

Abstract
Molecular recognition is fundamental to the specific interactions between molecules, of which the best known examples are antibody-antigen binding and complementary DNA hybridization. Reversible manipulation of the molecular recognition events is still a very challenging topic< and such studies are often performed at the molecular level. An important consideration is collection of changes at the molecular level to provide macroscopic observables. This research makes use of photo-responsive molecular recognition for the fabrication of novel photo-regulated dynamic materials. Specifically, a dynamic hydrogel was prepared by grafting azobenzene-tethered ssDNA and its complementary DNA to the hydrogel network. The macroscopic volume of the hydrogel can be manipulated through the photo-reversible DNA hybridization controlled by alternating irradiation of UV and visible light. The effects of synthetic parameters including the concentration of DNA, polymer monomer and permanent crosslinker are also discussed.
INTRODUCTION
Selective molecular recognition between specific molecules forms the basis of biological systems and is achieved through noncovalent interactions such as hydrogen bonding, hydrophobic forces, van der Waals forces, electrostatic effects, etc. Reversible manipulation of molecular recognition events is of crucial importance in the stimulus-responsive regulation of biological processes, for example the reversible enzyme and receptor recognition regulated by kinase and phosphatase through phosphorylation and dephosphorylation. Mimicking naturally occurring systems, different types of stimulus-responsive molecular recognition systems have been developed, such as the oxidation-responsive ferrocene/β-cyclodextrin and photo-responsive azobenzene/α-cyclodextrin interaction, and explored for their applications in construction of novel functional materials. One emerging area involves stimulus-responsive hydrogels, which show great potential in various applications, such as biosensing, drug delivery, microfluidics and self-healing materials.1–4 Based on the different designs, several classes of hydrogels have been developed with sensitivities towards temperature, pH, light, biomolecules, and other stimuli.5–9
There has been great interest in photo-responsive hydrogels for the development of “smart” systems. Among all different types of stimuli, light is one of the most attractive since it can be delivered remotely and instantly with high accuracy. Furthermore, light-responsive materials also remain a forefront topic because of their possible application in solar energy harvesting and utilization.10, 11 For example, Yamaguchi et al. developed photo-switchable gel assembly based on the photoresponsive azobenzene/α-cyclodextrin interaction.12. Matsubara et al. synthesized photochromic hydrogels based on the structural change of hydrogel network regulated by azobenzene isomerization.13 Our group recently engineered drug-releasing hydrogels based light-triggered sol-gel conversion.2 There are also reports of dynamic hydrogels with volume changes. However, they either focus on different properties such as self-assembly and photochromic properties,12, 13 or take advantage of effects other than molecular recognition14. To our best knowledge, there have not been reports about dynamic hydrogels with reversible volume changes based on photo-switchable molecular recognition.
With the aim of developing new photo-responsive dynamic materials, we have engineered a DNA-crosslinked hybrid acrylamide polymer hydrogel with light-induced reversible volume change. The basic functional modules of this photo-responsive hydrogel are the photo-switchable DNA duplex complexes regulated by azobenzene isomerization driven by light. It has been demonstrated that an azobenzene bearing DNA duplex dissociates when azobenzene coverts to the cis isomer upon UV light irradiation. However, the DNA duplex recovers when azobenzene converts back to the trans-form when visible light is applied.15, 16 This new type of light-responsive dynamic hydrogel has the potential to fabricate actuator devices which can convert light energy into a macroscopic volume change for different applications. Furthermore, the design of hydrogels can also be applied to other photo-responsive molecular recognition systems.
RESULTS AND DISCUSSION
Design of light-responsive dynamic hydrogels
Inspired by light-controlled DNA hybridization based on azobenzene isomerization15, 17, 18 (Fig. 1A), we engineered a hydrogel using DNA duplexes as photoreversible crosslinks. This photosensitive dynamic hydrogel was prepared by grafting azobenzene-tethered, single-stranded DNA and its complementary DNA to the hydrogel network, so that the degree of crosslinking could be regulated by light. With a two-step polymerization method8, we synthesized an acrylamide-backboned hydrogel network, in which linear acrylamide polymers are trapped, using N,N'-methylenebisacrylamide (MBAA) as a permanent crosslinker (Fig. 2A). The chemical structure of the resulting hydrogel is shown in Figure 2B. The azobenzene-modified DNAs are grafted onto the crosslinked network, while the complementary DNAs are modified on the linear polymer. When exposed to visible light, it is expected that the hybridization between the two complementary DNAs will serve as extra crosslinks in the network, so that the hydrogel will have a smaller volume (Fig. 1B). However, when the hydrogel is exposed to UV light, the disruption of DNA hybridization removes the extra crosslinks and triggers a volume increase of the hydrogel. By alternating irradiation of visible and UV light, a reversible volume transition is thus realized. Therefore, the reversible macroscopic volume change driven by UV and visible light can be viewed as a process of converting light energy to mechanical energy, meeting the initial goal of this work in converting photonic energy to usable forms of energy.
