

Abstract
MAP kinase Phosphatase 3 (MKP-3) was recently identified as an important regulator of glucose homeostasis in the liver and its expression can be repressed by insulin at post transcriptional level. In this study, the mechanism underlying insulin promoted decrease of MKP-3 protein was investigated by studying MKP-3 protein stability via immunoblot analysis in the presence of cycloheximide using cultured liver cells. Several pathways were examined and activation of the MEK/ERK pathway was found to mediate reduction of MKP-3 protein expression in response to insulin. MEK inhibitor markedly slowed down MKP-3 protein degradation. Mutation of two ERK phosphorylation sites on MKP-3 rendered it resistant to insulin and constitutively active MEK-induced MKP-3 protein degradation. To understand the biological effect of MKP-3 protein stability on liver cell glucose output, expression level of G6Pase gene, which encodes the key enzyme controlling the last step of de novo glucose synthesis in liver cells, was examined by real time PCR analysis upon manipulation of MEK signaling. Activation of MEK pathway in Fao cells resulted in decreased expression of G6Pase gene and lowered glucose output. Consistent with this result, MEK inhibitor increased expression of G6Pase gene and glucose output in Fao cells. In conclusion, insulin likely promotes MKP-3 protein degradation through activation of MEK/ERK pathway in liver cells and MKP-3 protein level affects the capability of Fao cells to output glucose.
Keywords: MAP kinase phosphatase, glucose output, protein degradation
1. Introduction
Mitogen-activated protein kinase phosphatase 3 (MKP-3, also known as DUSP6, PYST1 or rVH6) is a member of the dual specificity phosphatase family, which dephosphorylate phosphothreonine and phosphotyrosine residues of MAP kinases (Groom et al., 1996; Mourey et al., 1996; Wiland et al., 1996). MAP kinases are important cellular mediators for transducting signals from a variety of extracellular stimuli (Chang and Karin, 2001; Schaeffer and Weber, 1999). Activation of MAP kinases requires phosphorylation on both threonine and tyrosine residues located within the activation loop. There are 11 members in the MKP/DUSP family that contain a MAPK binding domain and they have different subcellular localization, tissue expression pattern or substrate preference (Lang et al., 2006; Owens and Keyse, 2007; Theodosiou and Ashworth, 2002). There are also additional 19 atypical DUSP family members which do not contain the MAPK binding domain and are smaller proteins (Lang et al., 2006). MKP-3 is a cytoplasmic phosphatase which is highly specific for dephosphorylation of ERK1/2 and plays an important role in attenuating ERK1/2 mediated mitogenesis (Camps et al., 1998; Fjeld et al., 2000; Zhao and Zhang, 2001). MKP-3 deficient mice displayed enhanced basal ERK1/2 phosphorylation and increased myocyte proliferation (Maillet et al., 2008). Interestingly, the MEK/ERK pathway itself also plays a role in down regulating MKP-3 expression, which forms a negative feedback loop between the kinases and the phosphatase (Jurek et al., 2009; Marchetti et al., 2005). In addition, the PI3K/mTOR pathway has also been reported to mediate growth factor induced MKP-3 protein degradation (Bermudez et al., 2008).
In addition to the role in attenuating MAP kinase signaling, our recent studies imply that MKP-3 has a novel role in regulating hepatic glucose homeostasis. We previously identified MKP-3 as a top hit in an expression screen of H4IIE hepatoma cells for genes antagonizing the effect of insulin on repressing Pepck gene transcription, a key enzyme controlling the first step of gluconeogenesis (Xu et al., 2005). MKP-3 was able to promote glucose output in cultured and primary liver cells as well as in the liver of lean mice. The expression of MKP-3 is markedly elevated in the liver of obese mice and knocking down MKP-3 expression in the liver of both lean and obese mice decreases fasting blood glucose levels (Wu et al., 2010). The action of MKP-3 on promoting hepatic glucose output is at least partially through dephosphorylation of phosphoserine 256 of FOXO1, a well known transcription factor for turning on the gluconeogenic program (the process of de novo glucose synthesis). Phosphorylated FOXO1 is inactive and retained in the cytosol. MKP-3 mediated dephosphorylation activates FOXO1 and subsequentially promotes its nuclear translocation and binding to the promoters of gluconeogenic genes, such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6pase). These two enzymes control the first and last step of glucose synthesis, respectively. The novel role of MKP-3 in regulating glucose metabolism makes it a potential therapeutic target for treating obesity-related hyperglycemia.
