

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection and associated diseases continue to represent major health problem worldwide. FOXO transcriptional factors play an important role in the regulation of cell apoptosis, cell cycle arrest, stress resistance, metabolism, and differentiation. This chapter will discuss the diverse functions of FOXO in different cell types including T cells, macrophages, neurons, and astrocytes within the context of HIV-1 infection. Given the overwhelming evidence that FOXO proteins influence the cell fate of immune cells, and involve in the homeostasis of the central nervous system (CNS), we will also discuss the potential role of FOXO factors in HIV-1-associated neurological disorders.
Keywords: FOXO, HIV-1, macrophage, astrogliosis, neurodegeneration
1. Introduction
The Forkhead Box O (FOXO) transcription factor family, the mammalian orthologs of Caenorhabditis elegans “forkhead protein” DAF-16, is characterized by a conserved DNA binding domain commonly known as a “forkhead box” or a “winged helix”.1-4 FOXO proteins play an essential role in the crosstalk between many signaling pathways, including cell cycle, metabolism, apoptosis, and cell survival. Regulation of FOXO transcription factors is carried out by a complex interplay of phosphorylation, acetylation, ubiquitination, and interaction with other protein partners. These post-translational modifications of FOXO proteins work in concert to determine the role that FOXO play in diverse cellular processes, which may be dependent on the microenvironmental cues and the appropriate downstream signals. The interplay of FOXO regulation may be involved in disease, which is the subject of intense investigation. Recently accumulating evidence shows that FOXO proteins play a critical role in the pathogenesis of HIV-1-infection. This chapter provides an overview of FOXO proteins and their potential roles in various pathological conditions such as HIV-1 infection and its associated neurological complications. Understanding the involvement of FOXO proteins in the pathogenesis of HIV-1 infection and other CNS-associated diseases may provide therapeutic targets for the treatment of HIV-1 infection and its associated neurodegenerative disorders and neuronal injury.
1.1. FOXO family members and general function
Four FOXO isoforms, namely FOXO1, FOXO3a, FOXO4, and FOXO6, have been identified in mammalian cells to date.1-5 The expression and function of FOXO isoforms have been investigated in great detail. Unlike other members of the family, FOXO6 is only detected in the developing brain and has different post-translational regulation mechanisms due to the lack of conserved C-terminus Akt-1 phosphorylation motif.5 FOXO1, FOXO3a, and FOXO4 are ubiquitously expressed and participate in diverse physiologic processes, including cell cycle regulation, differentiation, apoptosis, stress resistance, and metabolism. Notably, the cellular outputs and the cell fate decisions of FOXO are determined by a host of downstream factors that appear to be cell type- and microenvironment-specific. One important aspect of the FOXO family is that the gene expression pattern overlaps during development and in the adulthood, implying that FOXO proteins may bind to and regulate the same target genes. Thereby, FOXO isoforms display functional redundancy and compensation in vivo, whilst keeping isoform-specific function in some cell linages and tissues.
1.2. Regulation of FOXO protein activity
There are multiple post-translational modifications of FOXO proteins, including phosphorylation, acetylation, ubiquitination, and protein-protein interactions. Phosphorylation is the most critical modification as it essentially regulates the translocation of all FOXO proteins between the nucleus and cytoplasm. An exception to this is FOXO6, which is a nuclear factor and does not translocate out of the nucleus. FOXO phosphorylation primarily inhibits FOXO function with rare exceptions. Phosphatidylinositol 3-kinase (PI3K), which responds to growth factors and cytokines, has been known to regulate FOXO function.6-9 Akt-1 (also known as protein kinase B, PKB) phosphorylates FOXO3a at three residues and retains FOXO3a in the cytoplasm by promoting its association with adaptor molecule14-3-38, 10, 11, subsequent deactivation and degradation of FOXO3a. In the absence of Akt-1 phosphorylation, FOXO3a translocates to the nucleus, and controls cell cycle, apoptosis, and other processions through transcription of target genes.12-14 In addition to Akt-1, many kinases also phosphorylate FOXO. These include serum and glucocorticoid-regulated kinase (SGK), c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), dual specificity tyrosine-phosphorylated and regulated kinase (DYRK1A), mammalian Ste20-like kinase-1 (MST1) and IκB kinase (IKK) (for summary, see table 1).8, 11, 15 Though phosphorylation usually prevents activation of FOXO through cytoplasmic translocation, there are two exceptions to this rule. In response to stress stimuli, JNK and Mst1 phosphorylate FOXO at a distinct set of threonine residues and promote nuclear translocation leading to transcriptional activation (see table 1). Commonly, following cytoplasmic translocation, FOXO phosphorylation also results in FOXO ubiquitination and proteasomal degradation, further reducing the transcriptional activity of FOXO.10, 16-18
Table 1.
Summary of upstream kinases, phosphorylation sites, and the cellular outcomes for FOXO members.
| FOXO1 | FOXO3a | FOXO4 | Cellular outcome | References | |
|---|---|---|---|---|---|
| Akt-1 | T24, S256, S319 | T32*, S253*, S315 | T28, S193, S258 | Inactivation, cytoplasmic translocation | 19-22 |
| SGK | T24, S256, S319 | T32#, S253, S315# | T28, S193, S258 | Inactivation, cytoplasmic translocation | 11, 23-25 |
| CK1 | S322, S325 | S318, S321 | S261, S264 | 26, 27 | |
| CDK2 | S249 | 28 | |||
| MST1 | S212 | S207 | Activation, interact with JNK pathway | 12 | |
| DYRK1 | S329 | S325 | S268 | 29 | |
| ERK | S344, S294, S425 | Inactivation, cytoplasmic translocation | 30 | ||
| JNK | T447, T451 | Activation, nuclear translocation | 31 | ||
| IKKβ | S644 | Cytoplasmic translocation and ubiquitination | 32 |
indicates the phosphorylation preference of Akt-1, and
indicates the phosphorylation preference of SGK.
Another intriguing regulation method of FOXO is acetylation and/or deacetylation. Due to its influence on phosphorylation, acetylation can also affect FOXO subcellular localization. FOXO can be acetylated by the calcium response element-binding (CREB)-binding protein (CBP), p300, and p300/CBP-associated factor (PCAF); whereas FOXO can be deacetylated by histone deacetylases (HDACs) and NAD-dependent deacetylases.33-36 Acetylation may suppress FOXO proteins’ activity and serve as a negative feedback signal during FOXO activation. Acetylation-defective FOXO1 mutants have higher transcription than wild-type FoxO1, but the mutant FOXO1 tends to be rapidly ubiquitinated and degraded. However, mutations that mimic the acetylated state of FOXO increase stability but impair FOXO1 transcription.37 More specifically, acetylation of FOXO by CBP and/or p300 prevents the binding of FOXO to its target DNA, reduces the stability of the FOXO-DNA complex, and increases the phosphorylation of the non-essential Akt-1 phosphorylation site.38, 39 Deacetylation, on the other hand, may enhance the transcription activity of FOXO. During oxidative stress, SIRT2 expression increases and reduces the acetylation level of FOXO3a. As a consequence, FOXO transcriptional activity increases along with its target genes, p27Kip1, manganese superoxide dismutase (MnSOD), and Bcl-2-interacting mediator of cell death (Bim), which work in concert to induce oxidative stress-mediated apoptosis.40 Interestingly, in mouse pancreatic β cells, acetylation and deacetylation seems to reach equilibrium to maintain FOXO3a transcription activity. FoxO1 protects pancreatic β cells against oxidative stress by forming a complex with promyelocytic leukemia protein (Pml) and deacetylase SIRT1, subsequently activating the expression of NeuroD and MafA, two Insulin2 (Ins2) gene transcription factors that are known to alleviate oxidative stress.
