

Abstract
Hepatitis C virus (HCV) infection is a serious public health problem because of its worldwide diffusion and sequelae. It is not only a hepatotropic but also a lymphotropic agent and is responsible not only for liver injury—potentially evolving to cirrhosis and hepatocellular carcinoma—but also for a series of sometimes severely disabling extrahepatic diseases and, in particular, B-cell lymphoproliferative disorders. These latter range from benign, but prelymphomatous conditions, like mixed cryoglobulinemia, to frank lymphomas. Analogously with Helicobacter pylori related lymphomagenesis, the study of the effects of viral eradication confirmed the etiopathogenetic role of HCV and showed it is an ideal model for better understanding of the molecular mechanisms involved. Concerning these latter, several hypotheses have been proposed over the past two decades which are not mutually exclusive. These hypotheses have variously emphasized the important role played by sustained stimulation of the immune system by HCV, infection of the lymphatic cells, viral proteins, chromosomal aberrations, cytokines, or microRNA molecules. In this paper we describe the main hypotheses that have been proposed with the corresponding principal supporting data.
1. Introduction
Hepatitis C virus (HCV) infection is a major public health problem with an estimated 3-4 million people infected each year worldwide and about 170–200 million carriers. These latter are at risk of developing liver cirrhosis and/or liver cancer. More than 350,000 people die from HCV-related liver diseases each year. Moreover, these estimates do not take into account the extrahepatic aspects of HCV infection.
Early after its discovery, it was shown that HCV is also a lymphotropic virus [1]. As a consequence of the lymphatic infection, several lymphoproliferative disorders (LPDs) have been associated with this virus [2], including mixed cryoglobulinemia (MC), B-cell non-Hodgkin's lymphoma (NHL) [3–10] and monoclonal gammopathies [11–13].
Mixed cryoglobulinemia is the most frequent and well known LPD developing during HCV infection. Although clinically benign, MC is a prelymphomatous disorder leading to NHL in about 5–10% of cases. This makes MC a valuable model for study of pathogenetic mechanisms of HCV-related LPDs [2–14]. MC was previously interpreted as a lymphoma in situ, being characterized by bone marrow and/or liver infiltrates closely resembling NHL [15]. Therefore, it was hypothesized that HCV may be involved in the pathogenesis of NHL as well [1, 4]. This hypothesis was substantiated by several observations, including the significantly high prevalence of HCV infection in NHL patients in several studies [6, 7, 10, 12, 16–18]. A lot of data are presently available showing, in most cases, a significant association with B-cell NHL, even with a clear south-north gradient and involving different histopathological types of lymphoma, the most strictly associated being the lymphoplasmacytic, marginal zone and diffuse large B-cell lymphoma [19]. A case-control study has shown that HCV infection increases the risk for NHL involving the liver and major salivary glands by about 50-fold (i.e., a risk higher than that for hepatocellular carcinoma) and the risk for NHLs at other sites by about 4-fold [20].
The observation of the effect of viral eradication using antiviral agents strongly supports the etiopathogenetic link between HCV infection and lymphomagenesis. Analogously with what has been reported for MC, in the case of low grade B-cell lymphoma—and especially in cases of splenic lymphoma—clinical remission following effective antiviral therapy in HCV-associated cases has been observed [21–23].
Interestingly, in a recent Japanese study involving about 3,000 HCV-infected patients observed during a long-term follow-up, it was shown that the annual incidence of lymphoma was 0.23% and the cumulative rate of lymphoma development after 15 years was 2.6% in both the untreated and non-responder patients with persisting infection versus 0% in treated patients achieving viral eradication, strongly suggesting that antiviral therapy protects against the development of lymphoma [24].
2. Mechanisms of HCV-Related Lymphomagenesis
Several hypotheses, frequently interconnected with each other, have been proposed in regard to the possible mechanisms of HCV-related lymphomagenesis (Figure 1). These include a key role played by the sustained antigenic stimulation of the B-cell compartment, the role of viral lymphotropism and viral proteins, chromosomal aberrations, cytokines, and microRNAs.
Figure 1.
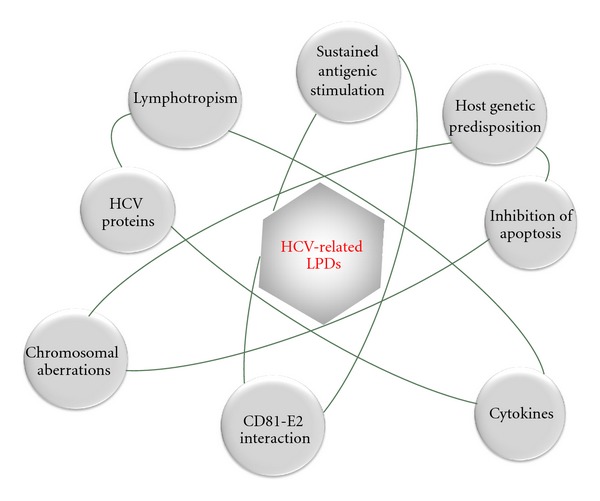
Pathogenesis of HCV-related lymphoproliferative disorders (LPDs). Main working hypotheses and their principal interconnections.