Figure 1.

(A) Light-controlled formation of DNA duplex based on azobenzene isomerization in the hydrogel. (B) Reversible volume transition of the DNA-crosslinked hydrogel regulated by UV and visible light.
Figure 2.

(A)Synthesis of light responsive dynamic hydrogels. (B) Chemical structure of the dynamic hydrogel.
Light induced volume changes of dynamic hydrogels
The ability of the hydrogels to undergo a light-induced volume transition was tested by monitoring the swelling upon UV light irradiation and shrinking with visible light irradiation. The hydrogels were synthesized under visible light in a shrunken state, in which all DNAs were hybridized. Before analysis of the photo-induced volume change, the hydrogels were soaked for three days in a large volume of buffer solution (10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, pH 8.0) and allowed to swell to equilibrium. Using a water bath, the temperature was maintained at 22°C throughout this process and the subsequent experiments. The percentage volume changes of the hydrogels were calculated from mass measurements considering the mass and density of polymer, DNA and water included.20–22
The light-responsive swelling was first analyzed by exposing the hydrogels to UV light (365 nm) at 2 hours after the start of monitoring under visible light. As shown by the black curve in Figure 3A, the volume of the hydrogel increased abruptly following the irradiation of UV light and reached equilibrium after 3 hrs. We attribute this volume increase to the photodissociation of DNA duplexes in the hydrogel network by UV light-induced trans-to-cis isomerization of azobenzene moieties. It is well known that azobenzene isomerizes from the trans- to cis- configuration upon UV light irradiation, and previous work has shown that the non-planar cis- azobenzene moieties can destabilize and dissociate the DNA duplex by steric hindrance.15–17 As a result of this isomerization, the DNA duplex crosslinkers are removed, thereby yielding a larger hydrogel volume.
Figure 3.

(A) UV light induced volume increase of the dynamic hydrogel containing complementary DNAs with azobenzene modification (black); (B) Volume changes of the dynamic hydrogel upon visible light irradiation (black) and in dark with no light (green) following UV light irradiation. Blue and red curves are for control experiments: Blue curve: control hydrogel (C1) containing complementary DNAs without azobenzene modification; Red curve: control hydrogel (C2) containing noncomplementary DNAs with azobenzene modification.
After the hydrogel swelled to equilibrium upon UV light irradiation, the light source was switched to visible light which caused the cis- azobenzene moieties to isomerize back to the trans form. Along with the cis- to trans- isomerization of azobenzene moieties, the two complementary ssDNA strands hybridized again to form the duplex, since the planar trans- azobenzene moieties could stabilize the duplex by stacking interactions.15–17 Therefore, the hydrogel shrank as the DNA duplex crosslinkers recovered (black curve, Fig. 3B).
To prove that the swelling and shrinking of the dynamic hydrogel is indeed regulated by photo-reversible DNA hybridization, we carried out control experiments using two control hydrogels (C1 and C2). Both control hydrogels shared the same structure with the dynamic hydrogel: all were synthesized through the two-step polymerization using two acrylate ssDNA monomers. However, no photoresponsive azobenzene moieties were modified in the DNA strands of control hydrogel C1. As expected, no volume change was observed upon either UV or visible light irradiation (blue curves in Fig. 3A and 2B). Several types of hydrogels were recently developed with volume changes based on an infrared (IR) light-induced photothermal effect which changed the local temperature of the hydrogels, 14, 23the results for control C1 clearly rule out thermal effects as the cause of the volume change.
Control hydrogel C2 had azobenzene moieties in one DNA strand, but the two DNA strands were not complementary to each other. Therefore, no photoreversible DNA duplex crosslinkers were possible, even though the azobenzene moieties could still isomerize upon UV and visible light irradiation. Results from volume change measurements shown by the red curves in Figure 3A and 3B indicated that the isomerization of azobenzene moieties themselves could not induce the volume change.