Ubiquitination of FOXO proteins provide another avenue to regulate their transcriptional activities. Notably, poly- and mono-ubiquitination result in different cellular outcomes for FOXO. Phosphorylation by Akt-1, ERK, or SGK not only retains FOXO in cytoplasm but also facilitates the polyubiquitination and degradation of FOXO. This polyubiquitination provides a potential negative feedback regulation to properly control FOXO activity in response to growth factor signaling.17, 30, 41-43 Phosphorylation of FOXO3a C-terminal residue Ser-644 by IKK also promotes polyubiquitination and degradation of FOXO3a through the proteasome pathway. Residue Ser-644 is exclusive to FOXO3a and is absent in other FOXO proteins.44 Though polyubiquitination promotes degradation of FOXO,41, 45 monoubiquitination of FOXO in response to cellular oxidative stress leads to the nuclear translocation and transcriptional activation of FOXO proteins.46-48 Monoubiquitination of FOXO is counteracted by USP7, a deubiquitinating enzyme that binds to FOXO proteins and represses FOXO’s activity. Surprisingly, neither monoubiquitination nor USP7-mediated deubiquitination affects FOXO protein stability.48
One crucial protein in the regulation of subcellular localization of FOXO is the chaperone protein 14-3-3, which plays a direct role in the phosphorylation and acetylation of FOXO. The 14-3-3 protein has a U-shaped structure that serves as a dock for the phosphorylated serine or threonine residues of FOXO. The 14-3-3 protein isoforms are also able to form stable homo- or heterodimers and thus could bind two ligands simultaneously.49, 50 Many of the phosphorylated serine or threonine residues are located in the FOXO nuclear localization sequence (NLS). Binding of 14-3-3 proteins to FOXO therefore masks or obscures the NLS and subsequently prevents FOXO proteins’ translocation into the nucleus.11, 12, 49, 51 In addition to binding to the phosphorylated residues, the 14-3-3 proteins seem to be required to bridge FOXO3a and SIRT1 together to facilitate the deacetylation process.
The fine-tuned regulation of FOXO transcriptional activity typically involves the interaction of FOXO and other partners. Depending on the cell type and cues from the microenvironment, these protein-protein interactions can either foster or suppress FOXO transcriptional activity and affect the subsequent cellular response. FOXO proteins play key roles in cell cycle control through p21Cip, p27Kip, and other genes by directly binding to their promoters. Furthermore, in the case of transforming growth factor beta (TGFβ) stimulation, FOXO requires additional proteins, namely Smads and FOXG1, to control the expression of the growth inhibitory gene p21Cip1. Smad2 and Smad3 hetero-oligomerize with Smad4, translocate to the nucleus, and form a complex with FOXO. This complex can bind to the promoter of p21Cip and turn on its expression. In contrast, FOXG1, a protein from a distinct FOX subfamily, inhibits p21Cip1 expression through binding FOXO/Smad complexes. These interactions enable the TGFβ/Smad pathway and the FOXG1 to delicately regulate the expression of downstream factors of FOXO.52-56 In addition, p15INK4b, another downstream factor of FOXO, requires CCAAT/enhancer binding protein β (C/EBPβ) and the FOXO-Smad complex to properly respond to TGFβ.52 RUNX3, a runt domain-containing transcription factor, is also required by FOXO to induce the expression of Bim. The Bim promoter contains one FOXO binding site and two RUNX3 binding sites in close proximity, and the interaction of FOXO and RUNX3 coordinately upregulates Bim expression and promotes apoptosis in gastric cancer cells.57
The interaction of FOXO proteins and their partners may release the transcriptional repressor from the promoter of target genes, and this removal leads to the expression of these genes. In this case, FOXO proteins serve as co-activators rather than specific transcription factors. For example, the tumor suppressor p53 normally inhibits Sirt1 expression by binding to two sites of Sirt1 promoter. Under nutrient deprivation, FOXO binds to p53 and releases p53 from the Sirt1 promoter, therefore activating Sirt1 expression. This interaction between FOXO and p53 is independent of the binding between FOXO and the Sirt promoter.58 Similarly, in muscle cells, the binding of FOXO with the transcriptional repressor Csl also releases Csl from Hes1 promoter and induces Hes1 expression. In addition, FOXO may interact with other proteins and inhibit FOXO transcription activity in return. For example, FOXG1 binds to FOXO-Smad complex and leads to FOXO target gene suppression. Furthermore, the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) may inhibit FOXO transcription through the formation of a protein complex in the FOXO promoter region.59
2. FOXO in HIV-1 infection and HIV-1 associated neurocognitive disorders
In HIV-1 pathogenesis, depletion of T cells and other immune cells is the most fundamental pathophysiological consequence. Notably, the immune activation may induce T cells and other cell loss in different stages of HIV-1-infection.60, 61 HIV-1 can directly kill the infected CD4+ T cells and destroy the uninfected and bystander cells simultaneously. In addition to the disruption of the peripheral immune system, HIV-1 also leads to a spectrum of viral-induced neurocognitive disorders. Although the mechanisms have yet to be fully elucidated, but HIV-1-mediated brain inflammation including overproduction of cytokines, chemokines, glutamate and others factors have been shown to play significant roles in disease progression.62-64 HIV-1 usually enters the brain shortly after initial infection, crossing the blood brain barrier via peripherally infected monocytes.65 Brain macrophages and microglia, unlike other cellular residents in the CNS, are able to sustain a productive HIV-1 infection within the brain.66 Therefore, while the rest of the body in general experiences a rise of viral load followed by a gradual decline, the isolated CNS maintains a low level, but persistent of HIV-1-infection. Although neurons are not infected by HIV-1, a dementia specific to HIV-1 has been described as HIV-1-associated dementia (HAD).67, 68
HAD is the clinical consequence of neuronal injury and dropout. The pathologic correlate to HAD, HIV encephalitis (HIVE), is characterized by HIV-1-infected and -activated macrophages and microglia, damage of neuronal dendrites and axons, and apoptotic neurons. As a result of HIV-1 infection, the immune cells are recruited to the proximate site and produce an array of factors including cytokines, proteases and other factors, yet they are unable to clear the infection. The difficulty in eradicating HIV-1 infection prolongs the immune response leading to a chronic inflammatory state. Chronic inflammation has both detrimental and beneficial effects. On one hand, these responses are essential in limiting viral spread; yet on the other hand, excessive inflammation is detrimental to resident cells such as neurons or neural stems cells that are important for the neuronal repair process.
HIV-1 infection and its consequence in the brain, macrophage/microglia infection and activation and its associated neuronal injury and/or loss, and the changes in neuronal repair process have been the focus of much investigation. As introduced previously, FOXO is a key factor in the determination of cell fate in different environments. In this section, we will discuss the potential roles of FOXO in HIV-1 infection, its associated immunodeficiency, and the long-term consequences on the host central nerve system during HIV-1 infection.
3. FOXO in T cell depletion
3.1. Potential signaling pathway of FOXO3a in T cell depletion
HIV-1 Infection leads to progressive CD4+ T cell depletion, resulting in AIDS (Acquired Immune Deficiency Syndrome) development. The mechanisms that trigger the CD4+ T cell death are not fully understood, but data indicate that apoptosis plays a major role in this cell loss. Both infected and uninfected CD4+ T-cells can die during HIV-1 infection by different cell death pathways, and bystander CD4+ T cell loss is now recognized as essential to the immunodeficiency.69-71
The general roles of FOXO proteins in the immune system might be relevant to immune homeostasis.72-74 Normally, FOXO inactivation is indispensable to maintain T cell survival and proliferation. Once FOXO is activated, FOXO triggers apoptosis in T cells by regulating the expression of several pro-apoptotic genes, such as FasL (Fas ligand, also known as CD95 ligand), Bcl-6 (B-cell lymphoma 6), Bim and Puma (p53 upregulated modulator of apoptosis).75-79 In HIV-1-induced T cell apoptosis, FOXO may also play an important role. Accumulating evidence suggests FOXO members participate in HIV-1-induced T cell apoptosis, directly or indirectly, through a differential regulation of apoptosis. Specifically, HIV-1-infection can trigger both intrinsic and extrinsic apoptotic pathways that are regulated via FOXO in infected and uninfected T cells during HIV-1 infection.