Interestingly, hepatic MKP-3 expression is regulated by several hormones critical for controlling blood glucose homeostasis. Glucagon, which stimulates hepatic glucose output and elevates blood glucose levels, upregulates MKP-3 protein expression in the liver of lean mice (Wu et al., 2010). Insulin is the major hormone repressing hepatic glucose output and lowering blood glucose levels. Insulin down-regulates MKP-3 protein expression in the liver of lean mice (Wu et al., 2010). These data indicate that MKP-3 might be a converging downstream mediator of multiple blood glucose controlling hormones. Insulin does not decrease MKP-3 gene expression, suggesting that the reduction of protein expression occurs at post-transcription level (Wu et al., 2010). In current study, we investigated the mechanism of insulin repressed MKP-3 protein expression using cultured liver cells, with a focus on protein degradation.
2. Materials and Methods
2.1. Reagents
Fao hepatoma cells were cultured in RPMI 1640 supplemented with 10%FBS. MKP-3 antibody was purchased from Santa Cruz (Santa Cruz, CA). Phospho-ERK, phospho-Akt, phospho-S6, total-ERK, Akt and S6 antibodies were purchased from Cell Signaling (Danvers, MA). Tubulin antibody was purchased from Abcam (Cambridge, MA). U0126 and rapamycin were purchased from Calbiochem (Rockland, MA). Insulin was purchased from Sigma (St Louis, MO).
2.2. Site-directed mutagenesis
MKP-3 S159A, MKP-3 S197A and MKP-3 S159AS197A were constructed by using QuickChange® II Site-Directed Mutagenesis Kit according to the manufacturer’s instruction (Stratagene, Santa Clara, CA). Two rounds of PCR reactions were performed to generate the double mutant. In the first round of PCR, serine 159 was mutated to alanine using primers 5'-gctcgtgcagcagcagtgccccgcctt-3' and 5'-aaggcggggcactgctgctgcacgagc-3'. On a separate wild type clone, serine 197 was mutated to alanine using primers 5'-cggactctgatggcgccccgctgtccaaca-3' and 5'-tgttggacagcggggcgccatcagagtccg-3. After sequence confirmation, serine 197 was then mutated to alanine in the second round of PCR by using MKP-3 S159A as a template followed by sequence confirmation.
2.3. Construction and purification of adenovirus
Adenovirus expressing mouse MKP-3 was constructed as described in previous publication (Wu et al., 2010). Adenoviruses expressing the constitutively active N-terminally myristoylation signal-attached human Akt1 in a tet-off manner and the tetracycline controlled transactivator (tTA) were purchased from Cell Biolabs, Inc (San Diego, CA). To construct adenoviral vectors for the constitutively active MEK (MEK S218DS222D), MKP-3 S159A, MKP-3 S197A and MKP-3 S159AS197A, the coding sequences of rat MEK S218DS222D and mouse MKP-3 S159A, MKP-3 S197A and MKP-3 S159AS197A were amplified by PCR, cloned into the entry vector and sequence confirmed. These coding sequences were then recombined into the Gateway-based pAd-CMV DEST™ vector (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Amplification of recombinant adenovirus was performed according to the manufacturer’s instructions (Invitrogen) using HEK 293A cells.
2.4. RNA extraction and real-time PCR analysis
RNA samples were extracted from Fao cells using the TRIzol reagent (Invitrogen, Carlsbad, CA) or the RNAeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNase I-treated RNA samples were reverse-transcribed with SuperScript III reverse transcriptase (Applied Biosystems, Carlsbad, CA) and random hexamers (Invitrogen) to generate cDNA. Real-time PCR analysis was performed using Power SYBR Green RT-PCR Reagent (Applied Biosystems) on ABI Prism thermal cycler model StepOnePlus (Applied Biosystems). Each 15-µl PCR reactions contained 1× reaction mix, 300nM forward primer, 300 nM reverse primer. The thermal cycling program was 50°C for 2 minutes, followed by 95°C for 10 minutes for 1 cycle, then 95°C for 15 seconds, followed by 60°C for 1 minute for 40 cycles. Melting curve analysis was performed to ensure the specificity of primers. 28S rRNA was used as a reference gene in each reaction. The delta delta ct method was used to quantify target gene expression and normalize it to reference gene.
2.5. Immunoprecipitation and western blot analysis
Cells were lysed with ice-cold lysis buffer supplemented with protease inhibitors. To immunoprecipitate endogenous MKP-3 from Fao cells, thirty microliters of Exactra D immunoprecipitation matrix (Santa Cruz Biotechnology) slurry was used to preclear cell lysates at 4 C for 30 min. Then forty microliters of MKP-3 antibody-bound Exactra D immunoprecipitation matrix slurry was added to pull down MKP-3. Protein lysates were separated on 4–12% Bis-Tris-PAGE gel (Biorad, Hercules, CA), transferred onto polyvinylidene difluoride membranes followed by blocking for 60 minutes in 1% bovine serum albumin/1×Tris-buffered saline with Tween-20 (TBST) or 5% nonfat dry milk/1×TBST. Membranes were then incubated with the corresponding primary antibodies in the presence of 1% bovine serum albumin/1×TBST or 5% nonfat dry milk/1×TBST overnight on a rocker at 4°C. After thorough washes in 1×TBST, membranes were exposed to corresponding horseradish peroxidase-linked secondary antibodies in 5% nonfat dry milk/1×TBST for 1 hour at room temperature. After thorough washes in 1×TBST, signals were detected with a chemiluminescence western blotting detection solution (PerkinElmer, Waltham, MA) on the Alpha-Inotech fluorochem image system.