2.1. The Role of Sustained Antigenic Stimulation
Sustained HCV-driven antigenic stimulation has been suggested to play a key role in inducing B-cell clonal expansion characterizing these disorders (Figure 2). The presence in the liver of lymphatic structures resembling lymphatic follicles is characteristic of HCV infection. It has been suggested that they represent an important site of B-cell clonal expansion, especially in patients with MC, where they have been found in almost all cases [25]. Furthermore, B lymphocytes isolated from hepatic follicles produced rheumatoid factor (RF) that most frequently display the WA cross-reactive idiotype, considered to be characteristic of MC [25]. In particular, it was observed that intrahepatic B-cell clonalities were invariably associated with extrahepatic manifestations of HCV infection, including high serum levels of RF activity, cryoglobulins, monoclonal gammopathy of undetermined significance (MGUS), and frank B-cell NHL [26]. The key role of antigen-driven stimulation in HCV-related lymphoproliferation was also supported by a study investigating mutations in the V(H) and V(K) genes of the B-cell clone inducing a frank NHL in an MC patient and producing an IgM homologous to a protein with RF specificity. The observation of an IgH ongoing mutation process and the expression of an Ig antigen receptor significantly homologous to an anti-HCV protein suggested that both MC and NHL were antigen-driven LPDs sustained by HCV [27]. The analysis of bone marrow specimens from the same patient taken at different times during the evolution from MC to NHL showed a marked reduction in intraclonal diversity at the stage of overt NHL, indicating a minor dependence of the cells on the antigen-driven mechanism. Such a progressive independence from the initial etiologic agent may be deduced also by the effects of viral eradication in patients with variable severity of the HCV-related LPD (see also the following). Analogously with Helicobacter pylori-related lymphomagenesis, it is conceivable that, during the multistep lymphomagenic process, progressive independence from the antigen-driven mechanism will develop, possibly due to the occurrence of chromosomal translocations or other genetic aberrations [14, 28] (Figure 2).
Figure 2.
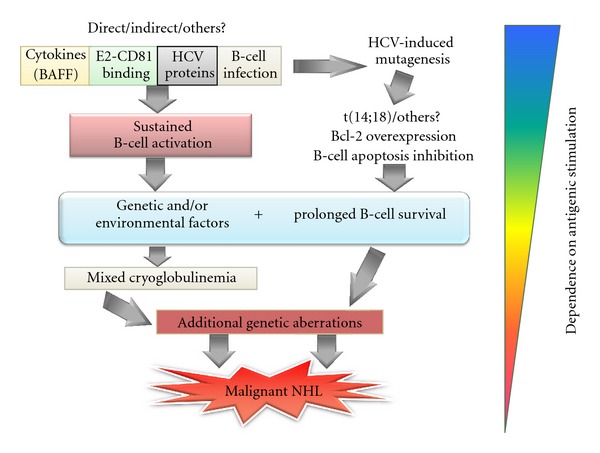
HCV-related LPD pathogenesis is a multifactorial and multistep process. Current data suggest that the starting points of this process are represented by the cooperation between a sustained and persistent stimulation—by direct or indirect action of viral particles or proteins—and antiapoptotic mechanisms acting on B-cell compartment. A predisposing genetic background would be responsible for the final evolution to a particular LPD (namely, MC). The progressive addition of genetic aberrations would lead to a frank neoplastic transformation, gradually making the process less dependent on the etiologic agent.
It has also been suggested that the same HCV antigens may be involved in the induction of both MC and lymphoma [27, 29]. The viral antigen/s responsible for B-cell clonal expansion are not perfectly defined yet. However, De Re and colleagues showed that, in patients with MC and immunocytoma, the B-cell receptor (BCR) of the monoclonal, overexpanded B-cell population, as well as the IgM-RF+ component of the cryoprecipitate, showed cross-reactivity against HCV NS3 antigen [30].
Other studies have focused on the possible role of proteins of the HCV envelope and mainly on HCV E2 protein (Figure 2). It has been shown that E2 interacts with the tetraspanin CD81, present also on the B-cell surface. This binding has been suggested to be responsible for sustained polyclonal B-cell activation essentially by lowering the B-cell activation threshold [31, 32].
Data supporting the hypothesis that some HCV-associated lymphomas could originate from B cells that were initially activated by the HCV-E2 protein have been provided. In fact, Quinn and coworkers showed that the immunoglobulin from one of two HCV-associated lymphomas they tested bound the E2 protein in a manner identical to a bona fide human anti-E2 antibody, hypothesizing that the B-cell activation derived from the dual binding of E2 to a cognate BCR and to the CD81 molecule, which is a component of a signaling complex [33].
Interestingly, the HCV-E2 protein appears to mimic human Ig. In fact, it was observed that the N-terminal region of E2 is antigenically and structurally similar to human Ig variable domains and could represent a target for anti-human IgG antibodies [34]. Consistently, the analysis of the CDR3 sequences of the IgM-RF+ purified from the cryoprecipitate in MC patients allowed the identification of HCV-E2 as the antigen driving the production of IgM-RF [35].