Several HIV-1 proteins have been shown to interfere with cellular proteins implicated in the control of cell cycle and apoptosis, particularly, cell cycle G2 arrest.80 HIV-1 protein Vpr induces cell cycle G2 arrest and blocks infected cells from proliferating.80 Vpr blocks cell cycle progression by activating the ATR (ataxia-telangiectasia and Rad3-related) complex (including ATR, Rad17 and Rad9-Hus1-Rad1), leading to Cdc25c functional suppression, Cdk1, and cyclin B downregulation. At the same time, activation of ATR complex also induces Gadd45a expression. Both G2 arrest and Gadd45a expression result in Bax activation, which induces apoptosis via the mitochondrial pathway.81-85 Interestingly, FOXO3a has been mechanistically linked to Vpr-induced cell cycle arrest and apoptosis.84, 86-88 Vpr may modulate FOXO’s function through two ways. First, Vpr is able to interfere with the association of FOXO3a with 14-3-3 and subsequently impede the shuttling of FOXO3a from the nucleus to the cytoplasm. Second, Vpr inhibits insulin/PI3K/Akt-1 signaling pathway, leading to FOXO3a activation and translocation to the nucleus.89, 90 The activation of FOXO may also induce G2/M cell cycle arrest through the upregulation of Gadd45a and cyclin G2.91, 92 In addition, FOXO may facilitate cell cycle arrest through the inhibition of the FOXM, which is known to positively drive G2/M phase transition.29, 93
FOXO is also involved in HIV-1 protein Tat-induced CD4+ T cell apoptosis. Tat triggers Egr1-PTEN-Akt-1 (early growth response-1/phosphate and tensin homolog deleted on chromosome 10/Akt-1) and p53 pathways, which converge on the regulation of FOXO3a transcription factor and result in FOXO3a activation. The FOXO3a target genes, FasL and TRAIL (Tumor necrosis factor(TNF)-related apoptosis-inducing ligand), are the primary TNF family members that engage in the extrinsic apoptotic pathway; while Puma, Noxa, and Bim are members of BCL-2 family that participate in the intrinsic apoptotic pathway. Therefore, HIV-1 protein Tat could induce apoptosis in CD4+ T cells through multiple pro-apoptotic target genes associated with FOXO3a.94 As Tat can be secreted by infected cells and taken up by uninfected cells, Tat-induced apoptosis could potentially affect both infected cells and uninfected cells.95, 96
As reviewed by Selliah, other HIV-1 proteins have been implicated in aspects of apoptosis, including Nef, Vpu, Env, and protease.80 Limited evidence is available to confirm the involvement of FOXO in apoptosis induced by those aforementioned proteins. However, the potential function of FOXO members through their target genes (such as FasL, TRAIL, and Bim), and the participation of these genes in the HIV-1-induced cell death, indicates that FOXO might contribute to the cell death induced by HIV-1-infection or HIV-1 encoded proteins.
3.2 FOXO3a commits to the survival of central memory CD4+ T cell in HIV-infection
CD4+ central memory (TCM) and effector memory (TEM) T cells are two distinct populations of memory T cells that can recognize foreign invaders such as bacteria or viruses upon second encounter. TCM and TEM have intrinsic different properties regarding proliferation, apoptosis and persistence. TCM cells are more resistant to apoptosis and have an increased capacity to proliferate or survive than TEM cells in vitro. These fundamental functional differences of TCM and TEM are conferred by the activation and phosphorylation status of two transcription factors, STAT5 and FOXO3a.97 In response to proliferative signals, TCM cells showed a significant increase in the levels of STAT5 phosphorylation compared with TEM cells; moreover, ex vivo TCM cells express higher levels of the inactive phosphorylated forms of FOXO3a and lower levels of the pro-apoptotic FOXO3a target protein Bim. The high level of active STAT5 and inactive FOXO3a ensure the TCM cell longevity and survival, which is critical in immunological memory. In HIV/AIDS, the persistence of TCM cells is critical to maintain proper immunological functions, as the rate of TCM cell decline predicts HIV disease progression. TCM and TEM cells from HIV+ elite controller (EC) subjects, who naturally control viral replication, are less susceptible to FasL-mediated apoptosis and survive longer after multiple rounds of T cell receptor activation when compared to TCM and TEM cells from successfully treated aviremic subjects or from HIV-1 seronegative donors. The persistence of TCM cells from EC subjects is a direct consequence of inactivation of the FOXO3a pathway. Silencing the FOXO3a by small interfering RNA or introducing a FOXO3a dominant-negative form extends the long-term survival of TCM cells from successfully treated subjects to a length of time similar to that of TCM cells from EC subjects. Therefore, inactivation of FOXO3a in both TCM and TEM cells of HIV patients may benefit the immune response specifically to HIV-1 and protect T cells from apoptosis. The crucial role of FOXO3a in the persistence of memory T cells provides a new prospect of therapeutic avenue to control the HIV-1 persistence.97, 98
4. FOXO in macrophage/monocyte pathology of HIV-1-infection
Macrophage represents early cellular target and a reservoir of HIV-1 in its natural host. Compared to T cells, macrophage/monocytes are more resistant to cytopathic effects of virus and sustain long-term productive infection throughout the disease course. Although the virus follows similar life cycle in macrophages and T lymphocytes, the infected macrophages are prone to evade the immunological attack, which results in the establishment of long-term reservoirs in macrophages and subsequently disseminates the virus to various tissues such as the brain and lung. The investigation of these tissue macrophages is often difficult because of their limited accessibility and inefficient recovery. Therefore, many in vitro studies of infection utilize monocyte-derived macrophages (MDMs), which provide a unique model for effective laboratory and primary HIV-1 infection. With this cellular model, we have found that FOXO3a contributes to HIV-1-mediated cell death of macrophages during productive infection. Similar role of FOXO3a has been identified in lymphocyte apoptosis. However, significant question remains to be answered regarding the exact role of FOXO3a in human macrophage in vivo. Whether the result in the in vitro cellular model could fully recapitulate the complexity of the viral replication, assemble, and transmission in vivo? The complex nature of viral infection requires an integration of viral proteins, RNA, and a range of host cellular factors. Therefore, multiple metabolic or cellular signaling pathways may participate and interfere with the regulation of transcription factors such as FOXO, which often affect the data interpretation and variable results may co-exist in the literature.99
4.1. The potential relationship of FOXO proteins and viral replication in macrophage
HIV-1 accessory protein Vpr, which arrests T cells in cell cycle G2 phase, has been found to disrupt the interaction between adaptor molecule 14-3-3 and FOXO3a. The transcriptional activity of FOXO3a is normally suppressed by insulin-induced sequestering of this protein in the cytoplasm. Vpr inhibited insulin-mediated Akt-1 phosphorylation and may change the subcellular localization of FOXO3a. Vpr may also interfere with insulin-induced co-precipitation of 14-3-3 and FOXO3a and antagonize the negative effect of insulin on FOXO3a transactivation on FOXO-responsive promoter. These results indicate that Vpr has the potential to activate FOXO3a and subsequent cause cell cycle arrest of HIV-1-infected cells.90 In contrast to CD4+ T cells, HIV-1-infected macrophages, which are terminally differentiated cell, typically resist cell death, support viral replication, and consequently, may facilitate HIV-1 transmission. There is evidence that shows HIV-1 accessory protein Vpr may also affect viral replication in macrophages through transcription factor FOXO3a. HIV-1 accessory protein Vpr has been found to regulate cyclin-dependent kinase inhibitor 1A (p21Cip), which is significantly upregulated during HIV-1 replication.100 The signaling pathway involved in Vpr-mediated p21 increase is unknown. It has been reported that transcription factor FOXO3a binds to p21 promoter and triggers p21 expression. Therefore, it is possible that Vpr activates FOXO3a through interfering with the 14-3-3-FOXO3a interaction and leads to p21 protein upregulation and contributing to viral replication.100
Another potential player interacting with FOXO3a in macrophage during HIV-1 infection is NF- κB. FOXO3a could inhibit NF-κB activity, because Foxo3a-deficient mice show increased NF- κ B activation.74 In HIV-1 infection, FOXO3a may also play a similar role in the inhibition of macrophage NF- κ B activation. It has been known that HIV-1 replication in macrophage requires NF- κB activity. In the early stage of HIV-1-infection, NF- κ B is activated by upstream kinases; and FOXO is functionally inhibited by its upstream kinases such as Akt-1 or ERKs. This functional inhibition is important for NF- κ B activation, which would promote HIV-1 replication and inflammatory cytokine production in HIV-1-infected macrophages.101 In the later stage of HIV-1-infection, productive HIV-1 infection attenuates PI3k/Akt-1 pathway, which lead to the activation of FOXO and translocation of FOXO into the nucleus. Activated FOXO may further inhibit NF-κB activity, preventing its pro-survival function as demonstrated in infected macrophages.