2.6. Glucose output assay
Fao hepatoma cells were seeded in 12-well plates. Forty-eight hours after infection with Ad-GFP or Ad-MEK CA, cells were incubated in serum free medium overnight. For MEK inhibitor study, cells were treated with U0126 or vehicle during the overnight incubation in serum-free DMEM. Next day, cells were incubated in 0.4 ml/well of phenol red-free, glucose-free DMEM containing 2 mM pyruvate and 20 mM lactate. Medium was collected three hours later and subjected to glucose measurement using the Amplex® Red Glucose/Glucose Oxidase Assay Kit (Invitrogen). Cells were lysed and protein concentration was determined for each lysate. The glucose output rate was normalized by cellular protein content.
2.7. Calculation of MKP-3 protein half life
Exogenous or endogenous MKP-3 protein levels were determined by immunoblot analysis. MKP-3 protein levels were then plotted against time points. For vehicle treated Fao cells over-expressing MKP-3, the equation is y = −0.0164x + 1.518 (y indicates MKP-3 protein level while × indicates time). When x=0, y=1.518. Half of MKP-3 is 0.759, therefore × (T1/2)=46.28 minutes. Similarly, the equation is: y = −0.0257x + 1.5879 for insulin treated Fao cells over-expressing MKP-3. For endogenous MKP-3 T1/2, Fao cells were treated with vehicle or insulin. The equations are y = −0.0103x + 0.8348 for vehicle treated cells and y = −0.0166x + 0.9272 for insulin treated cells.
2.8. Statistical analysis
Results are presented as mean ± SEM. Statistical significance was determined at P<0.05. Student’s two-tailed t-test was used to compare differences between groups.
3. Results
3.1. Activation of ERK pathway leads to decrease of MKP-3 protein expression
We previously reported that injection of insulin into lean male C57BL/6 mice markedly reduces expression level of MKP-3 protein in the liver (Wu et al., 2010). Our previous results indicated that MKP-3 mRNA level was not changed, indicating a potential mechanism at post-transcription level. In this study, we aim to investigate the pathway(s) involved in insulin repressed MKP-3 protein expression by using cultured Fao hepatoma cells. To study the kinetics of MKP-3 protein degradation without interference of protein synthesis, MKP-3 was over-expressed in Fao cells by adenovirus mediated gene transfer. Fao cells were then treated with insulin at the final concentration of 50nM for various time points in the presence of 10 M cycloheximide. Phosphorylation status of ERK, Akt and S6 were examined along with MKP-3 protein expression level. Insulin treatment activated the two classical pathways, Akt and ERK, at all time points examined (Figure 1A, supplemental figure 1). Phosphorylation status of S6 was not affected by insulin treatment in Fao cells (Figure 1A, supplemental figure 1). The expression level of exogenously expressed MKP-3 protein was significantly reduced 60 minutes after insulin treatment compared to vehicle control (Figure 1B). Insulin also similarly reduced expression of endogenous MKP-3 protein at the same time point (Figure 1C), therefore validates the use of exogenously expressed MKP-3 protein for mechanistic studies. The half life of exogenously expressed MKP-3 protein was reduced from 46 minutes to 31 minutes by insulin treatment (Figure 1D). The half life of endogenous MKP-3 protein was reduced from 41 minutes to 28 minutes by insulin treatment.
Figure 1. Insulin promotes MKP-3 protein degradation.

Fao cells were infected with Ad-MKP-3 for 48 hours followed by incubation in serum free medium overnight. Next day, cells were pretreated with 10 M cycloheximide for 30 min, followed by treatment with vehicle or 50nM insulin plus 10 M cycloheximide for indicated time points. A. Western blots of pS6, tS6, pAkt, tAkt, pERK, tERK, exogenously expressed MKP-3, endogenous MKP-3 and tubulin. B. Quantification of degradation of exogenously expressed MKP-3 protein in the presence of vehicle or insulin. C. Quantification of degradation of endogenous MKP-3 protein in the presence of vehicle or insulin. D. Calculation of half life of exogenously expressed MKP-3 protein in the presence of vehicle or insulin. E. Calculation of half life of endogenous MKP-3 protein in the presence of vehicle or insulin. *P<0.05, insulin treated vs. vehicle treated for the same time point. Veh, vehicle; Ins, insulin.