2.2. The Role of Viral Lymphotropism and Viral Proteins
The potential role of viral lymphotropism in the pathogenesis of HCV-related LPDs has been emphasized since the first evidence of the presence of viral replication in lymphatic cells [1] (Figure 2). The association between viral infection of peripheral blood mononuclear cells (PBMCs) and the presence of LPDs was initially shown in patients with MC, where more evident infection of PBMCs in comparison with HCV-positive patients without MC was observed [4]. HCV infection was then observed by Galli et al. in bone marrow cells from all patients with MC versus 43% of patients without MC [36]. After these pioneering reports, a large amount of data correlating the presence of HCV in the lymphatic compartment and the development of autoimmune/lymphoproliferative disorders has been produced. In a study using the model of injection of lymphoid cells from HCV-positive patients into SCID mice, it was shown that the samples derived from HCV patients with malignant LPD were characterized by positivity for HCV replicative intermediates, stronger signals when tested for HCV genomic sequences, and successful serial passage of infected cells in different animals [37]. In addition, Sung and coworkers showed the establishment of B-cell lines persistently producing infectious virus from an HCV-positive lymphoma [38].
One interesting point about the actual dimension of lymphocyte infection in HCV+ subjects has been provided by Pal and coworkers who showed HCV infection in 85% of lymph node specimens tested by in situ hybridization and HCV replication in 50% of cases by detection of HCV replicative intermediate [39]. Interestingly, quasispecies analysis in one case indicated that 68% of variants circulating in serum were also present in lymphoid tissues, and only 40% of serum variants were identified in liver, documenting a major contribution of lymphoid replication to HCV viremia [39].
The existence of lymphotropic viral quasispecies has also been demonstrated by elegant studies evaluating HCV IRES sequence in liver and PBMC and its ability to drive viral genome translation in different cell types [40, 41].
The M. Lai's group, using a model of in vitro HCV infection of B-cells, showed that the viral infection may induce an enhanced mutation rate of immunoglobulin genes and some oncogenes, possibly through induction of error-prone DNA polymerase and activation-induced cytidine deaminase (AID), suggesting that HCV may cause tumors by a hit-and-run mechanism [42]. More recently, Ito and coworkers observed a dramatically increased expression of AID in the B-cells of HCV patients, suggesting that this may be a key factor in the lymphomagenetic process mediated by HCV [43]. Furthermore, a significantly higher expression of several lymphomagenesis-related genes in the CD19+ of HCV patients than in controls was also shown [43].
In regard to viral proteins, particular attention has been focused on the HCV core protein due to previously shown pleiotropic effects on different cell signaling pathways modulating cell viability and proliferation [44]. Focusing on animal models, core transgenic mice developed lymphoma with a high frequency (80%) at ages over 20 months [45]. The core mRNA was shown in the enlarged lymph nodes of the transgenic mice which developed lymphoma. In another transgenic model, where the IFN signaling was disrupted, the inducible and persistent expression of the HCV core in the context of all structural proteins was associated with the development of lymphoid disorders including frank lymphoma, suggesting a synergistic action of the viral proteins with IFN signaling impairment in promoting lymphomagenesis [46].
More recently, the expression of the whole HCV genome, restricted to the B-cell compartment, resulted in a high prevalence of diffuse large B-cell lymphoma (DLBCL)(up to 29%) within 600 days after birth [47]. Interestingly, the HCV core gene was expressed in all lymphomas. A wide analysis of the cytokine and chemokine pattern showed elevated levels of serum IL-2Rα in mice with lymphoma, directly originating from lymphoma tissue [47].
The expression of the HCV core in primary B cells by an adenoviral vector significantly inhibited B-lymphocyte apoptosis and induced a dramatic down regulation of MHC class II molecules. Moreover, genes associated with leukemia and B-lymphoma were consistently up regulated by the HCV core in this model [48].
Finally, in a study performed on both B-cell lines expressing the HCV core protein and in primary B-cells from patients with LPDs, it was possible to show the altered expression of some isoforms of genes of the p53 family, the DNp63 and DNp73, previously shown to be overexpressed in human cancers, including lymphoma [49, 50].
Although ectopic protein expression in both in vivo and in vitro models does not perfectly reproduce the actual situation during chronic viral infection, the consistency and coherence of the data accumulated until now suggest a potential lymphomagenic effect of the core protein when expressed as a single protein or in the context of other viral proteins.
2.3. Chromosomal Aberrations
Interesting data are available about the role played by chromosomal aberrations in HCV-related LPDs. The most investigated genetic aberration was the (14;18) translocation—t(14;18)—that was found to be significantly associated with type II or monoclonal MC. The presence of t(14;18) in MC was correlated with the overexpression of the antiapoptotic bcl-2 gene in B-cells, resulting in an imbalance of the Bcl-2/Bax ratio and abnormal B-cell survival [51, 52] (Figure 2). The regression of the expanded B-cell clones following effective antiviral treatment and, in some relapsing patients, a new expansion of the same clones were also shown [53]. Furthermore, a long-term follow-up study allowed the identification of occult HCV persistence limited to the lymphatic compartment in some patients resulting sustained viral responders after antiviral therapy [54, 55]. More interestingly, such a persistent occult lymphatic infection was associated with the initial diagnosis of MC, the persistence of some MC symptoms after therapy, and the persistence of expanded t(14;18)+ B-cell clones [54, 55]. The observation of the possibility, even if rare, of a persisting MC disease in spite of complete viral eradication suggested the existence of points of no return in the evolution of the HCV-related lymphoproliferation.