4. 2. The dual role of FOXO3a in HIV-infection in macrophages
The exact molecular changes of protein profile in macrophages during HIV-1-infection in vivo remain to be fully understood. Studies have shown that HIV-1-encoded proteins are able to manipulate cellular pathways, modifying the apoptotic machinery that regulates host cell death in either a pro- or anti-apoptotic manner. This is critical during early stage of HIV-1 infection, whereas proper cell survival is needed to ensure viral replication. Akt-1 and NF-κB, both important for macrophage survival, are activated and cells are highly resistant to cell death compared with other tissue cell types. With infection progresses, RNA transcription in productively infected macrophages indicates a conflicted state where pro-apoptotic and anti-apoptotic cascades are modified as the cells respond to HIV-1. Death factors such as TRAIL, TNF, and Fas are upregulated and the anti-apoptotic factors Bcl-2, NAIP (neuronal apoptosis inhibitory protein), and Akt-3 are significantly downregulated, but survival factors including XIAP (X chromosome linked Inhibitor of apoptosis protein), MDM2 (murine double minute 2), and SOD2 (superoxide dismutase 2) are upregulated as well (N. Erdmann et al. manuscript in preparation). These data suggest that HIV-1 infection in macrophages is quite dynamic and HIV-1 may evolve by specifically modulating the survival-apoptotic equilibrium in favor of optimal viral replication. Disruption of the equilibrium in either way during viral life cycle has been proved to be detrimental. The role that FOXO3a plays in the HIV-1 infection course may also be bidirectional. First, during the early stage of infection, phosphorylation of Akt-1 and FOXO3a is increased and this ensures adequate cell survival for HIV-1 replication.102, 103 HIV-1 proteins, such as Tat, gp120 and Nef, have been shown to activate the PI3K/Akt-1-dependent survival pathway, which facilitates HIV-1 replication and viral particle production.102, 104-106 Second, PI3K/Akt-1 pathway is important for the resistance to cell death of HIV-1-infected macrophages. As the inhibition or attenuation of Akt-1 activity dramatically reduces the viability of long-living virus-infected macrophages. Alternatively, inhibition or attenuation of Akt-1 may sensitize infected macrophages to stresses or extracellular stimuli, which would otherwise not have caused cell death of macrophages.103 Indeed, both the phosphorylation of Akt-1 and FOXO3a decreased once productive infection established.107, 108 These evidences suggest that PI3K/Akt-1 activation contributes to viral replication and macrophage resistant to cell death in the early stage of infection. As a main downstream factor of PI3K/Akt-1 pathway, FOXO is regulated through phosphorylation on T32, S253, and S315 (FOXO3a) or on homologous sites (other family members). The detailed mechanism of how Akt-1 activation leads to FOXO3a inhibition and subsequent apoptosis-resistance has been well documented. However, the exact role of this signaling pathway play during HIV-infection has remained to be fully elucidated. HIV-1 does not induce significant apoptosis during early replication. Once the productive infection established, HIV-1 increases the activity of transcription factor FOXO3a by translocation to nucleus. Adenoviral delivery of constitutively active FOXO3a, which contains three mutated phosphorylation sites maintaining a transcriptional active FOXO3a was found to induce DNA fragmentation with decreased cell viability in MDM. Moreover, a dominant-negative mutant of FOXO3a, or small interfering RNA for FOXO3a in HIV-1-infected MDM decreased DNA fragmentation and protected macrophages in HIV-1-infected MDM, which suggests elevated FOXO3a activity promotes HIV-1-infected macrophage cell death.108 In addition, overexpression of constitutive active Akt-1 is sufficient to induce FOXO3a phosphorylation, suggesting that FOXO3a is a down stream of Akt-1 in macrophage.108. Comparison of primary HIV-1 isolates with laboratory strains also indicates that a similar infection course and cell loss during productive infection. The infection levels and cell loss are associated with the phosphorylation status of Akt-1 and FOXO3a, suggesting Akt-1/FOXO3a pathway plays an important role in HIV-1-induced cell death of human macrophage.
Based on the studies described above, we propose that FOXO3a may play a dual role in HIV-1-infected macrophages. In the early stage of infection, PI3K/Akt-1 are activated leading to FOXO3a inactivation and subsequent cell resistance to cell death; with the virus replicate and accumulate in macrophages, the PI3K/Akt-1 pathway is gradually downregulated and leads to FOXO3a activation. As a consequence of FOXO3a activation, cell death and apoptosis signaling pathways are triggered that result in macrophage cell death. Further investigation of this proposed model and the elucidation of the PI3K/Akt-1/FOXO3a pathway and its role in macrophages during HIV-1 infection should continue, as it will bring further understanding of HIV-1 pathogenesis.
5. FOXO and HIV-1 mediated central nervous system damage
HIV-1-infected monocytes or macrophages infiltrate into the CNS and may serve as a viral reservoir for persistent replication. Currently, about 40% to 70% of people infected with the HIV-1 develop CNS disorders and neurological complications.109, 110 More serious neurological symptoms typically present in patients with high HIV loads, generally when a person has advanced AIDS. HIV-associated neurocognitive disorder (HAND), which includes HIV-1-associated mild neurocognitive disorders and HIV-1 associated dementia (HAD), is frequently accompanied by neuronal injury and loss of neuronal subpopulations in the neocortex, limbic system, and basal ganglia in association with synaptic and dendritic damage, astrogliosis, and formation of microglial nodules and multinucleated giant cells.111 Although HIV cannot infect neurons, supportive cells of the nervous system, such as astrocytes and microglia, as well as monocyte/macrophages that have migrated to the brain, can be infected with the virus. Researches in this field suggest that HIV-1-infected cells can release proinflammatory cytokines, chemokines and some toxic products, and deliver aberrant signals, leading to neuronal toxicity and neuronal damage in the brain.
In this section, we will further discuss the role of FOXO members in HIV-1-induced neurological disorders. It is acknowledged that HIV-1-induced neurological disorders share some similar molecular mechanisms with other neurodegenerative diseases as it decreases neuronal survival, changes in neural stem/progenitor cells function, and causes astrogliosis. Thus, we will expand our topic to the potential function of FOXO in general neuronal stem/progenitor cell homeostasis, neuronal apoptosis and astrogliosis.
