

Drug delivery via liposomes has the potential to expand the therapeutic window by targeting and releasing drug at the disease site while minimizing drug concentrations elsewhere in the body.[1] In a liposome, a single bilayer defines the interior space, regulates release of the liposome contents, and protects the contents from the environment. However, finding a bilayer composition that provides the necessary physical integrity, drug retention, specific targeting and rapid contents release at the disease site has been problematic. Despite 40 years of research, only liposomal doxorubicin and amphotericin B are clinically available. Many drugs, including the antibiotic ciprofloxacin,[2,3] are retained in liposomes for weeks to months in buffer, but are released within minutes in serum.[1,4–9] Phospholipids and/or cholesterol can be removed from liposomes by high-density lipoproteins (HDL), leading to the formation of defects in the bilayer and release of small molecules.[5,9] Lipases and other enzymes can selectively break down lipids within the bilayer[7]. A possible solution is to protect the drug-encapsulating bilayer by surrounding it with a second, protective bilayer shell in a “vesosome”.[7,10] The second bilayer increases the serum half-life of ciprofloxacin from <10 minutes in liposomes to 6 hours in vesosomes. PEG-lipid coating prevents vesosome aggregation in blood, leading to a ~ 2 hour half-life in the circulation. The biodistribution of vesosomes is similar to unilamellar liposomes as shown by near-infrared full body images. The vesosome structure may be a viable alternative for targeted delivery of weakly basic drugs, such as ciprofloxacin, that leak too rapidly from liposomes.
To encapsulate liposomes within a bilayer, we take advantage of the ethanol-induced interdigitated phase of saturated phosphatidylcholines. As 100 nm dipalmitoylphosphatidylcholine (DPPC) liposomes formed by extrusion above 41 °C in the Lα phase are cooled to room temperature, the alkyl chains of the lipids “freeze” and tilt to accommodate the area mismatch between the phosphocholine headgroups and the frozen alkyl chains to form the Lβ' phase[10,11] (Fig. 1). Adding 3 molar ethanol to the Lβ' phase swells the headgroup region, resulting in the formation of the interdigitated Lbi phase.[12] Interdigitation increases membrane rigidity and ruptures highly curved liposomes, leading to the formation of bilayer sheets[7,10,11] as shown by freeze-fracture transmission electron microscopy (FFTEM, Fig. 1a).[13] The interdigitated LbI phase has a single X-ray diffraction reflection at 0.41 nm (Fig. 1b), consistent with an untilted, hexagonal packing.[12] After the bulk ethanol is removed, the diffraction pattern changes to that of the tilted Lβ' phase, with a sharp reflection at 0.42 nm and a broad shoulder at 0.41 nm.[12] The bilayer sheets remain open after the ethanol is removed, even though the bilayer is in the Lβ' phase (Fig. 1d).
Fig. 1.
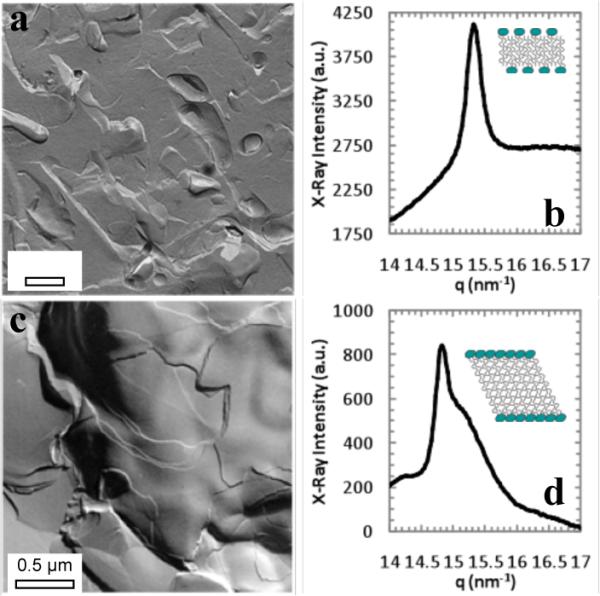
(a) FFTEM of interdigitated DPPC phase in 3M ethanol at room temperature. (b) X-ray diffraction of (a) shows a single reflection at q ≈ 15.3 nm−1 indicating an untilted hexagonal lattice (inset) with d ≈ 0.41 nm, consistent with the interdigitated LβI phase.[12] (c) FFTEM of Lβ' phase at room temperature following removal of ethanol from (a). The sheet structure remains. (d) A sharp reflection at q ≈ 14.8 nm−1 with a broad shoulder at q ≈ 15.25 nm−1, indicates a distorted hexagonal lattice with d-spacings of 0.41 nm and 0.42 nm, consistent with the tilted (inset) Lβ' bilayer phase re-forming after ethanol removal.[12]
The reduced bending rigidity that accompanies “melting” the lipid alkyl chains on heating above 41 °C into the Lα phase causes the bilayers to close; in the process, small vesicles and colloidal particles are encapsulated (Fig. S1).[7,10] On cooling to room temperature, the bilayers re-enter the Lβ' phase, but the vesosomes remain closed. This Lα - Lβ' - LbI - Lβ' - Lα - Lβ' phase progression for pure DPPC bilayers can accommodate small fractions (~ 6 mol%) of fluorescently labeled lipids and/or polyethylene glycol-lipids with PEG molecular weights varying from 750 to 2000 Da.