Recently, Goldberg-Bittman et al. reported an increased rate of aneuploidy in chronically infected HCV subjects versus healthy controls, with values similar to an NHL group. This observation suggests that HCV patients could be more prone to develop a lymphatic malignancy also because of bearing such alterations of the ploidy grade [56]. An important contribution to understanding the set of chromosome instability associated with HCV infection was provided by a study of Machida and coworkers. The authors observed a reduced expression of Rb protein—responsible for cell cycle arrest in case of DNA abnormalities—in various conditions including HCV-infected PBMCs isolated from patients, hepatocyte models infected in vitro with HCV or transfected with the viral core protein alone, and transgenic mice expressing core protein. In fact, lower levels of Rb could easily lead to skipping the mitosis checkpoints and contribute to generation of polyploid cells, a condition favoring neoplastic transformation [57].
2.4. Cytokines, Chemokines, and HCV-Related LPDs
Cytokines and chemokines are essential mediators of the immune response. A disturbance of the equilibrium between activating and repressing effects of these soluble molecules may be responsible for several autoimmune/lymphoproliferative disorders. Numerous reports have suggested that cytokines and chemokines are key factors in the pathogenesis of HCV-related LPDs. The MC model, as prelymphomatous condition, has been widely used to investigate the cytokine pattern characteristic of HCV-related LPDs. The role of Th1 cytokine profile (IFNγ and TNFα) and some chemokines (MIP-1α, MIP-1β, CXCL10, and CXCR3) in the pathogenesis of HCV-MC has been suggested by the elevated expression of these mediators observed in vasculitic lesions of MC patients [58]. CXCL13 was another chemokine reported as upregulated in MC patients [59]. This chemokine, also known as BCA-1 (B-cell-attracting chemokine-1) or BLC (B-lymphocyte chemoattractant), is a major regulator of B-cell trafficking, and its expression has been found to be significantly enhanced in microdissected samples from liver biopsy of patients with active cutaneous vasculitis. Antonelli and coworkers showed that serum concentrations of different cytokines and chemokines are significantly modified in MC patients [60] and have recently investigated the potential role of CXCL-10 and CXCL-11 in the pathogenesis of MC [61, 62]. In fact, high serum concentration of these soluble factors has been shown in HCV patients with MC when compared to patients without MC and healthy controls.
A growing body of evidence has accumulated in recent years showing the involvement of the B-cell-activating factor (BAFF or BLyS) in the pathogenesis of HCV-related LPDs (Figure 2). This B-cell-specific cytokine, belonging to the TNF-α family, is essential for B-lymphocyte development and survival. Several reports have shown a higher BAFF serum concentration in HCV patients than in healthy controls and, more significantly, in HCV patients with LPDs (for review, see [63]). The mechanisms of the enhanced serum concentration of BAFF in HCV-LPD patients have not been elucidated yet. A possible explanation has been recently suggested by the analysis of the polymorphic variants of the BAFF gene promoter. A particular allelic variant (−871 T), reported to induce an increased transcriptional activity of the BAFF gene [64], was significantly more frequent in patients with HCV-related MC than in HCV patients without MC. The genetic data were corroborated by the presence of higher levels of the cytokine in the serum of MC patients [65, 66].
Concerning HCV-positive subjects with NHL, Libra et al. showed an increase in the circulating levels of IL-1β [67]. As well, high serum osteopontin (OPN) levels were associated with B-cell NHL and HCV infection. Interestingly, the highest serum OPN concentrations were found among HCV-infected patients with concomitant type II MC with and without B-cell NHL [68].
2.5. MicroRNAs and HCV-Related LPDs
Increasing evidence supporting the role of microRNA (miRNA) deregulation in the pathogenesis of chronic hepatitis C infection and related disorders has been provided in the last years (for review: [69]). MiRNAs are small RNA molecules (19–22-mer noncoding RNAs) able to induce translational inhibition of target genes by partial base complementarity to the target mRNA 3′UTR. A liver-specific miRNA, miR-122, has been shown to be important for HCV replication [70]. It has been shown that interferon treatment can regulate miR-122 and other miRNA levels and that these latter mediate—at least in part—the antiviral effects of IFN [71]. These in vitro data have been partially corroborated by the observation of modified levels of miR-122 in patients unresponsive to the IFN therapy [72]. A few data are still now available concerning the involvement of miRNAs in the pathogenesis of HCV extrahepatic disorders. An interesting report recently analyzed the global miRNA expression profile in HCV-related and unrelated splenic marginal zone lymphomas (SMZLs). By using large-scale miRNA expression profiling analysis, Peveling-Oberhag and coworkers [73] quantitatively evaluated the expression of 381 miRNAs in microdissected SMZLs (HCV-positive and -negative) and in normal splenic tissues. The difference in expression profiles arose between SMZLs and normal spleens for 12 miRNAs, most of which were previously involved in various mechanisms of tumor formation for both lymphomas and other tumors. Only one miRNA, miR-26b, known to show tumor-suppressive activity, was significantly down regulated in HCV-positive lymphomas compared to HCV-negative SMZLs, suggesting a possible specific mechanism by which the virus might unfold its oncogenic potential in malignant lymphoma.
3. Conclusions
In conclusion, as summarized in Figure 2, current data suggest that HCV lymphomagenesis is a complex, multistep, multifactorial process, probably based on sustained B-cell-activation and the inhibition of B-cell apoptosis within a background of predisposing genetic factors and evolving through the progressive addition of genetic aberrations which allow the process to become progressively less dependent on the etiologic agent.
