

Sir, We read with great interest the report of a Tunisian family by Rouzier et al. (2011) describing the neurological disorder linked to a novel heterozygous missense mutation in MFN2 (1p36.2) (Rouzier et al., 2011). MFN2 mutations typically cause autosomal dominant axonal Charcot–Marie–Tooth disease (CMT2A, OMIM 609260), with peripheral nerve degeneration occasionally associated with visual failure and optic atrophy (Zuchner et al., 2004, 2006). Interestingly, the clinical manifestations among mutational carriers in this Tunisian family were even more variable, ranging from asymptomatic subclinical disease to an axonal sensorimotor neuropathy complicated by optic atrophy, deafness, cerebellar ataxia and proximal myopathy. Furthermore, the intriguing finding of cytochrome c oxidase (COX)-deficient fibres and multiple mitochondrial DNA deletions in skeletal muscle biopsies suggest that MFN2 mutations can result in disturbed mitochondrial DNA maintenance and an overt respiratory chain defect, in addition to marked fragmentation of the mitochondrial network. These deleterious consequences are strikingly reminiscent of the pathological features recently highlighted in Brain for autosomal dominant optic atrophy due to OPA1 mutations (Amati-Bonneau et al., 2008; Hudson et al., 2008; Yu-Wai-Man et al., 2010a). Here, we provide additional evidence that MFN2-associated neuropathy is a novel disorder of mitochondrial DNA maintenance in a study of 58 probands with CMT2A and confirmed MFN2 mutations (Table 1), compared with 131 age-matched normal controls.
Table 1.
MFN2 mutations identified in our patient cohort
| Mutation | Type | Functional domain | Number of patients |
|---|---|---|---|
| p.Glu65Stop | Nonsense | – | 3 |
| p.Arg94Trp | Missense | – | 8 |
| p.Arg94Gln | Missense | – | 2 |
| p.Ala100Gly | Missense | GTPase | 1 |
| p.Arg104Leu | Missense | GTPase | 3 |
| p.Arg104Trp | Missense | GTPase | 1 |
| p.Thr105Ala | Missense | GTPase | 1 |
| p.His165Tyr | Missense | GTPase | 6 |
| p.Gly202Ala | Missense | GTPase | 4 |
| p.Thr206Ile | Missense | GTPase | 2 |
| p.Thr232Asn | Missense | GTPase | 1 |
| p.Arg250Gln | Missense | GTPase | 1 |
| p.Arg259Cys | Missense | GTPase | 1 |
| p.Arg280His | Missense | GTPase | 1 |
| p.Gly298Arg | Missense | GTPase | 1 |
| p.Glu308Stopa | Nonsense | GTPase | 1 |
| p.Arg364Trp | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Arg364Pro | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Arg364Gln | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Met376Val | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Met376Ile | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Ala383Val | Missense | Coiled-Coil 1 (CC1) | 1 |
| p.Arg468His | Missense | – | 2 |
| p.Asp496Gly | Missense | – | 2 |
| p.Arg519Proa | Missense | – | 1 |
| p.Leu673Pro | Missense | – | 3 |
| p.Val705Ile | Missense | Coiled-Coil 2 (CC2) | 2 |
| p.Arg707Trp | Missense | Coiled-Coil 2 (CC2) | 1 |
| p.Arg707Pro | Missense | Coiled-Coil 2 (CC2) | 1 |
| p.Ala716Thr | Missense | Coiled-Coil 2 (CC2) | 1 |
| p.His750Pro | Missense | – | 1 |
| p.Gln751Stop | Nonsense | – | 1 |
| p.Tyr752Stop | Nonsense | – | 1 |
aOne patient harboured two heterozygous MFN2 mutations.
Total genomic DNA was extracted from the leucocyte fraction of venous blood samples. The average cellular mitochondrial DNA content was quantified with a SYBR Green™ quantitative polymerase chain reaction assay on a MyiQ™ real-time polymerase chain reaction detection system (Biorad), with MTND1 as the mitochondrial template and GAPDH as the nuclear-encoded housekeeping template (Yu-Wai-Man et al., 2010a). Relative mitochondrial DNA copy number was derived from the difference in threshold cycle (Ct) values obtained for MTND1 and GAPDH using the 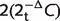 equation to account for two copies of GAPDH per cell nucleus.
equation to account for two copies of GAPDH per cell nucleus.
Mitochondrial DNA levels in the MFN2 group [mean mitochondrial DNA copy number = 195.7, standard deviation (SD) = 126.6, n = 58] were significantly higher compared with controls (mean mitochondrial DNA copy number = 60.9, SD = 42.3, n = 131, P < 0.0001) (Fig. 1A). Given the suggestion by Rouzier et al. (2011) that MFN2 mutations involving the functional GTPase domain were more likely to precipitate the severe multi-system phenotype documented in their family, we performed a subgroup analysis based on whether or not patients in our cohort harboured mutations within the highly conserved GTPase gene region. No significant difference was found between these two distinct mutational subgroups (P = 0.5957) (Fig. 1B).