We performed another control experiment, in which the dynamic hydrogel was left in the dark after swelling to equilibrium upon UV light irradiation, to determine if the shrinking process would be affected by thermal relaxation of the cis- azobenzene moieties. Previous work has shown that azobenzene can automatically isomerize from the cis- to trans- state by thermal relaxation at room temperature without any light irradiation, although this process can take up to days and is much slower than the visible light-activated isomerization, depending on the presence of different substituents.24, 25 To determine if thermal relaxation is important in the hydrogel volume changes, the volume were monitored after the UV light was removed at the 3-hour time point. As shown in Figure 3B (green curve), the shrinkage induced by thermal relaxation was much slower than that by the visible light, and only a small overall shrinkage was observed. Therefore, based on these extensive control experiments, the volume change of the dynamic hydrogel was induced solely by UV and visible light-regulated dissociation and association of azobenzene-modified DNA duplexes.
Reversible volume changes of light-responsive dynamic hydrogels
In order to make the light-responsive dynamic hydrogel useful for continuous harvesting and conversion of light energy, swelling and shrinking must be reversible. Therefore, we investigated the reversibility in response to alternate irradiation with UV (3hrs) and visible light (2hrs) for multiple cycles, as shown in Figure 4. In each cycle, the dynamic hydrogel swelled when the light source was switched from visible to UV and contracted when the light source was switched back to visible. For the control hydrogels C1 and C2, no volume change was observed. The volume change for the dynamic hydrogel was very reproducible, except that the expanded gel did not return to its original compact volume when irradiated with visible light. We attribute the decreased contraction to incomplete rehybridization of DNA upon visible light irradiation. The hydrogel is synthesized in the shrunken state with all ssDNA hybridize to form duplexes. In the first cycle, all the duplexes are dissociated by UV light, resulting in a large volume increase. However, not all of the dissociated ssDNAs are able to rehybridize as a result of the diffusion and rearrangement of the polymer networks and DNAs. Consequently, the reduced number of DNA duplex crosslinkers yields a larger volume in the compacted state, compared to the initial volume. However, after the first cycle, the degree of volume change becomes more reversible, because diffusion and rearrangement reach equilibrium, and the number of photoreversible DNA duplex crosslinkers stays the same.
Figure 4.
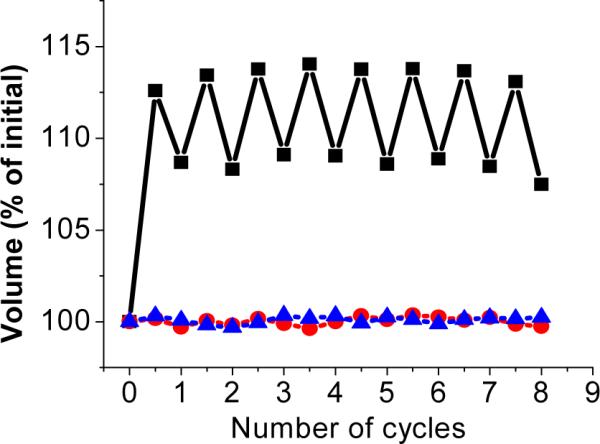
Reversible swelling and contracting of light-responsive dynamic hydrogels in response to alternate irradiation by UV and visible light. Black curve: dynamic hydrogel containing complementary DNAs with azobenzene modification; Blue curve: control hydrogel (C1) containing complementary DNAs without azobenzene modification; Red curve: control hydrogel (C2) containing noncomplementary DNAs with azobenzene modification.
Effects of synthesis parameters on volume changes
The percentage of reversible volume change was also determined to be dependent on conditions during hydrogel synthesis. We prepared dynamic hydrogels using varying concentrations of N, N'-methylenebisacrylamide (MBAA), acrylate DNA monomer, and acrylamide monomer (See supporting information, Fig. S3–5). We first changed the concentrations of MBAA and acrylate DNA monomer at fixed acrylamide concentration. Figure 5A shows that the percentage of reversible volume change decreased as the concentration of MBAA increased at fixed acrylate DNA monomer concentration. On the other hand, at fixed MBAA concentration, the reversible volume change increased as the DNA monomer concentration increased (Fig. 5B). According to the design of our dynamic hydrogel, the volume of hydrogel can be divided into two portions: the reversible portion and irreversible portion. The reversible portion of the hydrogel volume is determined by the photoregulated DNA duplex crosslinkers, while the irreversible portion depends on the permanent MBAA crosslinkers. As a result, the increase of MBAA concentration tends to increase the irreversible portion of volume and reduce the percentage of light-induced volume change. However, an increase in the acrylate DNA monomer concentration increases the reversible portion of volume and therefore enhances the percentage volume change. However, if the concentrations of both MBAA and acrylate DNA monomer are fixed, the acrylamide concentration within the range tested does not influence the percentage volume change, as shown in Figure 5C. We attribute this result to the constant ratio of DNA concentration to MBAA concentration, which results in similar percentage volume change.