Conflict of Interests
The authors declare the absence of any conflict of interests.
Acknowledgments
This work was supported by grants from the “Associazione Italiana per la Ricerca sul Cancro” (AIRC) Investigator Grant no. 1461, “Istituto Toscano Tumori” (ITT), “Fondazione Istituto di Ricerche Virologiche Oretta Bartolomei Corsi,” “Ente Cassa di Risparmio di Firenze,” and “Fondazione Cassa di Risparmio di Pistoia e Pescia.” Authors thank Mrs. Mary Forrest for the precious help in editing the paper.
References
- 1.Zignego AL, Macchia D, Monti M, et al. Infection of peripheral mononuclear blood cells by hepatitis C virus. Journal of Hepatology. 1992;15(3):382–386. doi: 10.1016/0168-8278(92)90073-x. [DOI] [PubMed] [Google Scholar]
- 2.Zignego AL, Giannini C, Monti M, Gragnani L. Hepatitis C virus lymphotropism: lessons from a decade of studies. Digestive and Liver Disease. 2007;39(supplement 1):S38–S45. doi: 10.1016/s1590-8658(07)80009-0. [DOI] [PubMed] [Google Scholar]
- 3.Ferri C, Marzo E, Longombardo G, et al. Interferon-α in mixed cryoglobulinemia patients: a randomized, crossover-controlled trial. Blood. 1993;81(5):1132–1136. [PubMed] [Google Scholar]
- 4.Ferri C, Monti M, La Civita L, et al. Infection of peripheral blood mononuclear cells by hepatitis C virus in mixed cryoglobulinemia. Blood. 1993;82(12):3701–3704. [PubMed] [Google Scholar]
- 5.Ferri C, Caracciolo F, Zignego AL, et al. Hepatitis C virus infection in patients with non-Hodgkin’s lymphoma. British Journal of Haematology. 1994;88(2):392–394. doi: 10.1111/j.1365-2141.1994.tb05036.x. [DOI] [PubMed] [Google Scholar]
- 6.Ferri C, La Civita L, Zignego AL. Non-Hodgkin’s lymphoma: possible role of hepatitis C virus. Journal of the American Medical Association. 1994;272(5):355–356. doi: 10.1001/jama.1994.03520050033023. [DOI] [PubMed] [Google Scholar]
- 7.Ferri C, La Civita L, Monti M, et al. Can type C hepatitis infection be complicated by malignant lymphoma? The Lancet. 1995;346(8987):1426–1427. doi: 10.1016/s0140-6736(95)92442-6. [DOI] [PubMed] [Google Scholar]
- 8.Zignego AL, Ferri C, Giannini C, et al. Hepatitis C virus genotype analysis in patients with type II mixed cryoglobulinemia. Annals of Internal Medicine. 1996;124(1 I):31–34. doi: 10.7326/0003-4819-124-1_Part_1-199601010-00006. [DOI] [PubMed] [Google Scholar]
- 9.Ferri C, Lo Jacono F, Monti M, et al. Lymphotropic virus infection of peripheral blood mononuclear cells in B-Cell non-Hodgkin’s lymphoma. Acta Haematologica. 1997;98(2):89–94. doi: 10.1159/000203597. [DOI] [PubMed] [Google Scholar]
- 10.Zignego AL, Ferri C, Giannini C, et al. Hepatitis C virus infection in mixed cryoglobulinemia and B-cell Non-Hodgkin’s lymphoma: evidence for a pathogenetic role. Archives of Virology. 1997;142(3):545–555. doi: 10.1007/s007050050100. [DOI] [PubMed] [Google Scholar]
- 11.Andreone P, Gramenzi A, Cursaro C, Bernardi M, Zignego AL. Monoclonal gammopathy in patients with chronic hepatitis C virus infection. Blood. 1996;88(3, article 1122) [PubMed] [Google Scholar]
- 12.Andreone P, Zignego AL, Cursaro C, et al. Prevalence of monoclonal gammopathies in patients with hepatitis C virus infection. Annals of Internal Medicine. 1998;129(4):294–298. doi: 10.7326/0003-4819-129-4-199808150-00005. [DOI] [PubMed] [Google Scholar]
- 13.Idilman R, Colantoni A, De Maria N, Alkan S, Nand S, Van Thiel DH. Lymphoproliferative disorders in chronic hepatitis C. Journal of Viral Hepatitis. 2004;11(4):302–309. doi: 10.1111/j.1365-2893.2004.00480.x. [DOI] [PubMed] [Google Scholar]
- 14.Zignego AL, Giannini C, Ferri C. Hepatitis C virus-related lymphoproliferative disorders: an overview. World Journal of Gastroenterology. 2007;13(17):2467–2478. doi: 10.3748/wjg.v13.i17.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zignego AL, Ferri C, Pileri SA, Caini P, Bianchi FB. Extrahepatic manifestations of hepatitis C virus infection: a general overview and guidelines for a clinical approach. Digestive and Liver Disease. 2007;39(1):2–17. doi: 10.1016/j.dld.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Ferri C, Pileri S, Zignego AL. Hepatitis C virus infection and non-Hodgkin’s lymphoma. In: Geodert J, NCI (NIH), editors. Infectious Causes of Cancer. Targets for Intervention. Totowa, NJ, USA: The Human Press; 2000. pp. 349–368. [Google Scholar]
- 17.Zignego AL, Ferri C, Innocenti F, et al. Lack of preferential localization of tumoral mass in B-cell non- Hodgkin’s lymphoma associated with hepatitis C virus infection. Blood. 1997;89(8):3066–3068. [PubMed] [Google Scholar]