Figure 1.

Comparison of mitochondrial DNA (MtDNA) blood copy number: (A) MFN2 mutational carriers compared with age-matched normal controls; (B) patients harbouring MFN2 mutations within the GTPase domain (mean mitochondrial DNA copy number = 207.9, SD = 150.2, n = 0 24) compared with those located outside this region (mean mitochondrial DNA copy number = 189.8, SD = 108.2, n = 34); ***P < 0.0001; ns = not significant at P = 0.5957.
Although we have previously shown that variation in the differential blood cell count can affect blood-derived mitochondrial DNA copy number (Pyle et al., 2010), the 3-fold increase in mitochondrial DNA content detected in our MFN2 cohort was substantially greater than the error attributable to this possible confounding factor. Mitochondrial proliferation is a well-recognized important diagnostic feature in skeletal muscle of patients with a range of mitochondrial cytopathies (Taylor et al., 2004; Aure et al., 2006). This compensatory mechanism is thought to occur in response to an underlying cellular bioenergetic crisis (the ‘sick mitochondrion hypothesis’), which leads to the classical ‘ragged-red fibre’ appearance on Gomori Trichrome or succinate dehydrogenase staining (Chinnery and Samuels, 1999; Capps et al., 2003; Durham et al., 2007). In this study, we have demonstrated the same phenomenon in blood leucocytes derived from patients with MFN2 mutations. Although Rouzier et al. (2011) determined mitochondrial DNA copy number in four skeletal muscle biopsies, they only report on the absence of mitochondrial DNA depletion in homogenate muscle extracts (Table 1). Pathologically increased mitochondrial DNA levels have been detected in laser-microdissected single skeletal muscle fibres from patients with OPA1 mutations, this effect being particularly marked in COX-deficient muscle fibres (Yu-Wai-Man et al., 2010b). Given the overlapping clinical, histological and molecular characteristics observed in these two primary disorders of mitochondrial dynamics, it would be of great interest to know whether mitochondrial DNA proliferation was also present in muscle fibres from patients with MFN2 mutations, especially in the two biopsies noted to have ragged-red fibres.
MFN2 is the newest member of an expanding group of nuclear mitochondrial disorders characterized by disturbed mitochondrial DNA maintenance, a process which, increasingly, seems to be intrinsically related to the state of the mitochondrial network (Chen et al., 2010; Elachouri et al., 2011). Our observation that MFN2 mutations cause mitochondrial proliferation in blood adds weight to the novel disease mechanism reported by Rouzier et al. (2011). Future work is needed to disentangle the complex interplay between disturbed mitochondrial fusion and fission, mitochondrial DNA instability and the eventual development of both neurological and visual deficits in patients with CMT2A and MFN2 mutations.
Funding
P.Y.W.M. is a Medical Research Council (MRC, UK) Clinician Scientist. R.H. is supported by the Academy of Medical Sciences (UK) and by the MRC (UK). P.S. is funded by IGA MH CR No 10554-3. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science and a UK National Institute of Health Senior Investigator who also receives funding from the MRC (UK), Parkinson's UK, the Association Francaise contre les Myopathies, and the UK NIHR Biomedical Research Centre for Ageing and Age-related Disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust.
References
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy plus phenotypes. Brain. 2008;131:338–51. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- Aure K, Fayet G, Lacene E, Romero NB, Lombes A. Apoptosis in mitochondrial myopathies is linked to mitochondrial proliferation. Brain. 2006;129:1249–59. doi: 10.1093/brain/awl061. [DOI] [PubMed] [Google Scholar]
- Capps GJ, Samuels DC, Chinnery PF. A model of the nuclear control of mitochondrial DNA replication. J Theor Biol. 2003;221:565–83. doi: 10.1006/jtbi.2003.3207. [DOI] [PubMed] [Google Scholar]
- Chen HC, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–9. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Samuels DC. Relaxed replication of mtDNA: a model with implications for the expression of disease. Am J Hum Genet. 1999;64:1158–65. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham SE, Samuels DC, Cree LM, Chinnery PF. Normal levels of wild-type mitochondrial DNA maintain cytochrome c oxidase activity for two pathogenic mitochondrial DNA mutations but not for m.3243A -> G. Am J Hum Genet. 2007;81:189–95. doi: 10.1086/518901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011;21:12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He LP, Schaefer AM, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–37. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- Pyle A, Burn DJ, Gordon C, Swan C, Chinnery PF, Baudouin SV. Fall in circulating mononuclear cell mitochondrial DNA content in human sepsis. Intensive Care Med. 2010;36:956–62. doi: 10.1007/s00134-010-1823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 2011 doi: 10.1093/brain/awr323. ; 135: 23–34. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord. 2004;14:237–45. doi: 10.1016/j.nmd.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010a;133:771–86. doi: 10.1093/brain/awq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, et al. OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Human Molecular Genetics. 2010b;19:3043–52. doi: 10.1093/hmg/ddq209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–81. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–51. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]