

Abstract
ATP citrate lyase (ACL) catalyzes the conversion of cytosolic citrate to acetyl-CoA and oxaloacetate. A definitive role for ACL in tumorigenesis has emerged from ACL RNAi and chemical inhibitor studies, showing that ACL inhibition limits tumor cell proliferation and survival and induces differentiation in vitro. In vivo, it reduces tumor growth leading to a cytostatic effect and induces differentiation. However, the underlying molecular mechanisms are poorly understood and agents that could enhance the efficacy of ACL inhibition have not been identified. Our studies focus on non-small cell lung cancer (NSCLC) lines, which show phosphatidylinositol 3-kinase (PI3K)/AKT activation secondary to a mutation in the K-Ras gene or the EGFR gene. Here we show that ACL knockdown promotes apoptosis and differentiation, leading to the inhibition of tumor growth in vivo. Moreover, in contrast to most studies, which elucidate how activation/ suppression of signaling pathways can modify metabolism, we show that inhibition of a metabolic pathway “reverse signals” and attenuates PI3K/AKT signaling. Additionally, we find that statins, inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which act downstream of ACL in the cholesterol synthesis pathway, dramatically enhance the anti-tumor effects of ACL inhibition, even regressing established tumors. With statin treatment, both PI3K/AKT and the MAPK pathways are affected. Moreover, this combined treatment is able to reduce the growth of EGF receptor resistant tumor cell types. Given the essential role of lipid synthesis in numerous cancers, this work may impact therapy in a broad range of tumors.
In tumor cells, de novo fatty acid synthesis occurs at high rates (McAndrew, 1986; Swinnen et al., 2006; DeBerardinis et al., 2008). A number of relevant enzymes show both increased expression and activity, including ACL, HMG-CoA reductase, and fatty acid synthase (FAS) (Swinnen et al., 2006). The mechanisms by which this occurs are being elucidated and include HIF activation of FAS (Menendez et al., 2005) and AKT activation of ACL (Migita et al., 2008).
Non-small cell lung cancer (NSCLC) is a leading cause of cancer deaths (Zhang et al., 2003). A549 cells are derived from a NSCLC patient and bear a point mutation in K-Ras, which activates the PI3K/AKT pathway (Okudela et al., 2004). These cells are a non-epidermal growth factor receptor (EGFR) mutant cell line (Costa et al., 2007) and have been used in many studies in tumor metabolism (Christofk et al., 2008) and differentiation (Rho et al., 2009). We chose this cell line because it is an established model for NSCLC, it demonstrates the Warburg effect, and its growth can be inhibited by blockade of ACL (Bauer et al., 2005; Hatzivassiliou et al., 2005). We also chose EGFR mutant cell lines (H1650, H1975), which are sensitive or resistant to EGFR inhibitors, respectively, to test whether our findings have validity in a larger set of NSCLC lines.
Growth factors (such as EGF, insulin, and PDGF) lead to activation of the PI3K/AKT pathway and this in turn leads to increased enzymatic activity of ACL via AKT mediated ACL phosphorylation. A seminal observation on the functional role of ACL in tumor growth was made by the Thompson group, who reported that decreasing the expression of ACL by shRNA or its activity by a small molecule inhibitor suppressed tumor growth and promoted differentiation in numerous glycolytic tumors (Hatzivassiliou et al., 2005). However, the in vivo effects were cytostatic at best and the underlying mechanisms remain to be elucidated.
The abnormal activation of the PI3K/AKT pathway in human and animal models of cancer has been validated by epidemiological and experimental studies. Somatic gene alterations leading to the inactivation of the tumor suppressor gene PTEN and gain-of-function mutations targeting PIK3CA (the gene encoding the catalytic phosphoinositide-3 kinase subunit p110α) have been described (Yuan and Cantley, 2008). Many of the intracellular components of this pathway are being targeted in anti-cancer drug discovery and clinical trials of PI3K and AKT inhibitors are in progress (Engelman et al., 2008). Thus, understanding what events can intercept this pathway is of paramount importance. We show that blocking lipid synthesis can dampen signaling through this key oncogenic pathway.
Various mechanisms for the effects of statins on tumor cells have been suggested. Statins function in the mevalonate pathway as small-molecule inhibitors of HMG-CoA reductase (Hanai et al., 2007). Inhibition of this enzyme results in decreased isoprenylation, which includes farnesylation and geranylgeranylation of several proteins (such as Ras family small GTPases) essential for cellular proliferation and survival. Statins also inhibit dolichol synthesis, which is known to stimulate DNA synthesis (Larsson, 1993). Systemic cholesterol lowering by statins may interfere with cell growth via the impairment of cell membrane synthesis. A key finding of this paper is that statins dramatically enhance the anti-tumor effects of ACL inhibition, perhaps by downregulating both the PI3K/AKT and MAPK pathways.
Experimental Procedures
Viral constructs, antibodies, and reagents
An empty shRNA vector was used as a control and three different ACL shRNA lentiviruses (designated as 284, 285, and 286) were obtained from Open Biosystems (now ThermoFisher Scientific, Huntsville, AL). Anti-ACL, phospho-ACL, phospho-AKT 308, phospho-AKT 473, cyclin D1, AKT1, AKT2, p-Bad (Ser136), and cleaved caspase 3 antibodies were purchased from Cell Signaling (Danvers, MA). Anti-E-cadherin, ZO-1, vimentin, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Lovastatin was obtained from Sigma Aldrich (St Louis, MO). Wortmannin and LY294002 (PI3K inhibitor) were from Cell Signaling.
Cells and cell culture
A549 cells were purchased from the American Type Culture Collection and A549-luc-C8 (P/N 119266) from Caliper Life Sciences. These cells were maintained in Ham’s F-12 medium (Mediatech, Inc., Herndon, VA) supplemented with 10% FCS and penicillin/streptomycin (P/S). H1650 and H1975 cells were maintained in RPMI medium (Mediatech, Inc.) supplemented with 10% FCS and P/S (Kobayashi et al., 2005). 293FT cells were purchased from Invitrogen and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Gaithersburg, MD) supplemented with 10% FCS and P/S supplemented with MEM non-essential amino acids 1 mM, L-glutamine 6 mM, sodium pyruvate 1 mM, and geneticin 500 μg/ml. All cell lines were grown at 37°C in a humidified incubator with 5% CO2. Cells were grown to 60–70% confluency, harvested with trypsin, and resuspended to the cell density required for each assay.
Generation of ACL knockdown cell lines
A549 cells were infected with an empty shRNA vector as a control and three different ACL shRNA lentiviruses designated as 284, 285, and 286 in Figure 1A, which target three different regions of the human ACL mRNA. Recombinant lentiviral particles were produced by transient transfection of 293FT cells according to a standard protocol. Subconfluent 293FT cells were co-transfected with 3 μg of an shRNA plasmid, and 9 μg Viral Power packaging mix (an optimized proprietary mix of three plasmids, pLP1, pLP2, and pLP/VSVG from Invitrogen) using lipofectamine 2000 (Invitrogen). After 16 h, the cells were switched to regular growth medium and were allowed to incubate for an additional 48 h. Conditioned cell culture media containing recombinant lentiviral particles was harvested and frozen. A549 cells were treated with the above cell culture supernatant containing lentiviral particles for 24 h. These cells were then selected in puromycin to generate stable cell lines with empty vector shRNA and ACL specific shRNA. Cell lines were validated for diminished ACL expression by western blot (WB) analysis.
Fig. 1.