- 18.Mele A, Pulsoni A, Bianco E, et al. Hepatitis C virus and B-cell non-Hodgkin lymphomas: an Italian multicenter case-control study. Blood. 2003;102(3):996–999. doi: 10.1182/blood-2002-10-3230. [DOI] [PubMed] [Google Scholar]
- 19.De Sanjose S, Benavente Y, Vajdic CM, et al. Hepatitis C and non-Hodgkin lymphoma among 4784 cases and 6269 controls from the international lymphoma epidemiology consortium. Clinical Gastroenterology and Hepatology. 2008;6(4):451–458. doi: 10.1016/j.cgh.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vita S, Zagonel V, Russo A, et al. Hepatitis C virus, non-Hodgkin’s lymphomas and hepatocellular carcinoma. British Journal of Cancer. 1998;77(11):2032–2035. doi: 10.1038/bjc.1998.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermine O, Lefrère F, Bronowicki JP, et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. The New England Journal of Medicine. 2002;347(2):89–94. doi: 10.1056/NEJMoa013376. [DOI] [PubMed] [Google Scholar]
- 22.Kelaidi C, Rollot F, Park S, et al. Response to antiviral treatment in hepatitis C virus-associated marginal zone lymphomas. Leukemia. 2004;18(10):1711–1716. doi: 10.1038/sj.leu.2403443. [DOI] [PubMed] [Google Scholar]
- 23.Saadoun D, Suarez F, Lefrere F, et al. Splenic lymphoma with villous lymphocytes, associated with type II cryoglobulinemia and HCV infection: a new entity? Blood. 2005;105(1):74–76. doi: 10.1182/blood-2004-05-1711. [DOI] [PubMed] [Google Scholar]
- 24.Kawamura Y, Ikeda K, Arase Y, et al. Viral elimination reduces incidence of malignant lymphoma in patients with hepatitis C. American Journal of Medicine. 2007;120(12):1034–1041. doi: 10.1016/j.amjmed.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 25.Sansonno D, De Vita S, Iacobelli AR, Cornacchiulo V, Boiocchi M, Dammacco F. Clonal analysis of intrahepatic B cells from HCV-infected patients with and without mixed cryoglobulinemia. Journal of Immunology. 1998;160(7):3594–3601. [PubMed] [Google Scholar]
- 26.Sansonno D, Lauletta G, De Re V, et al. Intrahepatic B cell clonal expansions and extrahepatic manifestations of chronic HCV infection. European Journal of Immunology. 2004;34(1):126–136. doi: 10.1002/eji.200324328. [DOI] [PubMed] [Google Scholar]
- 27.De Re V, De Vita S, Marzotto A, et al. Pre-malignant and malignant lymphoproliferations in an HCV-infected type II mixed cryoglobulinemic patient are sequential phases of an antigen-driven pathological process. International Journal of Cancer. 2000;87(2):211–216. doi: 10.1002/1097-0215(20000715)87:2<211::aid-ijc9>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 28.Sagaert X, Van Cutsem E, De Hertogh G, Geboes K, Tousseyn T. Gastric MALT lymphoma: a model of chronic inflammation-induced tumor development. Nature Reviews Gastroenterology and Hepatology. 2010;7(6):336–346. doi: 10.1038/nrgastro.2010.58. [DOI] [PubMed] [Google Scholar]
- 29.De Re V, De Vita S, Marzotto A, et al. Sequence analysis of the immunoglobulin antigen receptor of hepatitis C virus-associated non-Hodgkin lymphomas suggests that the malignant cells are derived from the rheumatoid factor-producing cells that occur mainly in type II cryoglobulinemia. Blood. 2000;96(10):3578–3584. [PubMed] [Google Scholar]
- 30.De Re V, Sansonno D, Simula MP, et al. HCV-NS3 and IgG-Fc crossreactive IgM in patients with type II mixed cryoglobulinemia and B-cell clonal proliferations. Leukemia. 2006;20(6):1145–1154. doi: 10.1038/sj.leu.2404201. [DOI] [PubMed] [Google Scholar]
- 31.Pileri P, Uematsu Y, Campagnoli S, et al. Binding of hepatitis C virus to CD81. Science. 1998;282(5390):938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 32.Rosa D, Saletti G, De Gregorio E, et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18544–18549. doi: 10.1073/pnas.0509402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinn ER, Chan CH, Hadlock KG, Foung SKH, Flint M, Levy S. The B-cell receptor of a hepatitis C virus (HCV)-associated non-Hodgkin lymphoma binds the viral E2 envelope protein, implicating HCV in lymphomagenesis. Blood. 2001;98(13):3745–3749. doi: 10.1182/blood.v98.13.3745. [DOI] [PubMed] [Google Scholar]
- 34.Hu YW, Rocheleau L, Larke B, et al. Immunoglobulin mimicry by hepatitis C Virus envelope protein E2. Virology. 2005;332(2):538–549. doi: 10.1016/j.virol.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 35.Ferri S, Dal Pero F, Bortoletto G, et al. Detailed analysis of the E2-IgM complex in hepatitis C-related type II mixed cryoglobulinaemia. Journal of Viral hepatitis. 2006;13(3):166–176. doi: 10.1111/j.1365-2893.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 36.Galli M, Zehender G, Monti G, et al. Hepatitis C virus RNA in the bone marrow of patients with mixed cryoglobulinemia and in subjects with noncryoglobulinemic chronic hepatitis type C. Journal of Infectious Diseases. 1995;171(3):672–675. doi: 10.1093/infdis/171.3.672. [DOI] [PubMed] [Google Scholar]