5.1. FOXO proteins in the regulation of stem cell homeostasis in the nervous system
FOXO proteins are homologous to C. elegans Daf-16, which determines metabolic insulin signaling and leads to lifespan extension. It is known that insulin-like signaling is essential for growth and metabolism in C. elegans. Inhibition of insulin-like signaling leads to Daf-16 activation and increase of stress resistance and longevity.112 Restored insulin-like pathway in neurons is sufficient to reinstate a wild-type life span.112 Further investigation revealed that Daf-16 is the key factor downstream of insulin-like pathways that controls the life span of C. elegans and regulates the expression of free radical-scavenging enzymes, catalase and SOD. In mammals, it has also been reported that FOXO proteins are important to maintain the stem cell pool. In Foxo3a-deficient mice, the proliferation and differentiation of hematopoietic progenitors were normal, but the number of colony formation cells was reduced. The ability of Foxo3a-/- hematopoietic stem cells (HSCs) to support long-term reconstitution of hematopoiesis was also impaired and was coupled to an elevation of ROS, defective maintenance of quiescence, and hypersensitive to cell-cycle-specific myelotoxic injury. Consequently, HSC frequencies were significantly decreased in aged Foxo3a-deficient mice.113 Considering the redundancy and compensability of three FOXO members, another group use conditional knockout of all FoxO1, FoxO3, and FoxO4 in the adult mouse hematopoietic system. FoxO-deficient mice exhibited the expansion of both the myeloid and lymphoid lineages accompanied with cell cycle progression of the long-term hematopoietic stem cells, suggesting that FoxO preoteins are important in maintaining HSCs in the quiescent state. The FoxO-deficient HSCs also display an increased level of apoptosis further contributing to the aberrant decrease in cell number.114 All these observations demonstrated that FOXO proteins are important in the maintenance of stem/progenitor cell homeostasis via cell cycle regulation and functional resistance to oxidative stress.
How FOXO regulates the function of neural stem cells or progenitor cells remains unclear, but evidence shows that FOXO proteins also play a role in the regulation of neuronal precursor cells. Erythropoietin (EPO), the traditional mediator of erythroid maturation, was found to modulate neural stem cell in the cellular protection and angiogenesis during development. EPO significantly increased neural progenitor cell proliferation and promoted neural progenitor cell differentiation into neurons, while it also functioned as a protective and an anti-inflammatory factor during oxidative stress.115 Further signaling studies demonstrate that EPO can activate Akt-1, JAK2, and negatively regulate downstream transcription factor FOXO3a.115-117 This study indicates that FOXO may play a similar role in neuronal stem cells and in hematopoietic stem cells as both share common properties of all stem cells. Based on this understanding and our observation on neuronal stem cells, we propose the following hypotheses. First, FOXO proteins play a role in the maintenance of neuronal stem/progenitor cell homeostasis. FOXOs control the cell cycle of stem cells and maintain the majority of stem cells in a quiescent state while a subset of them enter the cell cycle for self-renewal or differentiation. Second, FOXO proteins prevent neuronal stem/progenitor cells from oxidative stress-induced cell damage through scavenging free radicals, regulating SOD expression. Note that many aspects of this model have been derived from studies in non-neuronal stem cells, and have been extrapolated to neuronal stem/progenitor cells here.
5. 2. FOXO proteins are pivotal factors in neuronal apoptosis
In the nervous system, aberrant neuronal death is a feature of neurodegenerative diseases. Compared with other cell types, neurons are more sensitive to stress or other apoptotic stimuli than other cells of the brain. Indeed, oxidative stress-induced neuronal death is involved in Alzheimer’s disease, HIV-associated dementia, and other neuronal disease.118-120 In response to stress stimuli, FOXO-triggered expression of downstream factors, Bim, FasL, Puma, and TRAIL, may contribute to neuronal death. A recent study shows that oxidative stress elicits neuronal death through activation of FOXO by a dual process that involves timed activation of stress kinases and abrogation of IGF-I neuroprotection. On the one side, ROS includes activation of p38 MAPK to inhibit IGF-I signaling by interfering IGF-I receptor/IRS-1 interactions through phosphorylation of IRS-1. This leads to abrogation of AKT-1 inhibition of FOXO. On the other side, ROS recruits JNK2 to activate FOXO. These two pathways are independently activated in response to ROS; but both pathways inhibit FOXO3a trafficking from the nucleus to the cytoplasm and result in FOXO3a transcriptional activation and downstream pro-apoptotic factor Bim expression.121 Similarly, epileptic brain injury in rats leads to FOXO1 and FOXO3a activation in hippocampal neurons after Bim upregulation and neuronal apoptosis.122 Many other studies also demonstrated that FOXO transcriptional regulators provide an important link between stress signaling pathways and the neuronal cell death.122-125
5.3. FOXO3a in proinflammatory cytokine-induced astrogliosis
Reactive astrogliosis, including astrocyte proliferation and activation, is also one of the hallmarks of neurodegenerative diseases. Astrocytes proliferation in response to abnormal stimuli contributes to astrogliosis during brain disorders. The cell cycle inhibitors (cyclin-dependent kinase inhibitors), including flavopiridol, roscovitine, and olomoucine inhibit cell cycle progression at the G1/S and G2/M phases and reduce reactive astrogliosis initiated by ischemia or traumatic brain injury.126, 127 The PI3k/Akt-1 pathway seems to be important in the cell proliferation and cell cycle regulation in astrocytes. It has been reported that PI3K/Akt-pathway is involved in the process of injury-induced astroglial proliferation and anti-apoptosis in vivo. p-Akt1/2/3 increased in immunostaining in temporal correlation with the mechanical damage with a peak at 2 hours. The phosphorylated Akt positive-cells were often found co-labeled with GFAP around the stab wound.128 This indicated that the brain injury could activate Akt in astrocytes and subsequent astrocyte proliferation and result in astrogliosis.
Neurodegeneration In the diseases is often found to be accompanied with an increase of proinflammatory cytokines, such as IL-1β, TNF-α and IL-6, and these proinflammatory cytokines have been showed to mediate astrogliosis; however, the mechanisms by which this process occurs are not well defined. The investigation in the role of FOXO3a in inflammatory factor-mediated astrocyte proliferation has suggested that FOXO3a interfere astrogliosis via cell cycle regulation (Cui et al unpublished observation). IL-1β and TNF-α induced a significant increase of astrocyte proliferation as determined by Ki67 immunostaining. Cyclin D1, which marks the cell cycle progression, was also increased. FOXO3a, the main upstream regulator of cyclin D1, was phosphorylated and translocated from the nucleus to the cytoplasm with IL-1β and TNF-α stimulation. Wild-type FOXO3a (WT-FOXO3a) overexpression through adenovirus vector significantly upregulated downstream factor p27 and subsequently inhibited cyclin D1, which can induce cell cycle G1 phase arrest; at the same time, WT-FOXO3a upregulated Gadd45α expression, which can arrest cell cycle in G2 phase. In contrast, dominant-negative FOXO3a (DN-FOXO3a) decreased p27 and Gadd45α while upregulated cyclin D1. Consequently, WT-FOXO3a inhibited astrocyte proliferation. All these results demonstrated that FOXO3a is a pivotal transcriptional factor in proinflammatory cytokine-induced astrogliosis.
6. Summary and Future Directions
The regulation and function of the FOXO family have been extensively studied in the last decade and substantial progress has been made in understanding the signaling pathways and mechanisms involved in different cell types and systems. However, many questions remain to be answered about FOXO in HIV-diseases. FOXO appears to be important in the cell survival and the apoptosis in both HIV-1-infected cells and non-infected cells. Unraveling the multifaceted aspects of FOXO regulation will provide important insights to all the processes including T cell and macrophage apoptosis/survival in HIV-1 infection, neuronal apoptosis and astrogliosis in HIV-neurological diseases. Thus, a detailed understanding of FOXO proteins and their biology will provide new opportunities for developing more effective therapeutic approaches to treat HIV-diseases.
Fig. 1. Proposed mechanisms for how FOXO affects macrophage function during early and late stage of HIV-1 infection.