Characterization of ACL knockdown non-small cell lung cancer lines. (A–E, H, I, J: A549 cells; F, G, K: H1650 and H1975 cells). A: Western blot analysis of control shRNA cell line, ACL284 shRNA, ACL285 shRNA, and ACL286 shRNA cell lines by anti-ACL and β-actin antibodies. B: Morphology of control A549 cells and ACL knockdown cells (clone 285), showing mesenchymal spindle structure and epithelial cobble stone like-structure, respectively. C: Western blot analysis of control A549 cells and ACL knockdown cells (clone 285) by E-cadherin, ZO-1. Vimentin and GAPDH antibodies. D: Proliferation assay analysis of control A549 cells and ACL knockdown cells with or without lovastatin (1 μM). Error bar represent standard deviation and M denotes P-value < 0.01 when compared to control animal from the same day. E: Proliferation assay for H1650 cell lines (control, ACL284 shRNA, and ACL285 shRNA). Error bars represent standard deviation and M denotes P-value < 0.01 when compared to control cells from the same day. F: Proliferation assay of H1975 cell lines (control, ACL284 shRNA, and ACL285 shRNA). Error bar represent standard deviation and M denotes P-value < 0.01 when compared to control cells from the same day. G: Apoptosis assay analysis of control A549 cells and ACL knockdown cells. Error bar represent standard deviation and M denotes P-value < 0.01 when compared to control cells. H: Apoptosis assay analysis of control A549 cells and ACL knockdown cells with the indicated amount of lovastatin and with or without H2O2 incubation (1 μM). I: Cleaved caspase 3 and phospho-Bad (Ser136) expression in control A549 cells and ACL knockdown cells. J: Apoptosis assay for H1650 and H1975 cell lines (control, ACL284 shRNA, and ACL285 shRNA). [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcp]
Western blotting
Cultured cells after treatment were collected at specific times and solubilized in RIPA lysis buffer (Boston BioProducts) (Tris-HCl 50 mM, pH 7.4, NaCl 150 mM, NP-40 1%, sodium deoxycholate 0.5%, SDS 0.1%) or in Triton Lysis Buffer (Boston Bioproducts, Worcester, MA) (Tris-HCl 50 mM, pH 7.4, NaCl 150 mM, EDTA 5 mM, and TritonX 100 1%), with protease (Roche) and phosphatase (Active Motif) inhibitor cocktail. Proteins were separated by SDS-PAGE, transferred to PVDF membranes and detected using SuperSignal West Pico Chemiluminescent substrate (Pierce). For re-blotting, the membranes were stripped following the manufacturer’s protocol. Quantitative changes in protein phosphorylation were analyzed in triplicate from phospho-immuonoblot samples. Using densitometry software (ImageJ NIH) the signal intensities were quantitated along with each total protein blot (total AKT or AKT1+ AKT2, ERK, S6 protein, and ACL), which provided the baseline for signal normalization. Student’s t-tests were used to compare mean values as appropriate. The data are expressed accompanying each immunoblot as the mean values for a series of at least three experiments. The mean values are also shown in graphs as a % of maximum intensity along with the standard deviation. Each western blot shows representative data that was obtained from at least three independent experiments.
Apoptosis assay
Apoptosis in control and ACL knockdown cell lines was measured by harvesting cells and staining with Annexin-V-PE and 7AAD. Stained cells were analyzed by EasySite Plus Flowcytometer (Annexin assay, Guava Technologies, Hayward, CA). Apoptosis was also confirmed by cleaved caspase 3 blotting (western analysis).
Proliferation assay
Control and ACL knockdown cells lines were plated in 10 cm dishes at a density of 1 × 105 cells/dish in Ham’s F-12 medium supplemented with 10% FBS for 24 h at 37°C in 5% CO2. Cells were trypsinized 24, 48, 72, and 96 h after initial plating, and washed with PBS, resuspended in 1 ml of Hanks medium and counted in a hemocytometer. All samples were assayed in triplicate to generate proliferation curves.
Generation of tet-inducible ACL knockdown cell lines
We used the tet inducible expression vector (pTRIPZ) (Open BioSystems) that expresses the tetracycline transactivator and desired shRNA sequence under tetracycline response element (TRE) regulation. The pTRIPZ transactivator, known as the reverse tetracycline transactivator 3 (rtTA3) binds to and activates expression from TRE promoters in the presence of doxycycline. Unlike the original tetracycline transactivator, the rtTA3 is modified to bind to the TRE in the presence of doxycycline rather than in its absence. The TRE also drives the expression of a TurboRFP reporter in addition to the shRNAmir. The shRNAmir target sequences were cloned from pGIPZ into pTRIPZ by a simple restriction digest to generate the pTRIPZ ACL shRNAmir clones. Therefore, the target sequences used for generating the inducible shRNA were identical to those used in the non-inducible constructs. The constructs designed by this method required addition of doxycycline (1 μg/ml) for expression of tightly regulated induction of shRNAmir expression.
Tumor implantation
A549 control and ACL knockdown cells were trypsinized and re-suspended in PBS to a concentration of 5 × 106 cells in 100 μl. For some experiments, A549-luc-C8 cells (Bioware® cell line P/N 119266) were used. This is a luciferase expressing cell line derived from A549 cells by stable transfection of the North American firefly luciferase gene expressed from the CMV promoter (Caliper Life Sciences, Hopkinton, MA). We generated A549-luc control cells and A549-luc ACL knockdown cells with the 285 shRNA lentivirus. These cells were trypsinized and re-suspended in PBS to a concentration of 13 × 106 cells in 100 μl.
In handling the animals, we followed the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23 1996) and protocols were approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. On day 0, female athymic mice (Charles River, Wilmington, MA) were anesthetized by gas anesthesia (3% isoflurane) and tumor cells were injected subcutaneously in the flank. Ten mice were used in each treatment group for the initial experiment and 15 mice were used in each group for the second experiment.
Measurement of tumors
Tumor measurements were obtained using calipers every 7 days and tumor volume was calculated as follows: Tumor volume (V) =a × b × b/2, where a represents the minimum tumor diameter, and b represents the maximum tumor diameter (Hanai et al., 2005).
Statin feeding
Lovastatin was diluted in 0.5% methylcellulose (Fluka) and fed orally by disposable feeding sterile needles (Jorgensen Laboratories) at 50 mg/kg/day starting 2 weeks post tumor cell inoculation.
Tumor imaging (Contag et al., 2000; Edinger et al., 2002; Jenkins et al., 2003a,b; Murray et al., 2003; Scatena et al., 2004)
Mice bearing A549-luc cells were injected with firefly luciferin (150 mg/kg, Xenogen) by intraperitoneal injection using a 25 × 5/8″ gauge needle to image the luciferase signal at various time points. Mice were placed onto black paper in the IVIS® imaging box (Xenogen) and imaged dorsally 15 min after luciferin injection to assure a linear range of bioluminescence. At the end of the experiment, animals were euthanized as per the institutional animal protocol and tissue stored for immunohistochemical analysis.