- 37.Bronowicki JP, Loriot MA, Thiers V, Grignon Y, Zigneco AL, Bréchot C. Hepatitis C virus persistence in human hematopoietic cells injected into SCID mice. Hepatology. 1998;28(1):211–218. doi: 10.1002/hep.510280127. [DOI] [PubMed] [Google Scholar]
- 38.Sung MHV, Shimodaira S, Doughty AL, et al. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. Journal of Virology. 2003;77(3):2134–2146. doi: 10.1128/JVI.77.3.2134-2146.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pal S, Sullivan DG, Kim S, et al. Productive replication of hepatitis C virus in perihepatic lymph nodes in vivo: implications of HCV lymphotropism. Gastroenterology. 2006;130(4):1107–1116. doi: 10.1053/j.gastro.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 40.Di Liberto G, Roque-Afonso AM, Kara R, et al. Clinical and therapeutic implications of hepatitis C virus compartmentalization. Gastroenterology. 2006;131(1):76–84. doi: 10.1053/j.gastro.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Roque-Afonso AM, Ducoulombier D, Di Liberto G, et al. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. Journal of Virology. 2005;79(10):6349–6357. doi: 10.1128/JVI.79.10.6349-6357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machida K, Cheng KTN, Sung VMH, et al. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(12):4262–4267. doi: 10.1073/pnas.0303971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito M, Murakami K, Suzuki T, et al. Enhanced expression of lymphomagenesis-related genes in peripheral blood B cells of chronic hepatitis C patients. Clinical Immunology. 2010;135(3):459–465. doi: 10.1016/j.clim.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Giannini C, Bréchot C. Hepatitis C virus biology. Cell Death and Differentiation. 2003;10(supplement 1):S27–S38. doi: 10.1038/sj.cdd.4401121. [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa T, Shibuya K, Yasui K, Mitamura K, Ueda S. Expression of hepatitis C virus core protein associated with malignant lymphoma in transgenic mice. Comparative Immunology, Microbiology and Infectious Diseases. 2003;26(2):115–124. doi: 10.1016/s0147-9571(02)00038-3. [DOI] [PubMed] [Google Scholar]
- 46.Machida K, Tsukiyama-Kohara K, Sekiguch S, et al. Hepatitis C virus and disrupted interferon signaling promote lymphoproliferation via type II CD95 and interleukins. Gastroenterology. 2009;137(1):285.e11–296.e11. doi: 10.1053/j.gastro.2009.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasama Y, Sekiguchi S, Saito M. Persistent expression of the full genome of hepatitis C virus in B cells induces spontaneous development of B-cell lymphomas in vivo. Blood. 2010;116:4926–4933. doi: 10.1182/blood-2010-05-283358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu CG, Budhu A, Chen S, et al. Effect of hepatitis C virus core protein on the molecular profiling of human B lymphocytes. Molecular Medicine. 2006;12(1–3):47–53. doi: 10.2119/2006-00020.Wu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alisi A, Giannini C, Spaziani A, Caini P, Zignego AL, Balsano C. Involvement of PI3K in HCV-related lymphoproliferative disorders. Journal of Cellular Physiology. 2008;214(2):396–404. doi: 10.1002/jcp.21211. [DOI] [PubMed] [Google Scholar]
- 50.Alisi A, Giannini C, Spaziani A, et al. Hepatitis C virus core protein enhances B lymphocyte proliferation. Digestive and Liver Disease. 2007;39(supplement 1):S72–S75. doi: 10.1016/s1590-8658(07)80015-6. [DOI] [PubMed] [Google Scholar]
- 51.Zignego AL, Ferri C, Giannelli F, et al. Prevalence of bcl-2 rearrangement in patients with hepatitis C virus-related mixed cryoglobulinemia with or without B-cell lymphomas. Annals of Internal Medicine. 2002;137(7):571–580. doi: 10.7326/0003-4819-137-7-200210010-00008. [DOI] [PubMed] [Google Scholar]
- 52.Zignego AL, Giannelli F, Marrocchi ME, et al. T(14;18) translocation in chronic hepatitis C virus infection. Hepatology. 2000;31(2):474–479. doi: 10.1002/hep.510310230. [DOI] [PubMed] [Google Scholar]
- 53.Giannelli F, Moscarella S, Giannini C, et al. Effect of antiviral treatment in patients with chronic HCV infection and t(14;18) translocation. Blood. 2003;102(4):1196–1201. doi: 10.1182/blood-2002-05-1537. [DOI] [PubMed] [Google Scholar]
- 54.Giannini C, Giannelli F, Zignego AL. Association between mixed cryogtobulinemia, translocation (14;18), and persistence of occult HCV lymphoid infection after treatment. Hepatology. 2006;43(5):1166–1167. doi: 10.1002/hep.21132. [DOI] [PubMed] [Google Scholar]