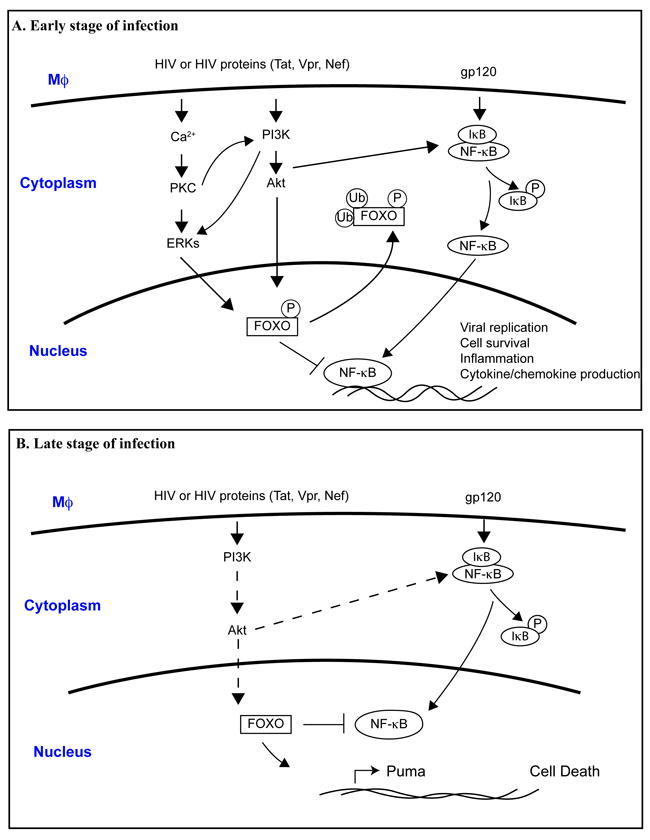
A. In the early stage of HIV-infection, the binding of HIV-1 or HIV-1 proteins with macrophage cell surface receptors induces intracellular signaling cascades such as Akt-1, ERKs, and NF-κB pathways. Activated Akt-1 and ERKs may inhibit FOXO function by phosphorylation. Subsequently, phosphorylated FOXO translocates to the cytoplasm and facilitates ubiquitination and degradation. As a consequence, inhibitory effect of FOXO to NF-κB was removed, which leads to enhanced NF-κB activation that promote viral replication, cell survival, inflammation and cytokine/chemokines production.
B. In the late stage of HIV-infection, it has been suggested that productive HIV-1 infection compromises PI3k/Akt-1 pathway, which lead to the activation of FOXO and translocation of FOXO in the nucleus. Activated FOXO triggers apoptosis pathways through increased expression of apoptotic proteins such as Puma. Activated FOXO can also inhibit NF-κB, preventing its pro-survival function. Dashed line indicates signal attenuation.
Fig. 2. Proposed mechanism for how FOXO influence neurons, astrocytes and neuronal progenitor cell function.
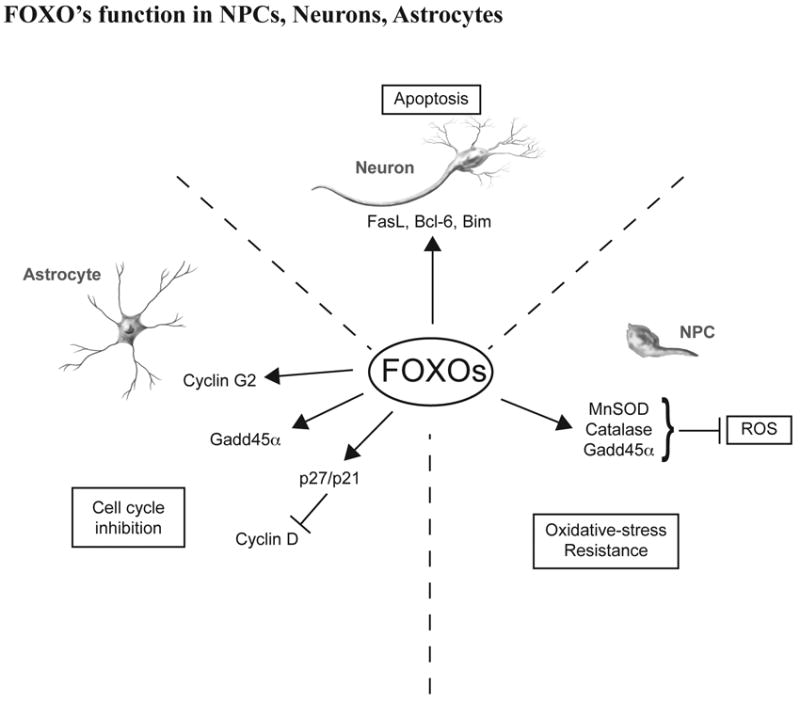
In response to oxidative stress or starvation, FOXO–dependent transcription in neurons serves to trigger apoptosis by inducing gene expression of FasL, BcL-6, Bim, etc. In astrocyte, FOXO suppress cell proliferation by inducing cell cycle regulatory proteins cyclin G2, Gadd45a and p27/p21. FOXO also play a crucial role in the homeostatic maintenance of neuronal progenitor cells by coordinating quiescence, stress resistance, and/or terminal differentiation.
References
- 1.Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993 Nov;5(3):230–235. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 2.Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997 Nov 1;90(9):3714–3719. [PubMed] [Google Scholar]
- 3.Anderson MJ, Viars CS, Czekay S, Cavenee WK, Arden KC. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics. 1998 Jan 15;47(2):187–199. doi: 10.1006/geno.1997.5122. [DOI] [PubMed] [Google Scholar]
- 4.Borkhardt A, Repp R, Haas OA, et al. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997 Jan 16;14(2):195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003 Sep 19;278(38):35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 6.Ho KK, Myatt SS, Lam EW. Many forks in the path: cycling with FoxO. Oncogene. 2008 Apr 7;27(16):2300–2311. doi: 10.1038/onc.2008.23. [DOI] [PubMed] [Google Scholar]
- 7.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007 Aug 1;120(Pt 15):2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 8.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE. 2003 Mar 4;2003(172):RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 9.Arden KC. FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene. 2008 Apr 7;27(16):2345–2350. doi: 10.1038/onc.2008.27. [DOI] [PubMed] [Google Scholar]
- 10.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008 Apr 7;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 11.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999 Mar 19;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 12.Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006 Jun 2;125(5):987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 13.Skurk C, Maatz H, Kim HS, et al. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004 Jan 9;279(2):1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- 14.Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 2004 Aug 1;104(3):784–787. doi: 10.1182/blood-2003-09-3071. [DOI] [PubMed] [Google Scholar]
- 15.Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf) 2008 Jan;192(1):19–28. doi: 10.1111/j.1748-1716.2007.01780.x. [DOI] [PubMed] [Google Scholar]
- 16.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008 Apr 7;27(16):2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Regan KM, Wang F, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005 Feb 1;102(5):1649–1654. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007 Aug 16;1(2):140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 19.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999 Jun 11;274(24):17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 20.Tomizawa M, Kumar A, Perrot V, Nakae J, Accili D, Rechler MM. Insulin inhibits the activation of transcription by a C-terminal fragment of the forkhead transcription factor FKHR. A mechanism for insulin inhibition of insulin-like growth factor-binding protein-1 transcription. J Biol Chem. 2000 Mar 10;275(10):7289–7295. doi: 10.1074/jbc.275.10.7289. [DOI] [PubMed] [Google Scholar]
- 21.Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999 Jun 11;274(24):17184–17192. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 22.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999 Apr 15;398(6728):630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 23.Richards JS, Sharma SC, Falender AE, Lo YH. Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-I, estrogen, and the gonadotropins. Mol Endocrinol. 2002 Mar;16(3):580–599. doi: 10.1210/mend.16.3.0806. [DOI] [PubMed] [Google Scholar]
- 24.Takaishi H, Konishi H, Matsuzaki H, et al. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci U S A. 1999 Oct 12;96(21):11836–11841. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001 Feb;21(3):952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rena G, Woods YL, Prescott AR, et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. Embo J. 2002 May 1;21(9):2263–2271. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Shibasaki F, Price R, et al. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell. 1998 May 29;93(5):851–861. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 28.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006 Oct 13;314(5797):294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 29.Woods YL, Rena G, Morrice N, et al. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001 May 1;355(Pt 3):597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008 Feb;10(2):138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essers MA, Weijzen S, de Vries-Smits AM, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J. 2004 Dec 8;23(24):4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finnberg N, El-Deiry WS. Activating FOXO3a, NF-kappaB and p53 by targeting IKKs: an effective multi-faceted targeting of the tumor-cell phenotype? Cancer Biol Ther. 2004 Jul;3(7):614–616. doi: 10.4161/cbt.3.7.1057. [DOI] [PubMed] [Google Scholar]
- 33.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004 Mar 26;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 34.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004 May 14;117(4):421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 35.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004 Jul 6;101(27):10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004 Jul 9;279(28):28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 37.Kitamura YI, Kitamura T, Kruse JP, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005 Sep;2(3):153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 38.van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends in biochemical sciences. 2005 Feb;30(2):81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005 Aug 9;102(32):11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007 Aug;6(4):505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 41.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003 Apr 4;278(14):12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 42.Yamagata K, Daitoku H, Takahashi Y, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008 Oct 24;32(2):221–231. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 43.Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008 Apr 1;4(3):378–380. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 44.Hu MC, Lee DF, Xia W, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004 Apr 16;117(2):225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 45.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003 Sep 30;100(20):11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brenkman AB, de Keizer PL, van den Broek NJ, et al. The peptidyl-isomerase Pin1 regulates p27kip1 expression through inhibition of Forkhead box O tumor suppressors. Cancer Res. 2008 Sep 15;68(18):7597–7605. doi: 10.1158/0008-5472.CAN-08-1059. [DOI] [PubMed] [Google Scholar]
