II receptor-transduced bone marrow leads to myeloid expansion and inflammatory disease. J Immunol. 2002;169:3485–3491. doi: 10.4049/jimmunol.169.7.3485. [DOI] [PubMed] [Google Scholar]
- Han Y, Guo Q, Zhang M, Chen Z, Cao X. CD69+ CD4+ CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol. 2009;182:111–120. doi: 10.4049/jimmunol.182.1.111. [DOI] [PubMed] [Google Scholar]
- Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L936–L946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Kumar RK, Zhou J, Foster PS, Webb DC. Ym1/2 promotes Th2 cytokine expression by inhibiting 12/15(S)-lipoxygenase: identification of a novel pathway for regulating allergic inflammation. J Immunol. 2009;182:5393–5399. doi: 10.4049/jimmunol.0803874. [DOI] [PubMed] [Google Scholar]
- Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F, Vanham G, Noel W, Bogaert P, Boonefaes T. et al. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol. 2005;77:321–327. doi: 10.1189/jlb.0304212. [DOI] [PubMed] [Google Scholar]
- Klass BR, Grobbelaar AO, Rolfe KJ. Transforming growth factor beta1 signalling, wound healing and repair: a multifunctional cytokine with clinical implications for wound repair, a delicate balance. Postgrad Med J. 2009;85:9–14. doi: 10.1136/pgmj.2008.069831. [DOI] [PubMed] [Google Scholar]
- Xu BJ, Yan W, Jovanovic B, An AQ, Cheng N, Aakre ME, Yi Y, Eng J, Link AJ, Moses HL. Quantitative analysis of the secretome of TGF-beta signaling-deficient mammary fibroblasts. Proteomics. 2010;10:2458–2470. doi: 10.1002/pmic.200900701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Komai-Koma M, Gilchrist DS, Hsu DK, Liu FT, Springall T, Xu D. Galectin-3 is a negative regulator of lipopolysaccharide-mediated inflammation. J Immunol. 2008;181:2781–2789. doi: 10.4049/jimmunol.181.4.2781. [DOI] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre: Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of T-cells in thymus, spleen and lymph nodes of WT and TβRII-/- mice. (A) FACS of WT and TβRII-/- thymus for CD4- and CD8-positive cells. (B) Cell counts of double negative (DN), double positive (DP), CD4 single positive and CD8 single positive cells in thymus. (C) Percentage of CD4 + CD25+ T-cells in thymus, lymph node and spleen of WT and TβRII-/- mice. Solid bar, WT; Open bar, TβRII-/- mice. (D) Left, single-cell suspensions of lymph nodes were gated for lymphocytes (P1) and right, CD4+ and CD8+ T cells. (E) Representative histogram of CD44 expression on CD4+ T cells of TβRII-/- (red line) and control (black line) mice. (F) Percentage of CD44+ cells in spleen and lymph nodes using the strategy in (D). (G) Percentage of CD69+ cells in lymph nodes. **, p < 0.05; ***p < 0.005. Figure S2 BMDMs with or without TβRII function. (A) Gating strategy for FACS on BMDMs of the indicated genotypes using CD11b and Ly6G. Right panel, the expression of F4/80, a pan-macrophage surface marker. (B) BMDMs were treated with the stimuli as indicated to the right for 24 hrs. Representative phase contrast images are shown. (C) Percentage BMDMs that had engulfed at least one apoptotic thymocyte (efferocytosis) in standard medium. (D) Efferocytosis of BMDMs with or without TβRII function treated for 24 hrs with medium alone (M0); 100 ng/ml LPS and 10 ng/ml IFNγ (M1), 10 ng/ml IL-4 (M2a), or 5 ng/ml hTGFβ1. M0 compared to M1, **p = 0.003. (E) Genomic DNA was extracted from WT and TβRII-/- BMDMs (n = 4). Real-time PCR was used to quantify the levels of TβRII. The level of TβRII in one WT sample was set to 1 and the levels of TβRII in other samples were normalized to that of the chosen WT sample. Figure S3 Expression of iNOS and Arg1 in WT and smad3-/- BMDMs. Western blot analysis of Arg1 and iNOS in BMDMs of WT and smad3-/- mice. Figure S4 IFNγ levels in the blood of WT and TβRII-/- mice. ELISA was used to measure IFNγ levels in the serum of WT and TβRII-/- mice on postnatal day 18-21 (n = 4).