Pulse Gradient Stimulated Echo (PGSE) NMR techniques[14] can distinguish otherwise identical ciprofloxacin inside and outside liposomes without the need for labeling or extractions (Fig. 2), which makes it ideal for release studies. The solution-state, isotropic 1H and 19F chemical shifts of specific moieties of ciprofloxacin (Fig. 2) depend on the local concentration. For resolved signals in a PGSE NMR measurement, the measured echo-signal intensities, S, in the presence of a magnetic field gradient decay exponentially with the ciprofloxacin diffusivity, D:[14]
| (1) |
δ is the duration of a field gradient pulse of amplitude, g, γ is the gyromagnetic ratio of the 1H or or 19F nuclei, and Δ is the time between the initial and reversed gradient pulses during which diffusion can occur. Ciprofloxacin inside liposomes diffuses with the liposome, so that Din~10−7 to 10−9 cm2/s. By comparison, ciprofloxacin in solution has Dfree~10−5 cm2/s. By appropriate choice of Δ, the amplitude of the 1H or 19F signals corresponding to the fast diffusing, free ciprofloxacin decay, while those of the slow diffusing, encapsulated ciprofloxacin are relatively unchanged (Fig. 2b).[14,15]
Fig. 2.

(a) Molecular structure of ciprofloxacin with protons labeled. (b) (top) Solution-state 1H NMR spectrum showing signal from aromatic protons 2, 5 and 8 in a liposome suspension and from histidine used as a pH marker (Fig. S2). (bottom) Diffusion-filtered 1H PGSE NMR spectrum that shows only one signal from slow-diffusing ciprofloxacin within the liposomes. (c) Solution-state 19F NMR spectra from ciprofloxacin (top) showing the concentration dependence of the 19F isotropic chemical shift and (bottom) in a liposome suspension with signals corresponding to ciprofloxacin inside and outside of the liposomes as determined from a diffusion-filtered 19F PGSE NMR spectrum (not shown).