- 47.Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG, Burgering BM. Mdm2 induces mono-ubiquitination of FOXO4. PLoS ONE. 2008;3(7):e2819. doi: 10.1371/journal.pone.0002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Horst A, de Vries-Smits AM, Brenkman AB, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006 Oct;8(10):1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 49.Brunet A, Kanai F, Stehn J, et al. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002 Mar 4;156(5):817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yaffe MB. How do 14-3-3 proteins work?-- Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002 Feb 20;513(1):53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 51.Dong S, Kang S, Gu TL, et al. 14-3-3 Integrates prosurvival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1-transformed hematopoietic cells. Blood. 2007 Jul 1;110(1):360–369. doi: 10.1182/blood-2006-12-065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massague J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006 Sep;10(3):203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 53.Gomis RR, Alarcon C, He W, et al. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006 Aug 22;103(34):12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007 Jan;292(1):C188–199. doi: 10.1152/ajpcell.00542.2005. [DOI] [PubMed] [Google Scholar]
- 55.Arden KC. FoxO: linking new signaling pathways. Mol Cell. 2004 May 21;14(4):416–418. doi: 10.1016/s1097-2765(04)00213-8. [DOI] [PubMed] [Google Scholar]
- 56.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004 Apr 16;117(2):211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 57.Yamamura Y, Lee WL, Inoue K, Ida H, Ito Y. RUNX3 cooperates with FoxO3a to induce apoptosis in gastric cancer cells. J Biol Chem. 2006 Feb 24;281(8):5267–5276. doi: 10.1074/jbc.M512151200. [DOI] [PubMed] [Google Scholar]
- 58.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004 Dec 17;306(5704):2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 59.Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J Biol Chem. 2003 Nov 14;278(46):45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- 60.Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol. 2003;21:265–304. doi: 10.1146/annurev.immunol.21.120601.141053. [DOI] [PubMed] [Google Scholar]
- 61.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med. 2006 Mar;12(3):289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 62.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7(1):75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 63.Mrak RE, Griffin WS. The role of chronic self-propagating glial responses in neurodegeneration: implications for long-lived survivors of human immunodeficiency virus. J Neurovirol. 1997;3(4):241–246. doi: 10.3109/13550289709029465. [DOI] [PubMed] [Google Scholar]
- 64.Zhang K, McQuibban GA, Silva C, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003 Oct;6(10):1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- 65.Koenig S, Gendelman HE, Orenstein JM, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 66.Eilbott DJ, Peress N, Burger H, et al. Human immunodeficiency virus type 1 in spinal cords of acquired immunodeficiency syndrome patients with myelopathy: expression and replication in macrophages. Proc Natl Acad Sci U S A. 1989;86(9):3337–3341. doi: 10.1073/pnas.86.9.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Annals of Neurology. 1986;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 68.McArthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore) 1987 Nov;66(6):407–437. doi: 10.1097/00005792-198711000-00001. [DOI] [PubMed] [Google Scholar]
- 69.Muthumani K, Choo AY, Hwang DS, et al. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood. 2005 Sep 15;106(6):2059–2068. doi: 10.1182/blood-2005-03-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bouzar AB, Villet S, Morin T, et al. Simian immunodeficiency virus Vpr/Vpx proteins kill bystander noninfected CD4+ T-lymphocytes by induction of apoptosis. Virology. 2004 Aug 15;326(1):47–56. doi: 10.1016/j.virol.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 71.Jekle A, Keppler OT, De Clercq E, Schols D, Weinstein M, Goldsmith MA. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J Virol. 2003 May;77(10):5846–5854. doi: 10.1128/JVI.77.10.5846-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng SL. Foxo in the immune system. Oncogene. 2008 Apr 7;27(16):2337–2344. doi: 10.1038/onc.2008.26. [DOI] [PubMed] [Google Scholar]
- 73.Coffer PJ, Burgering BM. Forkhead-box transcription factors and their role in the immune system. Nat Rev Immunol. 2004 Nov;4(11):889–899. doi: 10.1038/nri1488. [DOI] [PubMed] [Google Scholar]
- 74.Lin L, Hron JD, Peng SL. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004 Aug;21(2):203–213. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 75.Asselin-Labat ML, David M, Biola-Vidamment A, et al. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. 2004 Jul 1;104(1):215–223. doi: 10.1182/blood-2003-12-4295. [DOI] [PubMed] [Google Scholar]
- 76.Stahl M, Dijkers PF, Kops GJ, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002 May 15;168(10):5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 77.Tang TT, Dowbenko D, Jackson A, et al. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J Biol Chem. 2002 Apr 19;277(16):14255–14265. doi: 10.1074/jbc.M110901200. [DOI] [PubMed] [Google Scholar]
- 78.Dijkers PF, Birkenkamp KU, Lam EW, et al. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002 Feb 4;156(3):531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Erlacher M, Labi V, Manzl C, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006 Dec 25;203(13):2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Selliah N, Finkel TH. Biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001 Feb;8(2):127–136. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- 81.Li G, Elder RT, Qin K, Park HU, Liang D, Zhao RY. Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. J Biol Chem. 2007 Mar 9;282(10):7287–7298. doi: 10.1074/jbc.M607951200. [DOI] [PubMed] [Google Scholar]
- 82.Andersen JL, DeHart JL, Zimmerman ES, et al. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog. 2006 Dec;2(12):e127. doi: 10.1371/journal.ppat.0020127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lai M, Chen J. The role of Vpr in HIV-1 disease progression is independent of its G2 arrest induction function. Cell Cycle. 2006 Oct;5(19):2275–2280. doi: 10.4161/cc.5.19.3317. [DOI] [PubMed] [Google Scholar]
- 84.Andersen JL, Zimmerman ES, DeHart JL, et al. ATR and GADD45alpha mediate HIV-1 Vpr-induced apoptosis. Cell Death Differ. 2005 Apr;12(4):326–334. doi: 10.1038/sj.cdd.4401565. [DOI] [PubMed] [Google Scholar]
- 85.Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem. 2003 Jul 11;278(28):25879–25886. doi: 10.1074/jbc.M303948200. [DOI] [PubMed] [Google Scholar]
- 86.Fletcher TM, 3rd, Brichacek B, Sharova N, et al. Nuclear import and cell cycle arrest functions of the HIV-1 Vpr protein are encoded by two separate genes in HIV-2/SIV(SM) Embo J. 1996 Nov 15;15(22):6155–6165. [PMC free article] [PubMed] [Google Scholar]
- 87.Zimmerman ES, Sherman MP, Blackett JL, et al. Human immunodeficiency virus type 1 Vpr induces DNA replication stress in vitro and in vivo. J Virol. 2006 Nov;80(21):10407–10418. doi: 10.1128/JVI.01212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Belzile JP, Duisit G, Rougeau N, Mercier J, Finzi A, Cohen EA. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007 Jul;3(7):e85. doi: 10.1371/journal.ppat.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kino T, Chrousos GP. Human immunodeficiency virus type-1 accessory protein Vpr: a causative agent of the AIDS-related insulin resistance/lipodystrophy syndrome? Ann N Y Acad Sci. 2004 Jun;1024:153–167. doi: 10.1196/annals.1321.013. [DOI] [PubMed] [Google Scholar]