Immunohistochemical analysis of tumor tissue
Paraffin slides (5 μm thick) were deparaffinized with xylene and serial ethanol dilutions. Hematoxylin and eosin (H&E) staining was used to visualize cellular morphology in tissue sections. Slides were washed with xylene followed by rehydration in graded alcohols. After washing with H2O, slides were incubated with hematoxylin followed by a wash with H2O and ammonia water. Slides were then incubated with eosin followed by rehydration in graded alcohols and xylene incubation. For the E-cadherin staining, antigen retrieval was achieved with citrate in a pressure cooker for 5 min. Endogenous peroxidase activity was blocked for 30 min with a buffer solution containing peroxide (0.5% H2O2 in phosphate citrate). Slides were then incubated for 1 h at room temperature with E-cadherin antibody (1:500) or isotype-matched IgG as a negative control, followed by the secondary antibody (mouse, Envision PO system; DAKO) for 30 min. All slides were developed with diaminobenzidine followed by hematoxylin counterstaining. Before the slides were mounted, all sections were dehydrated in alcohol and xylene. For the mucicarmine staining, we followed the method mentioned at http://library.med.utah.edu/WebPath/HISTHTML/MANUALS/MUCICAR.PDF. This staining is based on the reaction of an aluminum–carmine chelate complex, which attaches to acid groups of mucin. Briefly, slides were deparaffinized and hydrated with distilled water followed by staining with Mayer’s hematoxylin for 10 min. Then slides were washed in running tap water for 5 min and stained with mucicarmine solution in a microwave at high power for 45 sec, followed by a quick rinse in distilled water. Metanil yellow stain was added for 1 min followed by quick dehydration using three changes of absolute alcohol.
Serum starvation
A549 control and ACL knockdown cells were plated in 6-well plates at approximately 50% confluency. Twenty four hours later, regular medium (10% serum) was changed to low serum medium (0.75%) and the cells were incubated for 14 h (serum starvation). Low serum medium was replaced by regular medium and cells were incubated for selected intervals. Cells were harvested for WB analysis.
Ras subcellular fractionation analysis
A549 shACL inducible cells were treated with 1 μM lovastatin and/or doxycycline for 48 h. Control and lovastatin lysates were isolated into cell membrane and cytosolic fractions using the Qproteome Cell Compartment kit (Qiagen) and prepared for WB analysis. Membranes were probed with pan-Ras (Calbiochem) antibody and, 14-3-3 (Cell Signaling Technologies) and Na-K-ATPase (Abcam) antibodies were used as cytosolic and membrane markers, respectively.
Acetate and citrate supplementation
Na-acetate and Na-citrate were dissolved in dH20 and added to cell culture media of A549 shACL inducible cells at designated concentrations for 48 h in conjunction with doxycycline. Cells were harvested for WB or apoptosis analysis as previously described.
Statistical analyses
Student’s t test was used to evaluate the statistical significance of the results. All values are expressed as mean ± S.E.
Results
ACL knockdown A549 cells show MET (reversal of EMT)
Because there are no established cancer cell lines that can be used to study metabolic alterations and signaling events associated with ACL knockdown, we have used the A549 lung cancer cells to generate ACL knockdown cell lines using RNA interference. ACL deficiency was confirmed by WB analysis (Fig. 1A). ACL knockdown A549 cells show epithelial cobblestone-like structure, compared to control A549 cells which show a mesenchymal spindle structure (Fig. 1B). The data shown is for cells infected with the 285 shRNA construct; however, the cells with the other two ACL specific hairpins showed similar results (data not shown). We hypothesized that the alteration in cell morphology may correlate with expression of a number of epithelial and mesenchymal markers and so we assessed expression of the epithelial markers (E-cadherin, ZO-1) and a mesenchymal marker (vimentin) by WB analysis (Fig. 1C, D). The increase in E-cadherin and ZO-1 levels and the decreased expression of vimentin are strong indicators that the ACL knockdown cells have undergone MET or a reversal of epithelial-mesenchymal transition (EMT). These data are consistent with the morphologic changes noted in the knockdown cells (Fig. 1B).
ACL deficiency affects proliferation, apoptosis, and cell cycle progression in A549 cells and cells with EGFR mutation
Next, we assessed the functional effects of ACL deficiency. We found that A549 cells and NSCLC lines harboring EGFR mutations when rendered ACL knockdown proliferate slower than control cells (Fig. 1D, E, F). The annexin-V and cleaved caspase assays indicate that ACL knockdown cells have higher rates of apoptosis than control cells (Fig. 1G, H, I, J) and cell cycle analysis shows that ACL deficiency causes a modest increase in the number of cells in the G1 phase of the cell cycle (data not shown). These data extend previous observations (Migita et al., 2008) by showing that ACL knockdown can cause similar phenotypic changes in several genetic backgrounds known to occur in NSCLC. These data point to two effects of ACL deficiency: Increased differentiation as exemplified by a reversal of EMT and a decreased growth rate, with apoptosis as the underlying mechanism. We also observed that phosphorylation of Bad, a pro-apoptotic member of the Bcl-2 family member, is decreased in the ACL knockdown cells (Fig. 1I). Bad is negatively regulated via phosphorylation (by AKT), suggesting that the ACL deficient state may be causing apoptosis through inhibition of Bad function. Moreover, the fact the ACL knockdown causes phenotypic changes in both K-Ras activated (A549) cells and in cells with EGFR mutations (our H series of cells with presumably wild type K-Ras) suggests that the mechanism(s) at play must act downstream of Ras activation. Since Bad is an AKT target, these data suggest that ACL knockdown may inhibit the PI3K/AKT pathway, a hypothesis that is explored below. Note that the anti-proliferative and apoptotic effects induced by ACL deficiency were neither observed in normal lung epithelial cells (BEAS-2B), nor were they seen in human endothelial cells (HUVEC) (Fig. 2).
Fig. 2.

Annexin assay for control and ACL knockdown lung epithelial (BEAS-2B) and human umbilical vein endothelial cells (HUVECs). Cells were treated with lovastatin at the indicated dose for 48 h and analyzed for apoptosis. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcp]
In vitro effects of ACL deficiency are enhanced by statin treatment
We hypothesized that a combination of statin treatment in the context of ACL deficiency in NSCLC cells would exert additional “anti-tumor” effects, perhaps by affecting multiple intracellular pathways. We began by examining effects on cell proliferation and apoptosis in vitro. Cell proliferation is downregulated with statins, an effect that is accentuated in the ACL deficient condition (Fig. 1D). Apoptosis is also activated in the ACL deficient condition compared to control cells (A549, H1650, and H1975) (Fig. 1G, H, I, J) and statin treatment augments this effect (Fig. 1H).
Next we asked what role ROS may play in the phenotypic effects noted with ACL knockdown. Incubation with H2O2 for 30 min did not affect control cells. However, in the ACL knockdown cells, H2O2 induced more apoptosis, which was further amplified with statin treatment (Fig. 1I). These data suggest that oxidant stress can tip ACL knockdown cells into apoptosis and that statin treatment magnifies this effect. Of importance, these statin effects were neither observed in normal lung epithelial cells (BEAS-2B) nor in human endothelial cells (HUVEC) (Fig. 2), suggesting selectivity of these treatments for tumor cells.
Synergistic effects on tumor growth of the ACL deficient condition and statin treatment
We hypothesized that the changes in cell growth and differentiation noted in vitro would lead to altered tumor growth and/or differentiation in vivo. A marked reduction of tumor size generated by the ACL knockdown cells compared to control cells was observed, an effect further augmented by statin feeding (Fig. 3A). We repeated this in vivo experiment with A549-luc cells. ACL knockdown A549-luc cells were generated and we first ascertained that they showed reduced ACL expression to undetectable levels (data not shown). To explore whether statin treatment might augment the effect of ACL knockdown, we focused on two treatment arms: The ACL knockdown cells and additional statin treatment. For this experiment, we injected 1.3 × 107 cells instead of 0.5 × 107 cells, as used earlier. Statin therapy dramatically enhanced the effects of ACL deficiency on tumor growth, even regressing established tumors (Fig. 3B). Nine of 15 tumors regressed (data not shown). In vivo tumor imaging data (Fig. 3C) show an example of tumor regression in the ACL knockdown plus statin treatment group.
Fig. 3.