Neutral ciprofloxacin (pK = 6.2), like many other weakly basic drugs, permeates within minutes through bilayers; however, if the liposome interior is acidified, ciprofloxacin is protonated, the bilayer permeability is negligible, and ciprofloxacin accumulates inside the liposomes[16]. To create a pH gradient, liposomes are hydrated with either a 150 or 300mM ammonium sulfate, 10mM histidine solution prior to extrusion[16]. Histidine is a NMR pH probe (Fig. S2) and 1H PGSE NMR was used to follow the ciprofloxacin concentration and pH gradient (Fig. 2b). Exchange of the external ammonium sulfate solution for phosphate buffered saline resulted in an liposome internal pHin ≈ 4.5 for 150mM ammonium sulfate, and pHin ≈ 4.0 for the 300mM ammonium sulfate; pHext ≈ 5.6 in both cases. These pH gradients (1.1 and 1.7 pH units, respectively) were stable to 5% for a month. Ciprofloxacin was added to the external solution at a drug/lipid molar ratio of 0.4 and heated at 60°C for 15 or 30 minutes. Ciprofloxacin concentrations inside and outside the liposomes were determined by averaging the peak areas from the ciprofloxacin H5, H8, and H2 protons before and after PGSE diffusion filtering (Fig. 2a,b). 85 – 95% of the available ciprofloxacin was concentrated in the liposomes containing 300 mM ammonium sulfate while 50% was encapsulated in liposomes containing 150 mM ammonium sulfate; there was little difference between heating 15 or 30 minutes at 60°C. The fraction encapsulated was stable to within 5% for a month at room temperature, as was the pH gradient.[16]
To load vesosomes, ciprofloxacin was added to a vesosome suspension (Fig. S1) with the internal compartments containing 300 mM ammonium sulfate and heated at 60°C for 30 minutes (the external vesosome compartment and the surrounding solution contained buffer at pH 5.5).1H NMR showed that 90% of the ciprofloxacin in solution was encapsulated. As was the case for unilamellar liposomes (Fig. 2b), two peaks (at similar chemical shifts as for the liposomes in Fig. 2a) were observed in the 1H NMR spectrum and only one remained after diffusion-filtering via PGSE, corresponding to the entrapped ciprofloxacin. Had a significant amount of ciprofloxacin accumulated in the space between the outer and inner vesosome bilayers, a third peak would appear at a chemical shift corresponding to that concentration. However, only two peaks were observed, confirming that ciprofloxacin accumulated in the internal ammonium sulfate-containing compartments by permeating through two bilayers. The fraction encapsulated and pH were stable to within 5% for a month.
19F PGSE NMR was used to determine the half-life of ciprofloxacin in serum to avoid the overlapping 1H signals of the serum components. Liposomes or vesosomes were combined with freshly thawed calf serum in a 1:1 volume ratio, and NMR measurements were immediately initiated at 37°C, yielding two 19F signals, one at 119.75 ppm corresponding to high ciprofloxacin concentrations shown to be inside the liposomes or vesosomes by PGSE, and a low concentration signal that ranged from 120 – 122 ppm for free ciprofloxacin (Fig. 2c). Within 30 minutes (Fig. 3), the liposomes were empty, consistent with literature results for ciprofloxacin (and similar weakly basic drug) release from phospholipid/cholesterol liposomes.[3,4,6,17–19] In comparison, vesosomes released ~ 20% of their initial contents after exposure to 50% serum in the first 30 minutes. More than 40% of the encapsulated ciprofloxacin was retained after 10 hours in serum at 37°C; the half-life for release increased from < 10 minutes in liposomes to ~ 6 hours in vesosomes. Again, the absence of a third 19F signal, intermediate in chemical shift between the inside and outside ciprofloxacin concentrations (Fig. 2c), show that the external vesosome bilayer did not provide a barrier to ciprofloxacin release. During serum exposure, the pH of liposomes containing ciprofloxacin remained < 5 (Fig. S3), while the ciprofloxacin exterior to the liposomes was at pH 7. No intermediate values of the liposome pH were seen. These results suggests that liposome release is due to failure of the bilayer barrier which led to rapid (~ 1 minute between NMR measurements) equilibration of the liposome or vesosome contents and pH with the surrounding solution, rather than a gradual neutralization of the liposome interiors by permeation of the ammonium sulfate.
Fig. 3.
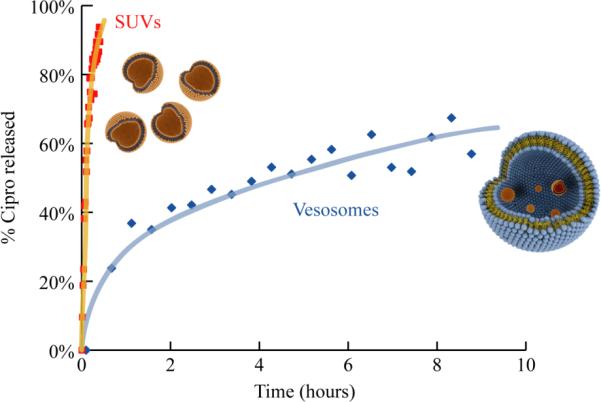
Fraction of ciprofloxacin released as determined from 19F NMR (Fig. 2c) at 37 °C in 50% calf serum from unilamellar DPPC/Chol liposomes and vesosomes. Ciprofloxacin release from liposomes is complete within 30 minutes while the half-life in vesosomes is ~ 6 hours. Lines are provided to guide the eye.