Figure 5.

Percentage reversible volume change of light-responsive dynamic hydrogels synthesized under different conditions: (A) percentage volume change as a function MBAA concentration; (B) percentage volume change as a function of DNA monomer concentration; (C) percentage volume change as a function of acrylamide concentration.
CONCLUSION
We have successfully constructed dynamic hydrogels with reversible volume changes based on photo-switchable DNA hybridization. The dissociation of DNA duplex crosslinkers in the hydrogels leads to the expansion of the hydrogel volume while the formation of DNA duplex crosslinkers causes the shrinkage of the hydrogel. With this approach, we are able to visualize the reversible dissociation and formation of the DNA duplexes through the volume change of hydrogels. Furthermore, during the last decade, considerable effort has been devoted to the development of DNA-based nanomachines.18, 26–30 However, most reported nanomachines fall short of practical use, as a consequence of their nanometer sizes and inconvenient energy sources. Our dynamic hydrogel takes advantage of the scaffold of the polymer network to convert molecular-level effects into macroscopic changes. The nanometer-sized, photoreversible DNA duplex crosslinkers are assembled into the hydrogel to create harvesting and conversion units for light energy. Macroscopic volume change of hydrogels is achieved by alternate irradiation with UV and visible light. The volume change could be potentially converted to mechanical or electrical energy by using proper device designs. There have been several examples where attempts were made to utilize azobenzenes to harvest solar energy, including an osmotic pressure-driven solar mechanical device31 and a solar thermal fuel system composed of azobenzene functionalized carbon nanotubes32. We believe that the light-reversible dynamic DNA hydrid hydrogels add a new concept to the solar energy field and could find applications in many areas, such as light-controlled actuators, micro-lenses and microvalves. These devices will be highly useful with remote control using photons in energy conversion. In addition, the DNA based photo-dynamic hydrogel can be extended to similar systems, such as photo-switchable azobenzene/α-cyclodextrin complexation, protein/substrate binding and protein/protein interaction.33–35
EXPERIMENTAL SECTION
DNA synthesis
All acrydite and azobenzene modified DNA oligonucleotides were synthesized via phosphoramidite chemistry on an ABI 3400 DNA synthesizer (Applied Biosystems, Inc., Foster City, CA). DNA sequences used in the experiments are shown in Table S1 (supporting information). After the synthesis, the DNA sequences were deprotected in concentrated AMA (1:1 mixture of ammonium hydroxide and aqueous methylamine) solution at 65 °C for 30 min, followed by further purification on a ProStar HPLC system (Varian, Palo Alto, CA) with a C-18 reversed-phase column (Alltech, 5μm, 250mm × 4.6 mm) using acetonitrile and 0.1 M triethylammonium acetate (TEAA) aqueous solution as mobile phases. The collected DNA products were dried and detritylated with acetic acid. The detritylated aptamers were precipitated with ethanol and dried using a vacuum drier. The purified aptamers were then dissolved in DNA grade water and quantified by determining the UV absorption at 260 nm using a UV-vis spectrometer (Cary Bio-300, Varian).