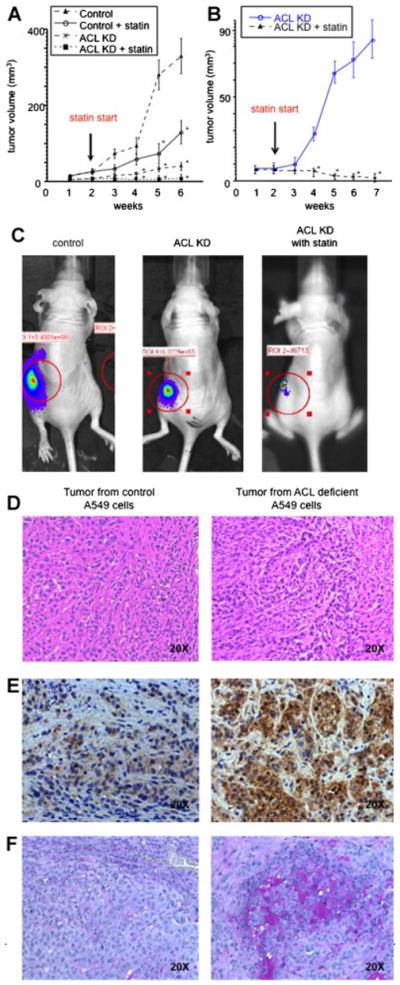
Xenograft tumor model. A: In vivo tumor growth of control A549 cells and ACL knockdown cells, feeding with or without lovastatin. Initial implanted cell number was 5 T106. Error bar represent standard deviation and M denotes P-value < 0.01 when compared to control animal from the same day. B: In vivo tumor growth of ACL knockdown cells feeding with or without lovastatin. Initial implanted cell number was 13 T106. Error bar represent standard deviation and M denotes P-value < 0.01 when compared to control animal from the same day. C: In vivo tumor imaging of control A549 cells and ACL knockdown cells. Immunohistochemistry of in vivo tumors. D: HE staining E: E-cadherin staining. F: Mucin staining.
Reversal of EMT and differentiation in ACL knockdown tumors
Tumor histology indicated that significant differentiation might have occurred in the ACL knockdown tumor, as evidenced by primitive glandular structures present as compared to their absence in the control tumor (Fig. 3D). In support of this, we found a marked increase in E-cadherin expression (Fig. 3E) in ACL knockdown tumors, suggesting that the differentiation caused by ACL inhibition is accompanied by reversal of EMT. Mucin is a marker of type II pneumocyte differentiation and A549 cells are thought to be derived from this cell type (Jarrard et al., 1998; Koparal and Zeytinoglu, 2003). Mucin staining in ACL knockdown tumors is markedly increased, further suggesting that differentiation is induced in this condition (Fig. 3F).
PI3K inhibition mimics the ACL deficient condition
We hypothesized that PI3K inhibition may affect A549 cells in a manner similar to that of ACL inhibition and that ACL inhibition may diminish PI3K/AKT signaling based on (a) the known effects of inhibition of the PI3K/AKT pathway on the processes of differentiation and apoptosis, (b) the observation by Thompson et al. that ACL inhibition seemed to work best only in cells that were glycolytic, an effect that is known to be mediated by AKT, and (c) the effects of ACL inhibition on Bad phosphorylation, an AKT target.
We found that treatment of control A549 cells with wortmannin showed a similar phenotype to that of ACL knockdown cells, namely, cobblestone morphology and an appositional growth pattern (Fig. 4A). Western blot analysis for E-cadherin indicates a dose-dependent increase of E-cadherin expression (Fig. 4B). Wortmannin also induces apoptosis of A549 cells in a dose dependent manner (Fig. 4C), data that is similar to the ACL deficient state. Similar data (not shown) was obtained with another PI3K inhibitor, LY294002. Importantly, apoptosis induction by PI3K inhibition was noted and it was reverted by addition of catalase (Fig. 4C), suggesting involvement of reactive oxygen species (ROS) in the induction of apoptosis by PI3K inhibitors.
Fig. 4.

PI3 kinase inhibitor mimics the ACL deficient condition. A: Cell morphology of control A549 cells treated with or without wortmannin (1 μM) for 48 h. B: E-cadherin western blot analysis of control A549 cells treated with the indicated amount of wortmannin for 48 h. C: Apoptosis assay analysis of control A549 cells treated with the indicated amount of wortmannin in the combination of with or without catalase for 24 h. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcp]
AKT signaling is downregulated in the ACL deficient state
Given the above data, we hypothesized that ACL might dampen PI3K/AKT signaling. Previous data demonstrated that AKT can upregulate ACL activity through phosphorylation (Berwick et al., 2002; Sale et al., 2006); here, we are postulating the reverse, namely that decreased ACL might inhibit PI3K/AKT signaling. We elected to first evaluate the effects of ACL inhibition on the phosphorylation status of AKT. The data in Figure 5A shows that AKT phosphorylation at both threonine 308 and serine 473 is markedly diminished in the ACL knockdown cells at baseline. To investigate the effects on activation of the PI3K/AKT pathway in a more “dynamic” manner, we serum starved two cell lines (A549 and H1650) and then refed them with serum (Fig. 5B, C). ACL knockdown cells show diminished phosphorylation of AKT over time at both phosphorylation sites.
Fig. 5.

Interception of PI3K/AKT signaling and MAPK signaling by ACL knockdown. A: Western blot of AKT phosphorylation in basal growth state (A549 cells). Densitometric quantification of phosphoblots (% of control) of AKT 308 and AKT 473 are shown on the right. Each band was scanned and subjected to densitometry. Each intensity of phospho AKT protein relative to the mean value of AKT1 and AKT2 were compared to control. M denotes P-value < 0.01 when compared to control. B: Left panel: Phosphorylation of AKT and MAPK in control and ACL KD cells after serum starved and serum refeeding (A549 cells). Right panel: Densitometric quantification of phosphoblots of AKT 308, AKT 473, and pERK is shown as a % of maximum value. Each band was scanned and subjected to densitometry. Each intensity of phospho AKT protein relative to the mean value of AKT1 and AKT2 or ERK was measured. C: AKT and MAPK phosphorylation in control and ACL KD cells after serum starvation (0.5%) and serum refeeding (H1650).
Statin treatment downregulates the phosphorylation of ACL and AKT
We speculated that statins may inhibit the PI3K/AKT pathway as has been described in other cell types (Graaf et al., 2004; Mistafa and Stenius, 2009). As shown in Figure 6A, statin treatment of ACL knockdown A549 cells, but not control A549 cells, caused dephosphorylation at threonine 308 and serine 473 in AKT in a time dependent manner, indicating that the PI3K/AKT pathway is impacted most dramatically by ACL inhibition in combination with statin treatment. In order to more fully assess the effects of statin alone on A549 cells, we treated the cells with statin for a longer time (6 h) and used various statin concentrations (Fig. 6B). These data indicate that statin treatment can diminish the amount of pAKT 308 and pAKT 473 in a dose dependent manner. We also observed that statin downregulated cyclin D1 expression, a target of the PI3K/ AKT pathway (Yu et al., 2001; Hulit et al., 2004). Disruption of cyclin D1 can cause cell cycle arrest, apoptosis, and differentiation. Interestingly, statin downregulated ACL phosphorylation, an effect that could be secondary to its effects on AKT. Statin treatment alone had a small effect on the phosphorylation state of MAPK (Fig. 6B) after 6 h of treatment.
Fig. 6.

In vitro synergystic effects of lovastatin and ACL KD. A: AKT phosphorylation with statin treatment (1 μM) at the indicated incubation time. The value of densitometric quantification of phosphoblots (% of maximum value) are below each blot. B: AKT and MAPK phosphorylation of ACL control A549 cells are statin dose dependent after 6 h of incubation. Values of densitometric quantification of phosphoblots (% of maximum value) are below each blot. C: AKT phosphorylation with statin (1 μM) preincubation followed by serum starving and EGF supplementation. Values of densitometric quantification of phosphoblots (% of maximum value) are below each blot.