The external membrane of the vesosome increases retention by being a barrier, not to small molecules, but to entry of larger molecular weight enzymes, lipases, or complexes that disrupt the interior compartments. Phospholipase A2 (PLA2, 14 kDa) releases carboxyfluorescein (and other small molecules) in hours from liposomes, but the same small molecules are retained in vesosomes for days.[7] In serum, HDL and other lipoproteins can form defects in the bilayer sufficiently large for small molecules to be released.[5,9] However, due to the vesosome structure, HDL or PLA2 can generate defects in the interior compartments only if they can find a sufficiently large defect in the exterior bilayer that they can pass through. By this time, the external bilayer is sufficiently degraded that it would no longer serve as a barrier to small molecule release, consistent with the NMR results that show no ciprofloxacin accumulates in the space between the interior compartments and the external bilayer.
There is disagreement concerning whether one of the main functions of a PEG-lipid coating is to reduce liposome aggregation via steric repulsion, or to reduce plasma protein adsorption, which prevents opsonization and removal by the reticuloendothelial system (RES).[8] Fig. 4 shows fluorescently labeled vesosomes with and without 4 mol% DPPC-PEG750 in the exterior bilayer after 30 minutes in human blood at 37°C. Without the PEG coating, the vesosomes form multimicron aggregates that are stable to vortexing (Fig. 4a). With the PEG layer (Fig. 4b), the vesosomes remain dispersed in blood. To determine the circulation lifetime of the PEG750 coated vesosomes, Texas-red DHPE-labeled vesosomes were injected into the tail vein of wild-type mice. Blood was drawn at regular intervals from the optic vein and images (Figure S4) at uniform magnification were quantified for fluorescence intensity as a function of time post-injection. The overall fluorescence intensity measured from the images decayed exponentially with a half-life of 2 hours (Figure S5). Fig. 5 shows the size distribution over time of vesosomes counted from a typical set of images. The initial distribution of the vesosome population was quite broad, with particles ranging from less than 0.5 μm to > 5 μm. Some of the larger particles were likely aggregates as seen in Fig. 4. Large particles were removed from the circulation faster than the smaller particles; after 5 hours, the mean size of vesosomes in the blood decreased from 2.2 to 1.3 μm and almost all the particles with size > 2.5 μm were removed.
Fig. 4.
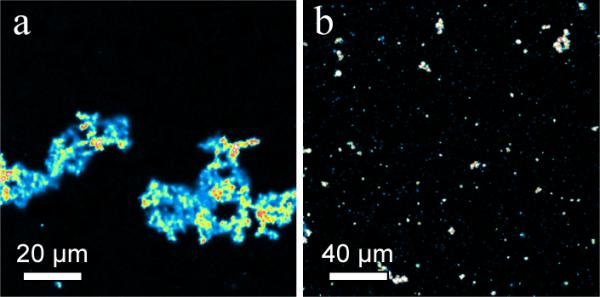
(a) Fluorescence image of aggregation of Texas-red DHPE labeled vesosomes after 30 minutes in human blood at 37 °C. (b) Fluorescence images of similar vesosomes as in (a) except for 4 mol% DPPE-PEG750 added to the outer bilayer shell. Aggregation is substantially reduced.
Fig. 5.