Synthesis of light-responsive dynamic hydrogels
The photoresponsive hydrogels were prepared by a two-step polymerization method in buffer solution (10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, pH 8.0) with a total volume of 30 μL (Fig. 2). In step I, the linear-backboned DNA hybrid polymer was synthesized by copolymerization of acrylamide (4%) and acrydite-DNA (A, 3 mM) using ammonium persulfate (APS) and tetramethylethylenediamine (TEMED). Afterwards, the resultant DNA hybrid linear polymer synthesized in step I was mixed with azobenzene-modified acrydite-cDNA (A') at a 1:1 ratio. The mixture was heated to 90 °C and slowly cooled to room temperature to allow complete hybridization between A and A'. Then, acrylamide and N,N'-Methylenebisacrylamide (MBAA) were added to the mixture, and step II of the polymerization process was carried out to form the dynamic hydrogel using APS and TEMED. After polymerization, the dynamic hydrogels were immersed in buffer to remove the unreacted monomers at 22 °C. The initial mass swelling ratios after washing were calculated and summarized in Table S2–S4 for hydrogels synthesized under different conditions. Control hydrogels were also prepared by the same method, but with different acrydite-DNA monomers. Control hydrogel (C1) was synthesized using complementary DNA monomers (A and ctrl-A') without azobenzene modification, while control hydrogel (C2) contained noncomplementary DNA monomers (B and A') with azobenzene modification.
Measurement of light-induced volume change
A 60 W table lamp and a 6 W handheld UV lamp (365 nm) were used as visible and UV light sources, respectively. During the light irradiation, the hydrogel was immersed in buffer (10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, pH 8.0) in a glass vial. The temperature was maintained at 22 °C using a water bath. The hydrogel mass was measured by taking out the hydrogel from the solution, removing excess solution from the gel surface, and weighing. The hydrogel was kept in darkness during the entire process. The volume of the hydrogels (V) can be viewed as the sum of the volumes of the polymer network (Vp) and the aqueous phase in the hydrogels (Vw). Assuming that the mass of polymer and aqueous phase are Mp and Mw and the densities are ρp and ρw,, respectively, then V= Vp+ Vw = (Mp/ ρp)+(Mw/ ρw). Since the change of volume and mass of the hydrogel is caused by absorption and release of aqueous phase, the change of hydrogel volume can be expressed by ΔV = ΔMw/ ρw = ΔM/ρw, where ΔM is the observed mass change. Therefore, the percentage volume change is equal to the percentage mass change of the hydrogel.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Jacob Jones and Dr. Kathryn Williams for insightful discussions. We also acknowledge the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. This work was supported by grants awarded by the National Institutes of Health (GM066137, GM079359 and CA133086). This work was also supported by the National Key Scientific Program of China (2011CB911001, 2011CB911003).
Footnotes
Supporting Information Available Experimental details including synthesis of acrydite and azobenzene phosphoramidite, DNA sequences used in the experiments, initial mass swelling ratios of hydrogels and the reversible volume change of hydrogels over multiple cycles. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Mohammed JS, Murphy WL. Adv Mater. 2009;21:2361–2374. [Google Scholar]
- (2).Kang H, Liu H, Zhang X, Yan J, Zhu Z, Peng L, Yang H, Kim Y, Tan W. Langmuir. 2011;27:399–408. doi: 10.1021/la1037553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dong L, Agarwal AK, Beebe DJ, Jiang H. Nature. 2006;442:551–554. doi: 10.1038/nature05024. [DOI] [PubMed] [Google Scholar]
- (4).Nakahata M, Takashima Y, Yamaguchi H, Harada A. Nature Communications. 2011;2:511. doi: 10.1038/ncomms1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tanaka T. Phys. Rev. Lett. 1978;40:820–823. [Google Scholar]