To determine whether the ERK pathway was responsible for promoting MKP-3 protein degradation, the constitutively active MEK (MEK S218DS222D, MEK CA) was over-expressed in Fao cells via adenovirus-mediated gene transfer to activate the ERK pathway. Overexpression of MEK CA significantly increased ERK phosphorylation and induced MKP-3 protein degradation at all time points examined (Figure 2A, supplemental figure 2A). Overexpression of MEK CA also significantly decreased expression of endogenous MKP-3 (Figure 2B, supplemental figure 2B). Consistent with this result, application of MEK inhibitor U0126 not only prevented insulin induced MKP-3 protein degradation but also markedly slowed down MKP-3 protein degradation in general compared to DMSO treated samples (Figure 2C, supplemental 2C), indicating that blockage of the ERK pathway also prevents MKP-3 protein degradation induced by factors other than insulin. In contrast, constitutive activation of the Akt pathway by over-expressing the N-terminally myristoylation signal-attached human Akt1 in a tetracyclin-off manner and the tetracycline controlled transactivator (tTA) does not affect MKP-3 protein degradation (Figure 3A, supplemental figure 3A). Basal phosphorylation level of S6, a substrate of the mTOR pathway, is high in Fao cells and cannot be further enhanced by insulin treatment (Figure 3B, supplemental figure 3B). Application of mTOR inhibitor rapamycin, which effectively blocked S6 phosphorylation, did not prevent insulin induced MKP-3 protein degradation (Figure 3B, supplemental figure 3B). These results suggest that effect of insulin on promoting MKP-3 protein degradation is most likely independent of the Akt and mTOR pathways.
Figure 2. Effect of the MEK/ERK pathway activation on MKP-3 protein degradation.

A. Activation of the ERK pathway and MKP-3 protein expression. Fao cells were co-infected with Ad-MKP-3 and Ad-MEK CA, the constitutively active MEK S218DS222D mutant, at 1:1 ratio for 48 hours followed by incubation in serum free medium overnight. Next day, cells were treated with 10 M cycloheximide for indicated time points. B. Activation of the ERK pathway and endogenous MKP-3 protein expression. Endogenous MKP-3 was immunoprecipitated from Ad-MEK CA infected cells 60 minutes after cycloheximide treatment. C. ERK inhibitor and MKP-3 protein expression. Fao cells were infected with Ad-MKP-3 for 48 hours followed by incubation in serum free medium overnight. Then cells were pre-treated with 10 M cycloheximide for 30 minutes, followed by treatment with insulin or vehicle plus cycloheximide for 60 minutes in the presence/absence of MEK inhibitor U0126.
Figure 3. Insulin-promoted MKP-3 protein degradation is independent of Akt and mTOR pathways.

A. Expression of the constitutively active N-terminally myristoylation signal-attached human Akt1 in a tet-off manner and the tetracycline controlled transactivator in Fao cells via adenovirus mediated gene transfer and the effect on MKP-3 protein degradation. B. Effect of mTOR inhibitor rapamycin on MKP-3 protein degradation.
3.2. ERK phosphorylation resistant MKP-3 mutant is not sensitive to insulin induced decrease of MKP-3 protein
It has been reported that ERK2 phosphorylates MKP-3 on serine residues 159 and 197 in response to serum in the Chinese hamster CCL39 fibroblast cell line (Marchetti et al., 2005). Mutation of these two serine residues to alanines protected MKP-3 protein from serum induced proteasomal degradation. To determine whether serine residues 159 and 197 are the target sites for insulin induced degradation of MKP-3 protein, adenoviruses expressing MKP-3 or MKP-3 S159AS197A were used to transduce Fao cells followed by treatment of insulin in the presence of 10 M cycloheximide. As shown in figure 4A–B, the wild type MKP-3 protein was significantly reduced by insulin treatment compared to vehicle control but MKP-3 S159AS197A protein level remained unchanged in response to insulin treatment. The protein level of MKP-3 S159AS197A is always higher than that of wild type MKP-3 when Fao cells were transduced with equal amount of viruses, indicating that the MKP-3 mutant may degrade slower compared to wild type even in the absence of insulin. Interestingly, insulin induced ERK phosphorylation was partially attenuated when Ad-MKP-3 S159AS197A was expressed (Figure 4A–B), which is possibly due to sustained feedback dephosphorylation by the mutant. The attenuated ERK phosphorylation possibly contributes to decreased degradation of MKP-3 S159AS197A. Next, the ERK pathway was activated by over-expressing MEK CA in Fao cells and the effect on protein degradation of MKP-3 S159AS197A was examined. The double mutant demonstrated complete resistance to MEK CA induced MKP-3 protein degradation while wild type MKP-3 was reduced by 82% (Figure 4C–D, the left panels). To determine which ERK phosphorylation site is important in mediating MEK CA induced MKP-3 protein degradation, MKP-3 S159A and MKP-3 S197A single mutants were expressed in Fao cells. Mutation of serine 159 to alanine resulted in a 45% reduction of MKP-3 protein upon MEK CA expression and mutation of serine 197 to alanine led to a 72% of MKP-3 protein reduction under identical condition (Figure 4C–D, the right panels). These results indicate that both residues are required for MEK CA induced MKP-3 protein degradation and serine 159 plays a bigger role than that of serine 197. Addition of U0126 slowed down degradation of wild type MKP-3 protein but did not have any significant effect on MKP-3 S159AS197A protein degradation (Figure 5, first four lanes/ bars). Consistently, U0126 also slowed down degradation of wild type MKP-3 protein when the ERK pathway was constitutively turned on but had no significant effect on MKP-3 S159AS197A protein degradation under identical condition (Figure 5, last four lanes/bars).