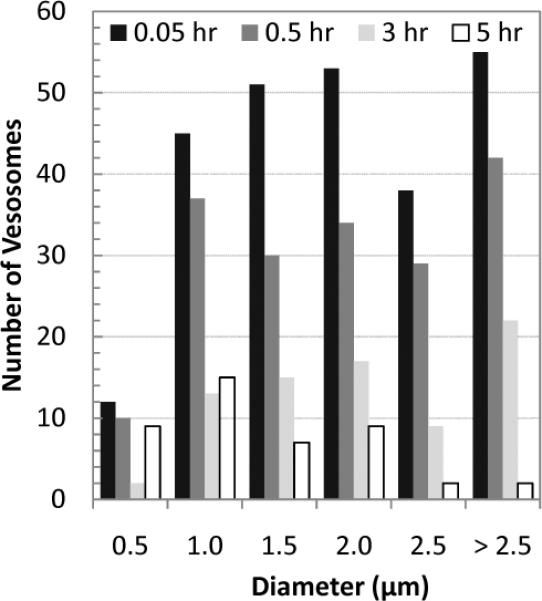
Size distribution of vesosomes with 4 mol% DPPE-PEG750 in the mouse circulation at different times after injection as measured from fluorescence images (Fig. S4). The half-life of > 2.5 μm diameter vesosomes is ~ 1 hour compared to the > 2 hours half-life for vesosomes < 2.5 μm.
To confirm the circulation lifetimes measured from the blood sampling, whole body, near-infrared fluorescence imaging was used to visualize the vesosome biodistribution in a living mouse (Fig. 6). Empty vesosomes were labeled on the exterior of the membrane with Alexa Fluor 750 (AF750, λex/λem 749nm/775nm) covalently linked to DSPE-PEG. Tissue autofluorescence was detected at 700 nm (red/orange) as shown in the pre-injection image; the AF750 labeled vesosomes were detected at 800 nm (green) in the post-injection images. The vasculature can be seen immediately following injection, and the green fluorescence intensity decreases as the vesosomes are cleared from the circulation. Highly-vascularized regions such as the face, paws, liver and spleen show the highest green fluorescence intensity. This biodistribution is similar to that reported for small unilamellar liposomes[1,4]. There is an absence of vesosome accumulation in the lungs, heart, and gut. The bladder is also visible in several of the images as the circular green region near the bottom, implying processing of the vesosomes through the kidneys (dorsal scans in Fig. S6). However, even at 3.5 hours, there is substantial green fluorescence visible throughout the larger vasculature, consistent with the ~2 hour half-life measured by blood sampling.
Fig. 6.
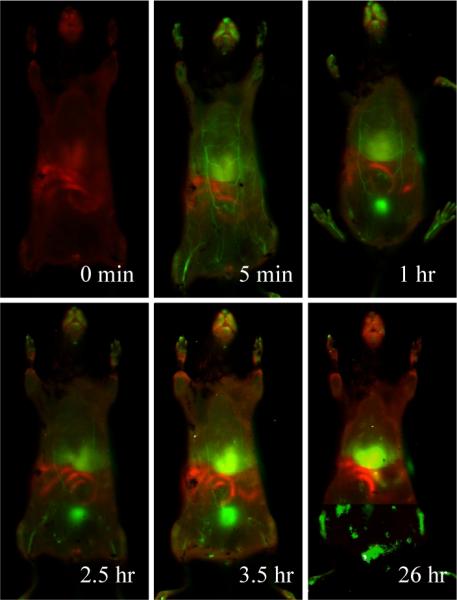
False-color near-infared fluorescence intensity maps of the ventral aspect of a mouse prior to injection with Alexa Fluor 750 labeled vesosomes and at 5 min, 1 hr, 2.5 hr and 3.5 hr, following injection. The red is body autofluorescence detected at 700 nm and the green is fluorescence detected at 800 nm due to the labeled vesosomes.
In summary, X-ray diffraction and freeze-fracture microscopy show that vesosome assembly occurs via a metastable bilayer phase progression of Lα - Lβ' - LbI - Lβ' - Lα - Lβ' as temperature and ethanol concentration are changed. In vesosomes, ciprofloxacin only accumulates in the interior compartments in response to a pH gradient. Both the encapsulated ciprofloxacin concentration and the pH gradient are stable for a month in buffer for both liposomes and vesosomes. In serum, the multilayered vesosome structure increased ciprofloxacin retention from < 10 minutes to ~ 6 hours compared to unilamellar liposomes of the same composition. Prior work on increasing ciprofloxacin (and similar weakly basic drugs) retention in liposomes has only achieved a maximum of ~1 hour,[6] which shows the advantage of the vesosome structure. PEG-lipid coating prevents the aggregation of vesosomes in blood,[8] leading to a half-life in the mouse circulation of 2 hours. Direct full body imaging of near infrared dye-labeled vesosomes show a similar circulation half-life and that the biodistribution of vesosomes is similar to that of conventional liposomes.