- (6).Tanaka T, Nishio I, Sun ST, Ueno-Nishio S. Science. 1982;218:467. doi: 10.1126/science.218.4571.467. [DOI] [PubMed] [Google Scholar]
- (7).Suzuki A, Tanaka T. Nature. 1990;346:345–347. [Google Scholar]
- (8).Miyata T, Asami N, Uragami T. Nature. 1999;399:766–769. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- (9).Beebe DJ, Moore JS, Bauer JM, Yu Q, Liu RH, Devadoss C, Jo BH. Nature. 2000;404:588–590. doi: 10.1038/35007047. [DOI] [PubMed] [Google Scholar]
- (10).Kanai Y, Srinivasan V, Meier SK, Vollhardt KPC, Grossman JC. Angew. Chem., Int. Ed. 2010;49:8926–8929. doi: 10.1002/anie.201002994. [DOI] [PubMed] [Google Scholar]
- (11).Cho J, Berbil-Bautista L, Pechenezhskiy IV, Levy N, Meier SK, Srinivasan V, Kanai Y, Grossman JC, Vollhardt KPC, Crommie MF. ACS Nano. 2011;5:3701–3706. doi: 10.1021/nn2000367. [DOI] [PubMed] [Google Scholar]
- (12).Yamaguchi H, Kobayashi Y, Kobayashi R, Takashima Y, Hashidzume A, Harada A. Nature Communications. 2012;3:603. doi: 10.1038/ncomms1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Matsubara K, Watanabe M, Takeoka Y. Angew. Chem., Int. Ed. 2007;46:1688–1692. doi: 10.1002/anie.200603554. [DOI] [PubMed] [Google Scholar]
- (14).Lo CW, Zhu D, Jiang H. Soft Matter. 2011;7:5604–5609. [Google Scholar]
- (15).Asanuma H, Ito T, Yoshida T, Liang X, Komiyama M. Angew. Chem., Int. Ed. 1999;38:2393–2395. doi: 10.1002/(sici)1521-3773(19990816)38:16<2393::aid-anie2393>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- (16).Liang X, Asanuma H, Kashida H, Takasu A, Sakamoto T, Kawai G, Komiyama M. J. Am. Chem. Soc. 2003;125:16408–16415. doi: 10.1021/ja037248j. [DOI] [PubMed] [Google Scholar]
- (17).Asanuma H, Takarada T, Yoshida T, Tamaru D, Liang X, Komiyama M. Angew. Chem., Int. Ed. 2001;40:2671–2673. [PubMed] [Google Scholar]
- (18).Kang H, Liu H, Phillips JA, Cao Z, Kim Y, Chen Y, Yang Z, Li J, Tan W. Nano Lett. 2009;9:2690–2696. doi: 10.1021/nl9011694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yuan Q, Zhang Y, Chen Y, Wang R, Du C, Yasun E, Tan W. Proc. Natl. Acad. Sci. U. S. A. 2011;108:9331–9336. doi: 10.1073/pnas.1018358108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Murphy WL, Dillmore WS, Modica J, Mrksich M. Angew. Chem., Int. Ed. 2007;46:3066–3069. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]
- (21).Sui Z, King WJ, Murphy WL. Adv. Mater. 2007;19:3377–3380. [Google Scholar]
- (22).Sui Z, King WJ, Murphy WL. Adv. Funct. Mater. 2008;18:1824–1831. doi: 10.1002/adfm.200800218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zeng X, Jiang H. Appl. Phys. Lett. 2008;93:151101. [Google Scholar]
- (24).Nishimura N, Tanaka T, Asano M, Sueishi Y. J. Chem. Soc., Perkin Trans. 1986;2:1839–1845. [Google Scholar]
- (25).Nishimura N, Sueyoshi T, Yamanaka H, Imai E, Yamamoto S, Hasegawa S. Bull. Chem. Soc. Jpn. 1976;49:1381–1387. [Google Scholar]
- (26).Li JJ, Tan W. Nano Lett. 2002;2:315–318. [Google Scholar]
- (27).Niemeyer CM, Adler M. Angew. Chem., Int. Ed. 2002;41:3779–3783. doi: 10.1002/1521-3773(20021018)41:20<3779::AID-ANIE3779>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- (28).Chen Y, Wang M, Mao C. Angew. Chem., Int. Ed. 2004;43:3554–3557. doi: 10.1002/anie.200453779. [DOI] [PubMed] [Google Scholar]
- (29).Bath J, Turberfield AJ. Nat. Nanotechnol. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- (30).Klapper Y, Sinha N, Ng TWS, Lubrich D. Small. 2010;6:44–47. doi: 10.1002/smll.200901106. [DOI] [PubMed] [Google Scholar]
- (31).Masiero S, Lena S, Pieraccini S, Spada GP. Angew. Chem., Int. Ed. 2008;47:3184–3187. doi: 10.1002/anie.200705313. [DOI] [PubMed] [Google Scholar]
- (32).Kolpak AM, Grossman JC. Nano Lett. 2011;11:3156–3162. doi: 10.1021/nl201357n. [DOI] [PubMed] [Google Scholar]
- (33).Harada A. Acc. Chem. Res. 2001;34:456–464. doi: 10.1021/ar000174l. [DOI] [PubMed] [Google Scholar]
- (34).Willner I, Rubin S, Wonner J, Effenberger F, Baeuerle P. J. Am. Chem. Soc. 1992;114:3150–3151. [Google Scholar]
- (35).Levskaya A, Weiner OD, Lim WA, Voigt CA. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.