ACL inhibition plus statin treatment impacts MAPK activation
We examined the effects of ACL inhibition plus statin treatment on both PI3K/AKT and MAPK pathways. We pretreated cells with lovastatin for 48 h, serum starved them, and then provided EGF supplementation (Fig. 6C). AKT phosphorylation was downregulated more by ACL inhibition plus statin treatment compared to ACL inhibition alone. Under these conditions, we noted markedly diminished phosphorylation of ERK by ACL inhibition in combination with statin treatment.
Generation of a tet-inducible ACL knockdown cell line
We also established a tet-inducible ACL knockdown system and used this system to confirm our observations made with the permanent ACL knockdown cells.
To validate our system, we first showed that ACL expression was decreased in a doxycycline dose-dependent manner. Paralleling this, we found upregulation of E-cadherin (Fig. 7A). Also, phospho-AKT and phospho S6 protein were decreased in parallel with this decrease of ACL levels (Fig. 7A). We noted minimal downregulation of ERK phosphorylation under the same conditions (Fig. 7A). We also confirmed that statin treatment amplifies the apoptotic effect of the ACL knockdown state (Fig. 7B). These data suggest that the effects seen with permanent ACL knockdown are not due to long-term adaptation of the cells but occur rapidly in response to ACL knockdown.
Fig. 7.

Doxycycline inducible ACL shRNA A549 cell system. A: Left panel: Doxycycline dose dependently leads to ACL knockdown. Right panel: Densitometric quantification of phosphoblots (% of maximum value) of AKT 308, AKT 473, S6 protein, and pERK is shown. Each band was scanned and subjected to densitometry. Each intensity of phospho protein relative to total AKT, S6 protein, or ERK was compared and shown as % of maximum value. Error bars represent standard deviation and * denotes P-value < 0.01 when compared to each control condition (doxycycline 0 μg/ml). B: Apoptosis assay of statin treated doxycycline inducible ACL shRNA A549 cells.
Acetate partially rescues the effects of the ACL deficient condition
The ACL knockdown state limits acetyl CoA synthesis from citrate in the cytoplasm. Acetate is the other source of cytoplasmic acetyl CoA, which is synthesized by the ACAS II enzyme. If cytoplasmic acetyl CoA depletion is the mechanism by which ACL knockdown is working, we might expect that supplementation with acetate would rescue the ACL knockdown phenotype. This was found to be the case for rescue of ACL function as it relates to histone acetylation (Wellen et al., 2009). We examined AKT phosphorylation using the tet (doxycycline) inducible ACL knockdown system with or without Na-acetate (Fig. 8A). The downregulated phosphorylation state of AKT 473 induced by ACL knockdown was clearly reversed by Na-acetate supplementation in a dose dependent manner. However, phosphorylation of AKT at residue 308 was not rescued. We also assessed apoptosis. Na-acetate supplementation partially rescued apoptosis induced by ACL knockdown (Fig. 8B).
Fig. 8.

Effects of acetate and citrate on the ACL knockdown phenotype. A: Na-acetate treatment in the doxycycline (1 mg/ml) inducible ACL shRNA A549 cell system rescues AKT phosphorylation. Values of densitometric quantification of phosphoblots (% of maximum value) are below each blot. B: Apoptosis assay following Na-acetate treatment in the doxycycline inducible ACL shRNA A549 cell system. C: Na-citrate treatment of doxycycline inducible ACL shRNA A549 cells diminishes Akt phosporylation. Values of densitometric quantification of phosphoblots (% of maximum value) are below each blot. D: Apoptosis assay for Na-citrate treatment in the doxycycline inducible ACL shRNA A549 cell system. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcp]
Citrate enhances the effects of ACL deficient condition
In the ACL knockdown cells, cytosolic citrate might be expected to increase. We hypothesized that this accumulation might be important for the ACL knockdown phenotype. If true, exogenous citrate supplementation might augment the effects on AKT phosphorylation induced in the ACL knockdown state. In A549 cells, Na-citrate supplementation caused a slight downregulation of AKT phosphorylation at both AKT 308 and 473 sites. These effects were more dramatic in ACL downregulated cells at the AKT 473 site (Fig. 8C). Next, we examined the effects of citrate on apoptosis induced by ACL knockdown (Fig. 8D). Citrate supplementation caused increased apoptosis in the A549 cells and induced more apoptosis in the ACL knockdown cells.
Ras distribution is unchanged in the ACL deficient state
To begin to define the point of intersection in the PI3K/AKT pathway that ACL knockdown impacts, we tested ras protein distribution in control and ACL knockdown cells (Fig. 9). Our goal was to eliminate the possibility that ACL knockdown leads to decreased production of mevalonate, which is necessary for ras prenylation. We isolated cytosolic and membrane fractions for each condition and analyzed these by western blotting. There was no significant change in ras distribution between control and ACL knockdown cells. Statin, as expected, slightly reduced membrane-localized ras, likely due to inhibition of ras prenylation. These data suggest that ACL knockdown does not affect PI3K/AKT signaling by diminishing ras targeting to the membrane through inhibition of ras prenylation. It is therefore likely that the effects of ACL knockdown on the PI3K/AKT pathway occur downstream of ras and studies are in progress to define this. These data are also consistent with the fact that the MAPK pathway was unaffected by ACL knockdown and consistent with the inability of mevalonate to rescue the phenotype of the ACL deficient state.
Fig. 9.

Ras localization under ACL knockdown and statin treatment conditions. The cytosolic and membrane fractions from each cell lysate were analyzed for ras expression.
Discussion
The ACL deficient condition has been reported to cause differentiation and apoptosis, leading to anti-tumor effects. The novel findings of this study are: (1) The ACL deficient state downregulates PI3K/AKT signaling in several different genetic backgrounds found in NSCLC cells, (2) ACL deficiency upregulates E-cadherin expression and impacts Bad phosphorylation likely contributing to MET and apoptosis, respectively, (3) a combination of ACL deficiency with statin treatment shows synergistic anti-tumor effects in vitro and in vivo, (4) statins downregulate ACL phosphorylation, (5) the ACL deficient state in combination with statin treatment downregulates both the PI3K/AKT and the MAPK pathways, (6) the anti-tumor effects of ACL deficient state are partially rescued by acetate and enhanced with citrate treatment.
ACL deficiency leads to interception of PI3K/AKT signaling
In the ACL deficient condition, Bad, a pro-apoptotic protein, is inactivated by phosphorylation. This factor is a target of PI3K/ AKT signaling via NFkB and AKT respectively. Moreover, PI3K inhibitors mimic the phenotype of ACL inhibition (differentiation and apoptosis). These data led us to hypothesize that ACL inhibition may intercept PI3K/AKT signaling.