Figure 4. Effect of insulin and ERK activation on degradation of MKP-3 mutant protein deficient in ERK phosphorylation site(s).
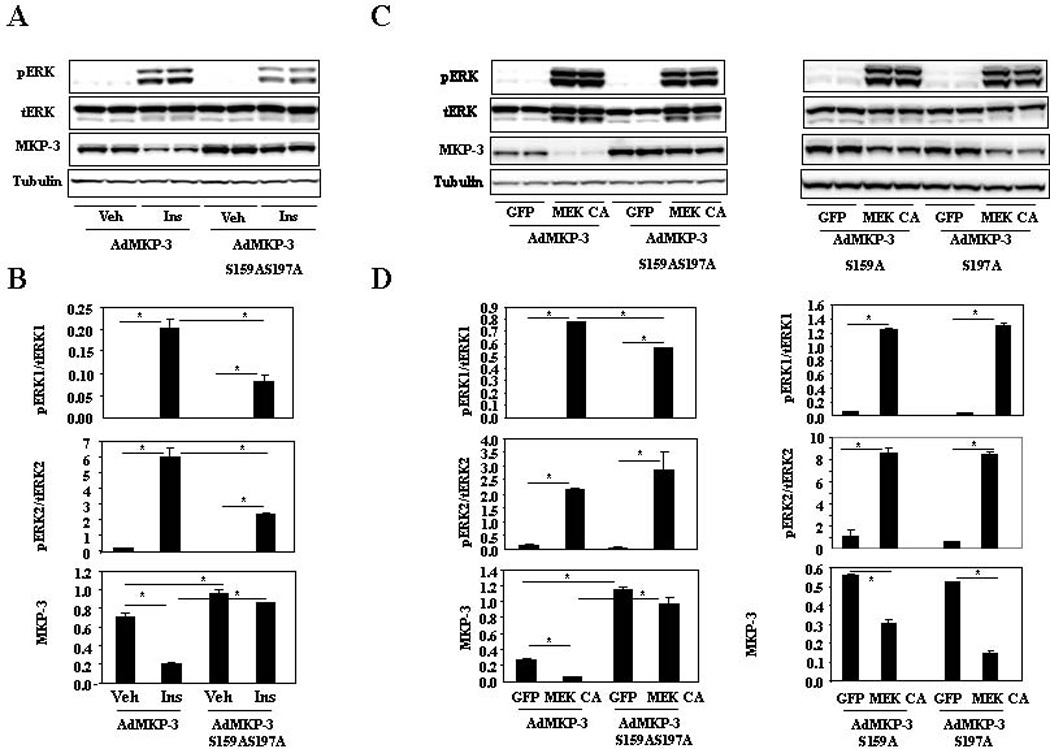
A. MKP-3 S159AS197A mutant is resistant to insulin-induced protein degradation. B. Quantification of western blots as shown in A. C. MKP-3 S159AS197A mutant is resistant to MEK CA induced protein degradation while MKP-3 S159A and MKP-3 S197A mutants can still be partially degraded. D. Quantification of western blots as shown in C. *, P<0.05 between groups as indicated on the graphs.
Figure 5. Effect of MEK inhibitor on degradation of MKP-3 mutant protein deficient in ERK phosphorylation sites.