- 55.Giannini C, Petrarca A, Monti M, et al. Association between persistent lymphatic infection by hepatitis C virus after antiviral treatment and mixed cryoglobulinemia. Blood. 2008;111(5):2943–2945. doi: 10.1182/blood-2007-09-112490. [DOI] [PubMed] [Google Scholar]
- 56.Goldberg-Bittman L, Kitay-Cohen Y, Hadari R, Yukla M, Fejgin MD, Amiel A. Random aneuploidy in chronic hepatitis C patients. Cancer Genetics and Cytogenetics. 2008;180(1):20–23. doi: 10.1016/j.cancergencyto.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 57.Machida K, Liu JC, McNamara G, Levine A, Duan L, Lai MMC. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. Journal of Virology. 2009;83(23):12590–12600. doi: 10.1128/JVI.02643-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saadoun D, Bieche I, Maisonobe T, et al. Involvement of chemokines and type 1 cytokines in the pathogenesis of hepatitis C virus-associated mixed cryoglobulinemia vasculitis neuropathy. Arthritis and Rheumatism. 2005;52(9):2917–2925. doi: 10.1002/art.21270. [DOI] [PubMed] [Google Scholar]
- 59.Sansonno D, Tucci FA, Troiani L, et al. Increased serum levels of the chemokine CXCL13 and up-regulation of its gene expression are distinctive features of HCV-related cryoglobulinemia and correlate with active cutaneous vasculitis. Blood. 2008;112(5):1620–1627. doi: 10.1182/blood-2008-02-137455. [DOI] [PubMed] [Google Scholar]
- 60.Antonelli A, Ferri C, Fallahi P, et al. High values of CXCL10 serum levels in mixed cryoglobulinemia associated with hepatitis C infection. American Journal of Gastroenterology. 2008;103(10):2488–2494. doi: 10.1111/j.1572-0241.2008.02040.x. [DOI] [PubMed] [Google Scholar]
- 61.Antonelli A, Fallahi P, Ferrari SM, et al. Circulating CXCL11 and CXCL10 are increased in hepatitis C-associated cryoglobulinemia in the presence of autoimmune thyroiditis. doi: 10.1007/s10165-011-0565-x. Modern Rheumatology. In press. [DOI] [PubMed] [Google Scholar]
- 62.Antonelli A, Ferri C, Ferrari SM, et al. High serum levels of CXCL11 in mixed cryoglobulinemia are associated with increased circulating levels of interferon-γ . Journal of Rheumatology. 2011;38(9):1947–1952. doi: 10.3899/jrheum.110133. [DOI] [PubMed] [Google Scholar]
- 63.De Vita S, Quartuccio L, Fabris M. Hepatitis C virus infection, mixed cryoglobulinemia and BLyS upregulation: targeting the infectious trigger, the autoimmune response, or both? Autoimmunity Reviews. 2008;8(2):95–99. doi: 10.1016/j.autrev.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 64.Novak AJ, Grote DM, Ziesmer SC, et al. Elevated serum B-lymphocyte stimulator levels in patients with familial lymphoproliferative disorders. Journal of Clinical Oncology. 2006;24(6):983–987. doi: 10.1200/JCO.2005.02.7938. [DOI] [PubMed] [Google Scholar]
- 65.Giannini C, Gragnani L, Piluso A, et al. Can BAFF promoter polymorphism be a predisposing condition for HCV-related mixed cryoglobulinemia? Blood. 2008;112(10):4353–4354. doi: 10.1182/blood-2008-07-170613. [DOI] [PubMed] [Google Scholar]
- 66.Gragnani L, Piluso A, Giannini C, et al. Genetic determinants in hepatitis C virus-associated mixed cryoglobulinemia: role of polymorphic variants of BAFF promoter and Fcγ receptors. Arthritis and Rheumatism. 2011;63(5):1446–1451. doi: 10.1002/art.30274. [DOI] [PubMed] [Google Scholar]
- 67.Libra M, Mangano K, Anzaldi M, et al. Analysis of interleukin (IL)-1beta IL-1 receptor antagonist, soluble IL-1 receptor type II and IL-1 accessory protein in HCV-associated lymphoproliferative disorders. Oncology Reports. 2006;15(5):1305–1308. [PubMed] [Google Scholar]
- 68.Libra M, Indelicato M, De Re V, et al. Elevated serum levels of osteopontin in HCV-associated lymphoproliferative disorders. Cancer Biology and Therapy. 2005;4(11):1192–1194. doi: 10.4161/cbt.4.11.2087. [DOI] [PubMed] [Google Scholar]
- 69.Kumar A. MicroRNA in HCV infection and liver cancer. Biochimica et Biophysica Acta. 2011;1809(11-12):694–699. doi: 10.1016/j.bbagrm.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 70.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Molecular biology: modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science. 2005;309(5740):1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 71.Pedersen IM, Cheng G, Wieland S, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449(7164):919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nature Medicine. 2009;15(1):31–33. doi: 10.1038/nm.1902. [DOI] [PubMed] [Google Scholar]
- 73.Peveling-Oberhag J, Crisman G, Schmidt A, et al. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia. 2012;26(7):1654–1662. doi: 10.1038/leu.2012.29. [DOI] [PubMed] [Google Scholar]