AKT activation is a multistep process involving both membrane translocation and phosphorylation. The pleckstrin homology domain of the AKT kinases has affinity for the 3′-phosphorylated phosphoinositides 3,4,5-triphosphate (PI 3,4,5-P3) produced by PI3K. Phospholipid binding triggers the translocation of AKT kinases to the plasma membrane. Upon membrane localization, AKT molecules are phosphorylated at threonine 308 (Thr-308) in the kinase activation loop and serine 473 (Ser-308) in the carboxyl-terminal tail. Thr-308 phosphorylation is necessary for AKT activation, and Ser-473 phosphorylation is required for maximal activity. Phosphorylation on these residues is induced by growth factors, such as EGF (Cook et al., 1993), and serum, probably due to LPA (Cook et al., 1993; Yart et al., 2002), and inhibited by the PI3K inhibitor (LY294002 and wortmannin). Indeed, the kinase responsible for Thr-308 phosphorylation, PDK1 (3-phosphoinositide-dependent kinase) is activated by the PI3K lipid product PI-3,4,5-P3 and phosphorylates Thr-308 in AKT upon PI3K activation by recognizing PI-3,4,5-P3. The identity of PDK2, the kinase(s) responsible for Ser-473 phosphorylation, is controversial. mTOR complex-2 (mTORC2) has been identified as the physiological PDK2 kinase (Sarbassov et al., 2005), and this fact is generally accepted in the field (Franke, 2008).
We observed that ACL inhibition diminished PI3K/AKT signaling at basal conditions in cell culture and during activation of this pathway following serum starvation and refeeding or EGF supplementation. Importantly, the effects of ACL inhibition on MAPK signaling were small.
Identification of the point in the PI3K/AKT signaling pathway that is affected by ACL knockdown is important for understanding the mechanism by which ACL inhibition leads to the changes in cell phenotype. We are currently examining this issue and considering various possibilities: The point of interception might be at level of a growth factor receptor, or at PI3K, PTEN, PDK1, or at AKT itself (action of a phosphatase). We have eliminated ras as a central point for ACL action. We have recently been able to show that ACL inhibition in a breast cancer cell line can alter the phenotype of cells deficient in PTEN and in cells in which the p110α catalytic unit is constitutively activated (data not shown), suggesting that the intersection point is either at PDK-1 or at AKT. Interestingly, AKT and ACL are part of a complex and AKT phosphorylates ACL (Berwick et al., 2002; Sale et al., 2006), which in turn is thought to induce its allosteric activation (Towle et al., 1997).
ACL deficiency causes tumor differentiation and MET
We found that ACL inhibition leads to differentiation and mesenchymal–epithelial transition (MET) in vivo and in vitro (Figs. 1B, C, D and 3E). Tumors from vector control cells were poorly differentiated and exhibited a disorganized cellular architecture. In contrast, tumors from ACL knockdown A549 cells displayed a more differentiated morphology marked by the presence of glandular structures bearing central lumens and intracytoplasmic and intraluminal mucin expression, suggesting differentiated respiratory epithelium. This indicates that ACL deficient state in vivo and in vitro shows an increased tendency toward epithelial cell differentiation. MET is characterized by the increase of epithelial markers and decrease of mesenchymal markers, as well as morphological change from a spindle cell phenotype to a cobblestone-like structure. Increased E-cadherin protein expression (and relocalization to the cell membrane) is a key feature of this transition, which is regulated tightly at transcriptional, post-translational, and protein stability levels.
ACL deficiency causes apoptosis involving the intrinsic pathway
There are two major signaling pathways causing apoptosis, the extrinsic death receptor mediated pathway, and the intrinsic mitochondria mediated pathway. The extrinsic pathway is initiated by ligation of transmembrane death receptors (Fas, TNF receptor, and TRAIL receptor) with their respective ligands (FasL, TNF, and TRAIL) to activate membrane-proximal caspases (caspase-8 and -10), which in turn cleave and activate effector caspases such as caspase-3 and -7. The intrinsic pathway requires disruption of the mitochondrial membrane and the release of cytochrome c, which works together with the other two cytosolic protein factors, Apaf-1 (apoptotic protease activating factor-1), and procaspase-9, to promote the assembly of a caspase-activating complex, which in return induces activation of caspase-9 and thereby initiates the apoptotic caspase cascade (Reed, 1994; Korsmeyer, 1995; Thornberry and Lazebnik, 1998; Wajant, 2002; Scorrano and Korsmeyer, 2003; Ghobrial et al., 2005). We found that phosphorylation of Bad protein, a pro-apoptotic member of the Bcl-2 family member, is decreased in ACL knockdown cells. Bad is negatively regulated by phosphorylation. Phosphorylated Bad associates with the 14-3-3 protein and is unable to activate pro-apoptotic members such as Bax and Bak. Bad is know to be phosphorylated by PI3K/AKT signaling and interception of this pathway by ACL knockdown could be the mechanism underlying the downregulation of Bad phosphorylation noted in ACL deficiency. These data also suggest that the intrinsic apoptosis pathway contributes to apoptosis caused by ACL deficiency.
Anti-tumor effects of ACL deficiency in vivo and enhanced effects with statin treatment
Statins can induce differentiation, affect tumor growth (via enhanced apoptosis and cell cycle arrest) and also affect the tumor microenvironment, influencing both angiogenesis and immune regulation (Tregs) (Meier et al., 2008). Multiple signaling pathways mediating these effects have been described (Graaf et al., 2004).
These effects are seen at various doses. Growth arrest and apoptosis occur in vitro at lovastatin concentrations ranging from 0.1 to 100 μM depending on the cell line used. A phase I trial revealed that administration of lovastatin in doses from 2 to 25 mg/kg daily results in drug plasma concentrations ranging between 0.1 and 3.9 μM (Thibault et al., 1996). These findings indicate that lovastatin induced anti-proliferative and proapoptotic effects occur at levels that are therapeutically achievable. However, statin monotherapy does not seem to impact clinical progression of cancer in humans and trials have been disappointing. Thus, combination therapies would be reasonable to consider. Indeed, synergistic effects of the combined use of statins with numerous drugs have been reported in preclinical studies both in vitro and in vivo. These drugs include Cox-2 inhibitors, tocotrienols, PPARγ agonists, bisphosphonates, and various chemotherapeutic drugs (cisplatin (Agarwal et al., 1999; Fromigue et al., 2008), 5-FU (Ahn et al., 2008), gemcitabine (Bocci et al., 2005), and paclitaxel (de Souza et al., 1997)). Also, statins can act as radiation sensitizers (Fritz et al., 2003).
Our tumor data show that statin treatment alone inhibits tumor growth and this effect is more dramatic in ACL knockdown cells (Fig. 3A, B). Interestingly, in contrast to the in vitro data which show that statin treatment of ACL knockdown cells does not diminish cell number, in vivo, we found that some tumors regressed. We repeated this in vivo experiment with A549-luc cells, focusing attention on only two treatment arms: The ACL knockdown cells and statin treatment of these tumors (Fig. 3B). These in vivo regression data are rather striking: Many mechanisms may be at play to explain why the in vivo data contrast to the modest effects seen in vitro. Studies to assess effects on the tumor microenvironment including angiogenesis and stromal responses are in progress. For example, one could speculate that since HIFs are downstream targets of the PI3K/ AKT pathway, HIF expression may be reduced by ACL knockdown and that this in turn could impact a number of well-known HIF targets such as VEGF, thus affecting angiogenesis.