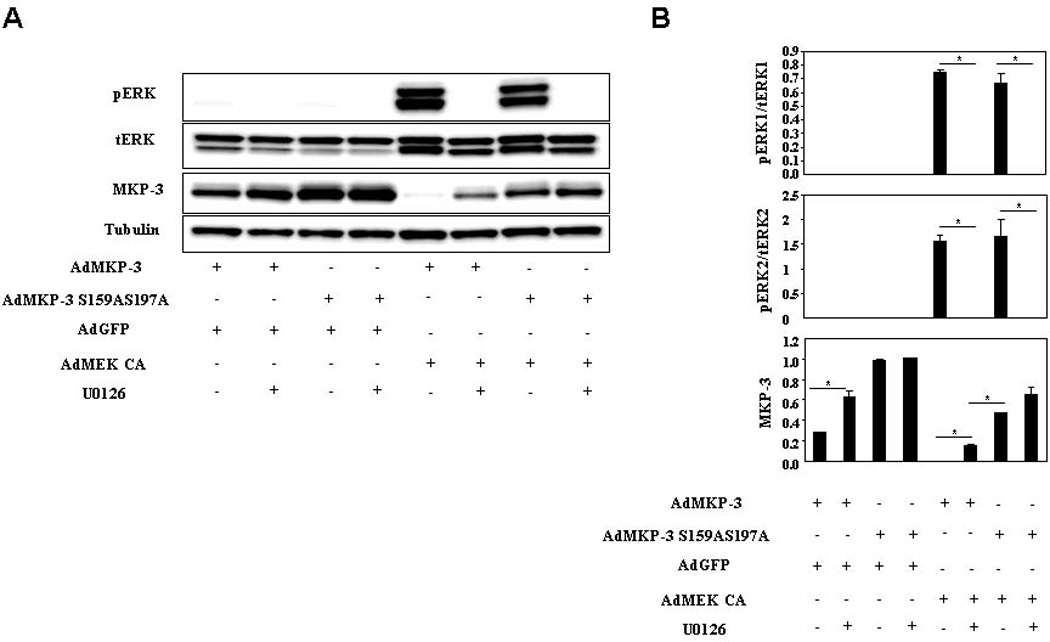
A. Fao cells were co-infected with Ad-MKP-3 or Ad-MKP-3 S159AS197A and Ad-GFP or Ad-MEK CA at 1:1 ratio. Forty eight hours after infection, cells were incubated in serum free medium overnight. Next day cells were treated with 10 M cycloheximide plus vehicle or 100 M U0126 for 30 minutes. B. Quantification of western blots as shown in A. *, P<0.05 between groups as indicated on the graphs.
3.3. Activation of ERK pathway represses expression of gluconeogenic genes and glucose output in Fao cells
We previously reported that MKP-3 is an important regulator in glucose homeostasis (Wu et al., 2010; Xu et al., 2005). To determine whether the ERK pathway affect glucose output in Fao cells by regulating MKP-3 protein degradation, MEK-CA was over-expressed in Fao cells. Constitutive activation of the MEK/ERK pathway significantly decreased the expression of G6pase gene and reduced glucose output (Figure 6A), consistent with our previous publication that knocking down MKP-3 expression in liver cells decreases glucose output. Furthermore, blockade of the ERK pathway by MEK inhibitor U0126 significantly increased the expression of G6Pase gene and promoted glucose output. (Figure 6B), consistent with our previous result that MKP-3 over-expression in this cell type promotes glucose output. Insulin is the major hormone suppressing hepatic glucose output. ERK phosphorylation deficient MKP-3 is resistant to insulin induced protein degradation, suggesting that MKP-3 S159AS197A may antagonize the effect of insulin on decreasing glucose output. To test this hypothesis, MKP-3 S159AS197A was over-expressed in Fao cells to examine G6Pase gene expression and glucose output. As shown in figure 6C, MKP-3 S159AS197A behaved like wild type MKP-3 in promoting G6Pase gene expression and glucose output in the absence of insulin (Figure 6C). When insulin was added, the effect of MKP-3 wild type on promoting G6Pase gene expression and glucose output was mostly repressed but the effect of MKP-3 S159AS197 was largely unaffected (Figure 6C).
Figure 6. Effect of MEK/ERK pathway on expression of G6Pase gene and glucose output.

A. Effect of ERK activation on expression of G6Pase gene and glucose output. *, P<0.05, cells expressing MEK CA vs. control cells expressing GFP. B. Effect of ERK inactivation on expression of G6Pase gene and glucose output. *, P<0.05, cells treated with U0126 vs. cells treated with DMSO. C. Effect of MKP-3 S159AS197A on expression of G6Pase gene and glucose output in the presence/absence of insulin. *, P<0.05, compared to cells expressing GFP under the same treatment condition; #, P<0.05, compared to cells expressing wild type MKP-3 under the same treatment condition.