- 90.Kino T, De Martino MU, Charmandari E, Ichijo T, Outas T, Chrousos GP. HIV-1 accessory protein Vpr inhibits the effect of insulin on the Foxo subfamily of forkhead transcription factors by interfering with their binding to 14-3-3 proteins: potential clinical implications regarding the insulin resistance of HIV-1-infected patients. Diabetes. 2005 Jan;54(1):23–31. doi: 10.2337/diabetes.54.1.23. [DOI] [PubMed] [Google Scholar]
- 91.Martinez-Gac L, Marques M, Garcia Z, Campanero MR, Carrera AC. Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol Cell Biol. 2004 Mar;24(5):2181–2189. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002 Apr 19;296(5567):530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 93.Delpuech O, Griffiths B, East P, et al. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol Cell Biol. 2007 Jul;27(13):4917–4930. doi: 10.1128/MCB.01789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dabrowska A, Kim N, Aldovini A. Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J Immunol. 2008 Dec 15;181(12):8460–8477. doi: 10.4049/jimmunol.181.12.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988 Dec 23;55(6):1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 96.Li CJ, Wang C, Friedman DJ, Pardee AB. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1995 Jun 6;92(12):5461–5464. doi: 10.1073/pnas.92.12.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Riou C, Yassine-Diab B, Van grevenynghe J, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 2007 Jan 22;204(1):79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Grevenynghe J, Procopio FA, He Z, et al. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med. 2008 Mar;14(3):266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]
- 99.Carter CA, Ehrlich LS. Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol. 2008;62:425–443. doi: 10.1146/annurev.micro.62.081307.162758. [DOI] [PubMed] [Google Scholar]
- 100.Vazquez N, Greenwell-Wild T, Marinos NJ, et al. Human immunodeficiency virus type 1-induced macrophage gene expression includes the p21 gene, a target for viral regulation. J Virol. 2005 Apr;79(7):4479–4491. doi: 10.1128/JVI.79.7.4479-4491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bren GD, Whitman J, Cummins N, et al. Infected cell killing by HIV-1 protease promotes NF-kappaB dependent HIV-1 replication. PLoS ONE. 2008;3(5):e2112. doi: 10.1371/journal.pone.0002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Francois F, Klotman ME. Phosphatidylinositol 3-kinase regulates human immunodeficiency virus type 1 replication following viral entry in primary CD4+ T lymphocytes and macrophages. J Virol. 2003 Feb;77(4):2539–2549. doi: 10.1128/JVI.77.4.2539-2549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chugh P, Bradel-Tretheway B, Monteiro-Filho CM, et al. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology. 2008;5:11. doi: 10.1186/1742-4690-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deregibus MC, Cantaluppi V, Doublier S, et al. HIV-1-Tat protein activates phosphatidylinositol 3-kinase/ AKT-dependent survival pathways in Kaposi’s sarcoma cells. J Biol Chem. 2002 Jul 12;277(28):25195–25202. doi: 10.1074/jbc.M200921200. [DOI] [PubMed] [Google Scholar]
- 105.Wolf D, Witte V, Laffert B, et al. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat Med. 2001 Nov;7(11):1217–1224. doi: 10.1038/nm1101-1217. [DOI] [PubMed] [Google Scholar]
- 106.Borgatti P, Zauli G, Colamussi ML, et al. Extracellular HIV-1 Tat protein activates phosphatidylinositol 3- and Akt/PKB kinases in CD4+ T lymphoblastoid Jurkat cells. Eur J Immunol. 1997 Nov;27(11):2805–2811. doi: 10.1002/eji.1830271110. [DOI] [PubMed] [Google Scholar]
- 107.Huang Y, Erdmann N, Peng H, et al. TRAIL-mediated apoptosis in HIV-1-infected macrophages is dependent on the inhibition of Akt-1 phosphorylation. J Immunol. 2006 Aug 15;177(4):2304–2313. doi: 10.4049/jimmunol.177.4.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cui M, Huang Y, Zhao Y, Zheng J. Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages. J Immunol. 2008 Jan 15;180(2):898–906. doi: 10.4049/jimmunol.180.2.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boisse L, Gill MJ, Power C. HIV infection of the central nervous system: clinical features and neuropathogenesis. Neurol Clin. 2008 Aug;26(3):799–819. x. doi: 10.1016/j.ncl.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 110.Gonzalez-Duarte A, Robinson-Papp J, Simpson DM. Diagnosis and management of HIV-associated neuropathy. Neurol Clin. 2008 Aug;26(3):821–832. x. doi: 10.1016/j.ncl.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 111.Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1 associated neurodegeneration. Crit Rev Neurobiol. 1996;10(1):57–67. doi: 10.1615/critrevneurobiol.v10.i1.30. [DOI] [PubMed] [Google Scholar]
- 112.Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans lifespan by insulinlike signaling in the nervous system. Science. 2000 Oct 6;290(5489):147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- 113.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007 Jun 7;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 114.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007 Jan 26;128(2):325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 115.Wang L, Zhang ZG, Gregg SR, et al. The Sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J Biol Chem. 2007 Nov 2;282(44):32462–32470. doi: 10.1074/jbc.M706880200. [DOI] [PubMed] [Google Scholar]
- 116.Bakker WJ, van Dijk TB, Parren-van Amelsvoort M, et al. Differential regulation of Foxo3a target genes in erythropoiesis. Mol Cell Biol. 2007 May;27(10):3839–3854. doi: 10.1128/MCB.01662-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sathyanarayana P, Dev A, Fang J, et al. EPO receptor circuits for primary erythroblast survival. Blood. 2008 Jun 1;111(11):5390–5399. doi: 10.1182/blood-2007-10-119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994 Jun 17;77(6):817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 119.Wu Y, Wu Z, Butko P, et al. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci. 2006 Dec 13;26(50):13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang H, Wu YC, Nakamura M, et al. Parkinson’s disease genetic mutations increase cell susceptibility to stress: mutant alpha-synuclein enhances H2O2- and Sin-1-induced cell death. Neurobiol Aging. 2007 Nov;28(11):1709–1717. doi: 10.1016/j.neurobiolaging.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 121.Davila D, Torres-Aleman I. Neuronal death by oxidative stress involves activation of FOXO3 through a two-arm pathway that activates stress kinases and attenuates insulin-like growth factor I signaling. Mol Biol Cell. 2008 May;19(5):2014–2025. doi: 10.1091/mbc.E07-08-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shinoda S, Schindler CK, Meller R, et al. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004 Apr;113(7):1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003 Aug 18;162(4):613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sanderson TH, Kumar R, Murariu-Dobrin AC, Page AB, Krause GS, Sullivan JM. Insulin activates the PI3K-Akt survival pathway in vulnerable neurons following global brain ischemia. Neurol Res. 2009 Feb 6; doi: 10.1179/174313209X382449. [DOI] [PubMed] [Google Scholar]
- 125.Obexer P, Geiger K, Ambros PF, Meister B, Ausserlechner MJ. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007 Mar;14(3):534–547. doi: 10.1038/sj.cdd.4402017. [DOI] [PubMed] [Google Scholar]
- 126.Zhu Z, Zhang Q, Yu Z, et al. Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia. 2007 Apr 1;55(5):546–558. doi: 10.1002/glia.20476. [DOI] [PubMed] [Google Scholar]
- 127.Di Giovanni S, Movsesyan V, Ahmed F, et al. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Franke H, Sauer C, Rudolph C, Krugel U, Hengstler JG, Illes P. P2 receptor-mediated stimulation of the PI3-K/Akt-pathway in vivo. Glia. 2008 Dec 29; doi: 10.1002/glia.20827. [DOI] [PubMed] [Google Scholar]