To elucidate some of the mechanisms by which statins might be enhancing the effects of ACL knockdown, we assessed the impact on PI3K/AKT and MAPK signaling. As shown in Figure 6A, B, statin treatment diminished AKT phosphorylation in a time and dose dependent manner and the effect was more dramatic in the ACL deficient state. However, we observed only slight downregulation of ERK phosporylation after 6 h of statin treatment. We examined the effects of long term treatment with statin on MAPK signaling. As shown in Figure 6C, a 24 h incubation with statin caused obvious downregulation of MAPK phosphorylation in the ACL deficient state comparing to control A549 cells, suggesting that the combination of ACL inhibition and statin treatment diminished both PI3K/AKT signaling and MAPK pathway. These data may explain the significant anti-tumor effects of this combination in vivo. Indeed both pathways are activated in A549 cells, since they contain K-ras activating mutation in an LKB1 deficient background. PI3K/AKT and MAPK signaling are two of the most important signaling cascades dysregulated in cancers (Grant, 2008). Moreover, inhibition of PI3K signaling at the level of mTORC1 has been shown to activate a feedback loop in Ras-MAPK signaling through an S6K1 and PI3K-dependent process. Thus, dual blockade of PI3K and MAPK signaling is often required to obtain substantial anti-tumor effects both in vitro and in vivo (Carracedo et al., 2008; Kinkade et al., 2008). Indeed such dual blockade is effective in several cancer models, including breast cancer (Ripple et al., 2005; Mirzoeva et al., 2009), melanoma (Merighi et al., 2005; Matsuoka et al., 2009), leukemia (Champelovier et al., 2008), ovarian carcinoma (Kawaguchi et al., 2007), mesothelioma (Cole et al., 2006), Ewing sarcoma (Yamamoto et al., 2009), and in lung cancer, where an engineered mouse lung tumor was driven by mutant K-ras (Engelman et al., 2008).
Interestingly, statin treatment also diminished ACL phosphorylation (Fig. 6B), indicating that statin itself can exert inhibitory effects on ACL function. Whether this is dependent on inhibition of the PI3K/AKT pathway or independent of it remains to be ascertained.
Our observations have clinical relevance. As noted, cancer trials with statins have been unimpressive and it is unlikely that the use of ACL inhibitors alone would produce more than a cytostatic response. A combination of the type described here, potentially in conjunction with traditional chemotherapies or ideally with targeted therapies used for NSCLC (such as EGFR inhibitors for tumors with EGFR mutations and wild type K-ras) might produce additional benefit. Also, as noted above, the concentration of statin used in our in vitro studies has been achieved in clinical trials (Thibault et al., 1996).
Anti-tumor effects of ACL deficient state is partially diminished by acetate and enhanced by citrate treatment
Since acetyl-CoA cannot move freely from mitochondria to cytosol, mitochondrially derived citrate is transported into the cytosol where it is cleaved by ACL and cytosolic acetyl-CoA is produced. Cytosolic acetyl-CoA is the requisite building block for endogenous synthesis of fatty acids, cholesterol and isoprenoids as well as for acetylation reactions that modify proteins. Therefore, ACL is located upstream of the other lipogenic enzymes and connects glucose metabolism (a way of generating acetyl CoA) and lipogenesis (Hatzivassiliou et al., 2005). ACL inhibition should result in the cytosolic accumulation of citrate, and diminished production of acetate. Acetate treatment partially diminished the anti-tumor effects of ACL deficient state, suggesting the amount of cytosolic acetyl-CoA might be important for the anti-tumor effects of the ACL deficient condition. How the diminished acetyl CoA or the potentially increased citrate leads to inhibition of PI3K/AKT signaling is not understood but it is conceivable that these molecules interact with a member of the PI3K/AKT signaling pathway and modify kinase activity of one or more of its members.
In summary, we have shown that combination of both ACL knockdown and statin treatment diminishes tumor growth in vivo and in vitro, through inhibiting both PI3K and MAPK signals, two major survival pathways for cancer cells. The effects in vivo are more impressive than in vitro, suggesting that this combination may have additional effects on the tumor microenvironment. We have shown that ACL blockade can impact both K-ras mutant and EGFR mutant lung cancer cell lines. Our studies in a tet-inducible ACL knockdown system corroborate these findings. The precise mechanisms underlying ACL knockdown induced apoptosis and differentiation are being elucidated and the point of interception in PI3K/AKT pathway at which ACL knockdown acts is the subject of ongoing studies. Indeed, the regulation of PI3K/AKT signaling by ACL may represent a way of synchronizing nucleotide, lipid and protein synthesis. The latter is known to be stimulated by mTORC1, and former is enhanced by increased glycolysis due to AKT activation and increasing flux through the pentose phosphate pathway. Thus, our studies point to a deep connection between metabolic and canonical signaling pathways and suggest that each can impact the other.
Acknowledgments
The authors thank members of the Sukhatme Laboratory and Dr. Alex Toker for useful discussions. They also thank Han Xie for technical support. This work was supported by seed funds from Beth Israel Deaconess Medical Center to VPS.
Abbreviations
- ACL
ATP–citrate lyase
- EMT
epithelial–mesenchymal transition
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- PI3K
phosphatidylinositol 3-kinase
- NSCLC
non-small cell lung cancer
Footnotes
Conflict of Interest: L. Cantley is a cofounder of Agios, a company that is developing inhibitors for metabolic targets in cancer.
Literature Cited
- Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res. 1999;5:2223–2229. [PubMed] [Google Scholar]
- Ahn KS, Sethi G, Aggarwal BB. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem Pharmacol. 2008;75:907–913. doi: 10.1016/j.bcp.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277:33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- Bocci G, Fioravanti A, Orlandi P, Bernardini N, Collecchi P, Del Tacca M, Danesi R. Fluvastatin synergistically enhances the antiproliferative effect of gemcitabine in human pancreatic cancer MIAPaCa-2cells. Br J Cancer. 2005;93:319–330. doi: 10.1038/sj.bjc.6602720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champelovier P, El Atifi M, Pautre V, Rostaing B, Berger F, Seigneurin D. Specific inhibition of basal mitogen-activated protein kinases and phosphatidylinositol 3 kinase activities in leukemia cells: A possible therapeutic role for the kinase inhibitors. Exp Hematol. 2008;36:28–36. doi: 10.1016/j.exphem.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- Cole GW, Jr, Alleva AM, Zuo JT, Sehgal SS, Yeow WS, Schrump DS, Nguyen DM. Suppression of pro-metastasis phenotypes expression in malignant pleural mesothelioma by the PI3K inhibitor LY294002 or the MEK inhibitor UO126. Anticancer Res. 2006;26:809–821. [PubMed] [Google Scholar]
- Contag CH, Jenkins D, Contag PR, Negrin RS. Use of reporter genes for optical measurements of neoplastic disease in vivo. Neoplasia (New York, NY) 2000;2:41–52. doi: 10.1038/sj.neo.7900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SJ, Rubinfeld B, Albert I, McCormick F. RapV12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibroblasts. Embo J. 1993;12:3475–3485. doi: 10.1002/j.1460-2075.1993.tb06022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza PL, Castillo M, Myers CE. Enhancement of paclitaxel activity against hormone-refractory prostate cancer cells in vitro and in vivo by quinacrine. Br J Cancer. 1997;75:1593–1600. doi: 10.1038/bjc.1997.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Edinger M, Cao YA, Hornig YS, Jenkins DE, Verneris MR, Bachmann MH, Negrin RS, Contag CH. Advancing animal models of neoplasia through in vivo bioluminescence imaging. Eur J Cancer. 2002;38:2128–2136. doi: 10.1016/s0959-8049(02)00410-0. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF. PI3K/Akt: Getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- Fritz G, Brachetti C, Kaina B. Lovastatin causes sensitization of HeLa cells to ionizing radiation-induced apoptosis by the abrogation of G2 blockage. Int J Radiation Biol. 2003;79:601–610. doi: 10.1080/09553000310001609233. [DOI] [PubMed] [Google Scholar]