4. Discussion
Our recent publications demonstrate that MKP-3 has a previously unrecognized role in promoting glucose output in liver cells and in the liver of lean and obese mice. Manipulation of MKP-3 expression in the liver of lean and obese mice has an impact on blood glucose levels, therefore making MKP-3 a potential therapeutic target for treating obesity-related hyperglycemia. The fact that MKP-3 protein expression can be regulated by hormones critical for maintaining blood glucose levels within a normal range makes it an interesting protein to study the mechanism of expression regulation. In our current study, we focused on investigating the mechanism of insulin repressed MKP-3 protein expression in cultured liver cells, a cell type important for glucose metabolism but in which regulation of MKP-3 expression has never been investigated. Unlike some of the early genes of the DUSP family, such as MKP-1 and MKP-2 which can be rapidly induced by growth factors and stress in an ERK dependent manner (Brondello et al., 1999; Hirsch and Stork, 1997; Misra-Press et al., 1995), the regulation of MKP-3 expression is complicated and different results have been reported for different cell lines. In PC12 neurons, MKP-3 expression can be rapidly induced by nerve growth factor (NGF) and fibroblast growth factor (FGF). Upregulation of MKP-3 in this cell type plays a critical role in inactivating ERK in cytosol during essential stages of NGF-mediated PC12 differentiation (Camps et al., 1998). MKP-3 expression can also be rapidly induced by FGF in NIH3T3 cells (Ekerot et al., 2008). In embryonic stem cell line P12, induction of MKP-3 parallels ERK activation (Reffas and Schlegel, 2000). In contrast, MKP-3 is constitutively expressed in human skin fibroblasts and can not be induced by stress or mitogens (Groom et al., 1996). In addition to upregulation by growth factors or constitutive expression, MKP-3 can also be down-regulated by growth factors. In hamster fibroblast cells, serum promotes MKP-3 protein degradation in both MEK/ERK and PI3K/mTOR dependent pathways (Bermudez et al., 2008; Marchetti et al., 2005). Interestingly, platelet-derived growth factor (PDGF) BB regulates MKP-3 protein in a biphasic manner, promoting MKP-3 protein degradation in the early phase and inducing MKP-3 protein synthesis in the later phase (Jurek et al., 2009).
We chose to study the stability of MKP-3 protein upon insulin treatment due to two reasons: 1) Decreased MKP-3 protein expression upon insulin treatment is not accompanied by decreased MKP-3 gene expression, excluding the possibility of reduction of transcription; 2) Previous reports showed that serum repressed MKP-3 protein expression was attributable to decreased MKP-3 protein stability (Bermudez et al., 2008; Marchetti et al., 2005). Therefore, MKP-3 was over-expressed in Fao cells and degradation was followed upon blockade of protein synthesis by addition of cycloheximide.
The effect of insulin on promoting MKP-3 protein degradation was first confirmed in cultured Fao hepatoma cells. We want to point out that MKP-3 is a fast turn over protein and the majority of degradation occurred in our system is due to absence of new protein synthesis due to addition of cycloheximide, supported by evident MKP-3 protein degradation in vehicle treated cells. However, insulin did accelerate MKP-3 protein degradation under identical conditions as evidenced by a shorter half life and lower MKP-3 protein levels 30 and 60 minutes post insulin treatment. Then, three classical insulin stimulated pathways, MEK/ERK, PI3K/Akt and mTOR/S6 were examined individually for their potential involvement in insulin promoted MKP-3 degradation by either constitutive activation of the pathways or blockade of the pathways. Our results indicate that the MEK/ERK pathway is mainly responsible for stimulating MKP-3 protein degradation upon insulin treatment. This is different from a previous publication, in which both MEK/ERK and PI3K/mTOR pathways were reported to be involved in serum induced MKP-3 protein degradation in hamster fibroblast cells (Bermudez et al., 2008). The discrepancy may be due to differential activation of signaling pathways in two completely different cell types. In CCL39 hamster fibroblast cells, insulin alone actually was not able to induce ERK phosphorylation. In Fao hepatoma cells, insulin stimulates strong ERK phosphorylation but does not activate mTOR pathway as reflected by unchanged S6 phosphorylation.
We further confirmed that the MEK/ERK pathway is responsible for insulin promoted MKP-3 protein degradation by demonstrating that the MKP-3 mutant deficient in serine residues 159 and 197, two sites for ERK dependent phosphorylation, is resistant to protein degradation induced by insulin or constitutive activation of the ERK pathway. Both serine residues are required for this action since mutation of individual residue protected MKP-3 protein from degradation to a lesser extent compared to the double mutant. Although it has been reported that serine 174 and serine 300 are important for PDGF promoted MKP-3 protein degradation in a MEK dependent manner (Jurek et al., 2009), we did not follow up further on these two residues since MKP-3 S159AS197A is completely resistant to insulin and MEK CA induced degradation. Consistently, application of U0126 only increases wild type MKP-3 protein in the presence of insulin or under the condition of MEK CA over-expression but has no effect on ERK phosphorylation resistant MKP-3 S159AS197A mutant. In addition, ERK dependent regulation of MKP-3 degradation has an effect on gluconegenic gene expression and glucose output in Fao cells. These results provide novel information regarding regulation of MKP-3 expression in a cell type important for glucose metabolism.
Highlights.
In this study, we examined the mechanism of insulin promoted MKP-3 protein degradation in cultured liver cells.
We used both gain and loss-of-function experiments to demonstrate that the MEK/ERK pathway, not the PI3/Akt or the mTOR/S6 pathway, mediates the effect of insulin on degrading MKP-3 protein.
We further confirmed that ERK phosphorylation deficient MKP-3 mutant is resistant to insulin promoted protein degradation.