These results show a distinct advantage to adding a second bilayer to protect drug carrying compartments from blood components. The current generation of vesosomes would benefit from efforts to decrease the mean size, which would likely extend the in vivo circulation time. However, as ciprofloxacin retention (6 hr half-life) in vesosomes is significantly greater that the circulation half-life (2 hr), the vesosome might be useful as an actively targeted carrier. Non-specific vesosomes would have sufficient time to clear from the circulation and drug release would occur primarily from vesosomes retained at the target site.
Experimental
Materials and Methods
DPPC, DPPE-PEG750, DPPE-PEG2000, and cholesterol were purchased from Avanti Polar Lipids (Alabaster, Al). Lipid-conjugated fluorescent dyes NBD-HPC, Oregon Green DHPE, and Texas Red DHPE were purchased from Invitrogen. Alexa Fluor 750 was obtained from Invitrogen in its succinimidyl ester form, and conjugated to DSPE-PEG2000-NH2 followed by purification by HPLC. Ammonium sulfate and ciprofloxacin (98%) were purchased from Sigma-Aldrich (St. Louis). Newborn calf serum (Sigma #N4887) was thawed upon reception, divided into 10mL aliquots and stored frozen at −20°C.
Vesosome Preparation
DPPC and any necessary cholesterol, dyes or PEG-lipids were dissolved in chloroform in glass vials and the solvent removed by evaporation. The lipids were hydrated with the appropriate buffer at 55 °C, and 50 or 100 nm liposomes were prepared by standard extrusion methods using a Lipex Biomembranes Extruder (Vancouver, Canada).[16] DPPC or modified DPPC liposomes were transformed into interdigitated bilayer sheets by dropwise addition of ethanol (3 molar net ethanol concentration) to the liposome suspension at room temperature.[7,11] The interdigitated sheets were washed with buffer, followed by centrifugation. 55/45 DPPC/Chol liposomes to be encapsulated were added to the washed bilayer sheets and held at 55 °C for 20 minutes to induce encapsulation and form vesosomes. Vesosomes were separated from unencapsulated vesicles by size exclusion chromatography using Sephacryl S-1000 beads (GE Healthcare).
PGSE NMR
1H spectra were acquired at room temperature using a Varian Unity Inova 500 MHz NMR spectrometer. To eliminate the contribution from the water protons in the 1H spectra, the W5 water suppression sequence was used[20]. For following ciprofloxacin release in serum, the samples were spun in the 19F NMR probehead at ~ 20Hz and allowed to equilibrate at 37 °C for 4 minutes before acquisition began. Each time point corresponded to 16 scans accumulated over 53 seconds for the liposome samples, and 128 scans accumulated over 7 minutes for the vesosome samples.
Fluorescence microscopy
Whole human blood from healthy volunteers was mixed with EDTA before vesosomes were added to a final concentration of 1 mg/mL. The mixture was vortexed and left in a shaking water bath at 37°C for 30 minutes. Aliquots were drawn, placed on glass slides and imaged under a fluorescence microscope.
Animal Studies
All animal studies were conducted under a protocol approved by the University of California, Santa Barbara Institutional Animal Use and Care Committee (Santa Barbara, CA). 100 uL of a 2.5 mg/ml dispersion of Texas-red DHPE labeled vesosomes filtered through a 5 μm filter was administered through the tail vein of wild-type mice. Blood was drawn from the optic vein under full anesthesia at various intervals, chelated with EDTA, and a 5–10 μL drop was placed on a microscope slide and sealed with a cover slip. Fluorescence images at fixed magnification were quantified by total fluorescence intensity and by counting individual vesosomes.
For full body imaging, wild-type mice were limited to a high-rat, no cellulose diet to minimize background fluorescence. Hair was removed with a commercial hair removal agent prior to vesosome administration through the tail vein. After being anesthetized, the mouse was placed on an Odyssey Infrared Imaging System at regular intervals post-injection.