- Fromigue O, Hamidouche Z, Marie PJ. Statin-induced inhibition of 3-hydroxy-3-methyl glutaryl coenzyme a reductase sensitizes human osteosarcoma cells to anticancer drugs. J Pharmacol Exp Therap. 2008;325:595–600. doi: 10.1124/jpet.108.136127. [DOI] [PubMed] [Google Scholar]
- Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA: Cancer J Clin. 2005;55:178–194. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar HJ. The risk of cancer in users of statins. J Clin Oncol. 2004;22:2388–2394. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Grant S. Cotargeting survival signaling pathways in cancer. J Clin Invest. 2008;118:3003–3006. doi: 10.1172/JCI36898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, Lecker SH. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. 2007;117:3940–3951. doi: 10.1172/JCI32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai J, Mammoto T, Seth P, Mori K, Karumanchi SA, Barasch J, Sukhatme VP. Lipocalin 2 diminishes invasiveness and metastasis of Ras-transformed cells. J Biol Chem. 2005;280:13641–13647. doi: 10.1074/jbc.M413047200. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, Russell R, Pestell RG. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–7611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrard JA, Linnoila RI, Lee H, Steinberg SM, Witschi H, Szabo E. MUC1 is a novel marker for the type II pneumocyte lineage during lung carcinogenesis. Cancer Res. 1998;58:5582–5589. [PubMed] [Google Scholar]
- Jenkins DE, Oei Y, Hornig YS, Yu SF, Dusich J, Purchio T, Contag PR. Bioluminescent imaging (BLI) to improve and refine traditional murine models of tumor growth and metastasis. Clin Exp metastasis. 2003a;20:733–744. doi: 10.1023/b:clin.0000006815.49932.98. [DOI] [PubMed] [Google Scholar]
- Jenkins DE, Yu SF, Hornig YS, Purchio T, Contag PR. In vivo monitoring of tumor relapse and metastasis using bioluminescent PC-3M-luc-C6cells in murine models of human prostate cancer. Clin Exp Metastasis. 2003b;20:745–756. doi: 10.1023/b:clin.0000006817.25962.87. [DOI] [PubMed] [Google Scholar]
- Kawaguchi W, Itamochi H, Kigawa J, Kanamori Y, Oishi T, Shimada M, Sato S, Shimogai R, Terakawa N. Simultaneous inhibition of the mitogen-activated protein kinase kinase and phosphatidylinositol 3′-kinase pathways enhances sensitivity to paclitaxel in ovarian carcinoma. Cancer Sci. 2007;98:2002–2008. doi: 10.1111/j.1349-7006.2007.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Koparal AT, Zeytinoglu M. Effects of carvacrol on a human non-small cell lung cancer (NSCLC) cell line A549. Cytotechnology. 2003;43:149–154. doi: 10.1023/B:CYTO.0000039917.60348.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- Larsson O. Cell cycle-specific growth inhibition of human breast cancer cells induced by metabolic inhibitors. Glycobiology. 1993;3:475–479. doi: 10.1093/glycob/3.5.475. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Tsubaki M, Yamazoe Y, Ogaki M, Satou T, Itoh T, Kusunoki T, Nishida S. Tamoxifen inhibits tumor cell invasion and metastasis in mouse melanoma through suppression of PKC/MEK/ERK and PKC/PI3K/Akt pathways. Exp Cell Res. 2009;315:2022–2032. doi: 10.1016/j.yexcr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- McAndrew PF. Fat metabolism and cancer. Surg Clin North Am. 1986;66:1003–1012. doi: 10.1016/s0039-6109(16)44037-5. [DOI] [PubMed] [Google Scholar]
- Meier P, Meier R, Blanc E. Influence of CD4+/CD25+ regulatory T cells on atherogenesis in patients with end-stage kidney disease. Expert Rev Cardiovasc Ther. 2008;6:987–997. doi: 10.1586/14779072.6.7.987. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Vellon L, Oza BP, Lupu R. Does endogenous fatty acid metabolism allow cancer cells to sense hypoxia and mediate hypoxic vasodilatation? Characterization of a novel molecular connection between fatty acid synthase (FAS) and hypoxia-inducible factor-1alpha (HIF-1alpha)-related expression of vascular endothelial growth factor (VEGF) in cancer cells overexpressing her-2/neu oncogene. J Cell Biochem. 2005;94:857–863. doi: 10.1002/jcb.20367. [DOI] [PubMed] [Google Scholar]
- Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. 2005;280:19516–19526. doi: 10.1074/jbc.M413772200. [DOI] [PubMed] [Google Scholar]
- Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, Hu Z, Knight Z, Feiler HS, Gascard P, Parvin B, Spellman PT, Shokat KM, Wyrobek AJ, Bissell MJ, McCormick F, Kuo WL, Mills GB, Gray JW, Korn WM. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistafa O, Stenius U. Statins inhibit Akt/PKB signaling via P2X7 receptor in pancreatic cancer cells. Biochem Pharmacol. 2009;78:1115–1126. doi: 10.1016/j.bcp.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, Keast PK, Brassard JA, O’Farrell AM, Cherrington JM, Pryer NK. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis. 2003;20:757–766. doi: 10.1023/b:clin.0000006873.65590.68. [DOI] [PubMed] [Google Scholar]
- Okudela K, Hayashi H, Ito T, Yazawa T, Suzuki T, Nakane Y, Sato H, Ishi H, KeQin X, Masuda A, Takahashi T, Kitamura H. K-ras gene mutation enhances motility of immortalized airway cells and lung adenocarcinoma cells via Akt activation: Possible contribution to non-invasive expansion of lung adenocarcinoma. Am J Pathol. 2004;164:91–100. doi: 10.1016/S0002-9440(10)63100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, Kim CH, Lee JC. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer (Amsterdam, Netherlands) 2009;63:219–226. doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Ripple MO, Kalmadi S, Eastman A. Inhibition of either phosphatidylinositol 3-kinase/Akt or the mitogen/extracellular-regulated kinase, MEK/ERK, signaling pathways suppress growth of breast cancer cell lines, but MEK/ERK signaling is critical for cell survival. Breast Cancer Res Treat. 2005;93:177–188. doi: 10.1007/s10549-005-4794-6. [DOI] [PubMed] [Google Scholar]
- Sale EM, Hodgkinson CP, Jones NP, Sale GJ. A new strategy for studying protein kinase B and its three isoforms. Role of protein kinase B in phosphorylating glycogen synthase kinase-3, tuberin, WNK1, and ATP citrate lyase. Biochemistry. 2006;45:213–223. doi: 10.1021/bi050287i. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Scatena CD, Hepner MA, Oei YA, Dusich JM, Yu SF, Purchio T, Contag PR, Jenkins DE. Imaging of bioluminescent LNCaP-luc-M6 tumors: A new animal model for the study of metastatic human prostate cancer. Prostate. 2004;59:292–303. doi: 10.1002/pros.20003. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: New players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, Trepel J, Liang B, Patronas N, Venzon DJ, Reed E, Myers CE. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2:483–491. [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Towle HC, Kaytor EN, Shih HM. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu Rev Nutr. 1997;17:405–433. doi: 10.1146/annurev.nutr.17.1.405. [DOI] [PubMed] [Google Scholar]
- Wajant H. The Fas signaling pathway: More than a paradigm. Science. 2002;296:1635–1636. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Ohno T, Wakahara K, Nagano A, Kawai G, Saitou M, Takigami I, Matsuhashi A, Yamada K, Shimizu K. Simultaneous inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. J Cancer Res Clin Oncol. 2009;135:1125–1136. doi: 10.1007/s00432-009-0554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yart A, Chap H, Raynal P. Phosphoinositide 3-kinases in lysophosphatidic acid signaling: Regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim Biophys Acta. 2002;1582:107–111. doi: 10.1016/s1388-1981(02)00144-0. [DOI] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: Variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Gao WY, Turner S, Ducatman BS. Gleevec (STI-571) inhibits lung cancer cell growth (A549) and potentiates the cisplatin effect in vitro. Mol Cancer. 2003;2:1. doi: 10.1186/1476-4598-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]