We also showed that the MEK/ERK pathway affects glucose output, possibly through regulating MKP-3 protein level.
Supplementary Material
Supplemental figure 1. Quantification of S6, Akt and ERK phosphorylation by normalization to total protein.
{kind=link}
Supplemental figure 2, quantification of western blots shown in figure 2.
{kind=link}
Supplemental figure 3, Quantification of western blots shown in figure 3
{kind=link}
Acknowledgements
This work was supported by NIDDK R01 DK080746 awarded to H.Xu. B. Feng received a scholarship from China Scholarship Council.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
No conflict of interest exists. The funding resources play no role in the manuscript.
References
- Bermudez O, Marchetti S, Pages G, Gimond C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene. 2008;27:3685–3691. doi: 10.1038/sj.onc.1211040. [DOI] [PubMed] [Google Scholar]
- Brondello JM, Pouyssegur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999;286:2514–2517. doi: 10.1126/science.286.5449.2514. [DOI] [PubMed] [Google Scholar]
- Camps M, Chabert C, Muda M, Boschert U, Gillieron C, Arkinstall S. Induction of the mitogen-activated protein kinase phosphatase MKP3 by nerve growth factor in differentiating PC12. FEBS Lett. 1998;425:271–276. doi: 10.1016/s0014-5793(98)00250-6. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, Keenan ID, Dickinson RJ, Storey KG, Keyse SM. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412:287–298. doi: 10.1042/BJ20071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjeld CC, Rice AE, Kim Y, Gee KR, Denu JM. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J Biol Chem. 2000;275:6749–6757. doi: 10.1074/jbc.275.10.6749. [DOI] [PubMed] [Google Scholar]
- Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. Embo J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- Hirsch DD, Stork PJ. Mitogen-activated protein kinase phosphatases inactivate stress-activated protein kinase pathways in vivo. J Biol Chem. 1997;272:4568–4575. doi: 10.1074/jbc.272.7.4568. [DOI] [PubMed] [Google Scholar]
- Jurek A, Amagasaki K, Gembarska A, Heldin CH, Lennartsson J. Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J Biol Chem. 2009;284:4626–4634. doi: 10.1074/jbc.M808490200. [DOI] [PubMed] [Google Scholar]
- Lang R, Hammer M, Mages J. DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol. 2006;177:7497–7504. doi: 10.4049/jimmunol.177.11.7497. [DOI] [PubMed] [Google Scholar]
- Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008;283:31246–31255. doi: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti S, Gimond C, Chambard JC, Touboul T, Roux D, Pouyssegur J, Pages G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol Cell Biol. 2005;25:854–864. doi: 10.1128/MCB.25.2.854-864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra-Press A, Rim CS, Yao H, Roberson MS, Stork PJ. A novel mitogen-activated protein kinase phosphatase. Structure, expression, and regulation. J Biol Chem. 1995;270:14587–14596. doi: 10.1074/jbc.270.24.14587. [DOI] [PubMed] [Google Scholar]
- Mourey RJ, Vega QC, Campbell JS, Wenderoth MP, Hauschka SD, Krebs EG, Dixon JE. A novel cytoplasmic dual specificity protein tyrosine 21 phosphatase implicated in muscle and neuronal differentiation. J Biol Chem. 1996;271:3795–3802. doi: 10.1074/jbc.271.7.3795. [DOI] [PubMed] [Google Scholar]
- Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- Reffas S, Schlegel W. Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem J. 2000;352(Pt 3):701–708. [PMC free article] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-reviews3009. REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiland AM, Denu JM, Mourey RJ, Dixon JE. Purification and kinetic characterization of the mitogen-activated protein kinase phosphatase rVH6. J Biol Chem. 1996;271:33486–33492. doi: 10.1074/jbc.271.52.33486. [DOI] [PubMed] [Google Scholar]
- Wu Z, Jiao P, Huang X, Feng B, Feng Y, Yang S, Hwang P, Du J, Nie Y, Xiao G, Xu H. MAPK phosphatase-3 promotes hepatic gluconeogenesis through dephosphorylation of forkhead box O1 in mice. J Clin Invest. 2010;120:3901–3911. doi: 10.1172/JCI43250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang Q, Shen M, Huang X, Dembski M, Gimeno R, Tartaglia LA, Kapeller R, Wu Z. Dual specificity MAP kinase phosphatase 3 activates PEPCK transcription and increases gluconeogenesis in rat hepatoma cells. J Biol Chem. 2005 doi: 10.1074/jbc.M508027200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zhang ZY. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J Biol Chem. 2001;276:32382–32391. doi: 10.1074/jbc.M103369200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. Quantification of S6, Akt and ERK phosphorylation by normalization to total protein.
Supplemental figure 2, quantification of western blots shown in figure 2.
Supplemental figure 3, Quantification of western blots shown in figure 3