Supplementary Material
Acknowledgements
The work was supported in part by the US National Institutes of Health grants HL-080718 and EB-012637 and by the US National Science Foundation grant CBET-0829182. We are grateful to E. Ruoshlati for generous use of his laboratory for the in vivo fluorescence imaging.
References
- [1].Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. J. Pharm. Sci. 2008;97:4696. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- [2].Webb MS, Boman NL, Wiseman DJ, Saxon D, Sutton K, Wong KF, Logan P, Hope MJ. Antimicrobial Agents and Chemotherapy. 1998;42:45. doi: 10.1128/aac.42.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maurer-Spurej E, Wong KF, Maurer N, Fenske DB, Cullis PR. Biochimica et Biophysica Acta. 1999;1416:1. doi: 10.1016/s0005-2736(98)00204-1. [DOI] [PubMed] [Google Scholar]
- [4].Noble CO, Guo Z, Hayes ME, Marks JD, Park JW, Benz CC, Kirpotin DB, Drummond DC. Cancer Chemother. Pharmacol. 2009;64:741. doi: 10.1007/s00280-008-0923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Senior J, Gregoriadis G. Life Sciences. 1982;30:2123. doi: 10.1016/0024-3205(82)90455-6. [DOI] [PubMed] [Google Scholar]
- [6].Johnston MJW, Semple SC, Klimuk SK, Ansell S, Maurer N, Cullis PR. Biochimica et Biophyisca Acta. 2007;1768:1121. doi: 10.1016/j.bbamem.2007.01.019. [DOI] [PubMed] [Google Scholar]
- [7].Boyer C, Zasadzinski JA. ACS Nano. 2007;1:176. doi: 10.1021/nn7002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dos Santos N, Allen C, Doppen AM, Anantha M, Cox KAK, Gallagher RC, Karlsson G, Edwards K, Kenner G, Samuels L, Webb MS, Bally MB. Biochimica et Biophyisca Acta. 2007;1768:1367. doi: 10.1016/j.bbamem.2006.12.013. [DOI] [PubMed] [Google Scholar]
- [9].Rothblat GH, Phillips MC. Current Opinion in Lipidology. 2010;21:229. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kisak ET, Coldren B, Zasadzinski JA. Langmuir. 2002;18:284. [Google Scholar]
- [11].Ahl PL, Perkins WR. Methods in Enzymology. 2003;367:80. doi: 10.1016/S0076-6879(03)67007-2. [DOI] [PubMed] [Google Scholar]
- [12].Adachi T, Takahashi H, Ohki K, Hatta I. Biophysical Journal. 1995;68:1850. doi: 10.1016/S0006-3495(95)80361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zasadzinski JA, Bailey SM. J. Electron Microsc. Technique. 1989;13:309. doi: 10.1002/jemt.1060130406. [DOI] [PubMed] [Google Scholar]
- [14].Antalek B. Concepts in Magnetic Resonance. 2002;14:225. doi: 10.1002/cmr.10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Momot KI, Kuchel PW. Concepts in Magnetic Resonance Part A. 2006;28A:249. [Google Scholar]
- [16].Fenske DB, Cullis PR. Meth. Enzymology. 2005;291:7. doi: 10.1016/S0076-6879(05)91001-X. [DOI] [PubMed] [Google Scholar]
- [17].Maurer N, Wong KF, Hope MJ, Cullis PR. Biochimica et Biophysica Acta. 1998;1374:9. doi: 10.1016/s0005-2736(98)00125-4. [DOI] [PubMed] [Google Scholar]
- [18].Bakker-Woudenberg I, Schiffelers RM, Storm G, Becker MJ, Guo L. Methods in Enzymology. 2005;391:228. doi: 10.1016/S0076-6879(05)91014-8. [DOI] [PubMed] [Google Scholar]
- [19].Bakker-Woudenberg I, ten Kate MT, Guo L, Working P, Mouton JW. Antimicrobial Agents and Chemotherapy. 2001;45:1487. doi: 10.1128/AAC.45.5.1487-1492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu ML, Mao XA, Ye CH, Huang H, Nicholson JK, Lindon JC. J. Magnetic Resonance. 1998;132:125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.