

Abstract
Prostacyclin, or PGI2, is an end product derived from the sequential metabolism of arachidonic acid via cyclooxygenase and PGI synthase (PGIS). The receptor for PGI2, IP, can be found on a variety of cell types and signaling through this receptor exhibits broad physiological effects. Historically, PGI2 has been understood to play a role in cardiovascular health, specifically having powerful vasodilatory effects via relaxation of smooth muscle and inhibiting of platelet aggregation. For these reasons, PGI2 has a long history of use for the treatment of pulmonary arterial hypertension (PAH). Only recently, its importance as an immunomodulatory agent has been investigated. PGI2 regulates both the innate and adaptive immune systems and its effects are, for the most part, thought to be anti-inflammatory or immunosuppressive in nature, which may have implications for its further clinical use.
1. Introduction
Prostacyclin, or PGI2, was first reported by Needleman and Vane in 1976 and is an end product derived from the sequential metabolism of arachidonic acid via cyclooxygenase-2 (COX-2) and prostacyclin synthase (PGIS) [1]. COX-2 is expressed upon specific stimulation such as cytokines, growth factors, bacterial endotoxins, tumor promoters, and hormones by macrophages, neutrophils, and activated mesenchymal cells [2–4]. There is rare expression of COX-2 in unstimulated tissues [5–7], but it can be present at low basal levels in endothelium and the renal macula densa [2, 5]. COX-2 is typically associated with proinflammatory conditions such as atherosclerotic lesions, aortic aneurysms, or vascular damage where COX-2 derived products likely provide a protective effect [8–10]. COX-2 is inhibited by nonsteroidal anti-inflammatory (NSAIDS) and specific COX-2 inhibitors, which may have tissue specific effects.
Several additional cells types have been shown to express COX-2 and PGIS and they include fibroblasts, follicular dendritic cells, endothelial cells, smooth muscle cells, and thymic nurse cells. Production of PGI2 is decreased by the inhibition of PGIS by tyrosine-nitrating agents such as peroxynitrite [11] and tetranitromethane [12]. Lastly, PGIS can be limited by substrate-dependent suicide inactivation if there is adequate conversion of PGH2, the substrate for PGIS, which causes accumulation of inactivated enzyme [13].
PGI2 is primarily produced in mammalian vasculature with elevated levels in pulmonary arterial segments when compared to systemic circulation [14]. As such, PGI2 has been understood to play a role in cardiovascular health specifically inhibiting platelet aggregation and having powerful vasodilatory effects via relaxation of smooth muscle [5, 15]. PGI2 analogues have been successfully used for therapy in pulmonary arterial hypertension, peripheral occlusive disease, vascular complication of diabetes mellitus, and treatment of reperfusion injury. Only recently, its importance as an immunomodulatory agent has been investigated.
2. PGI2 Receptor Signaling
The cell surface receptor for PGI2 is a seven transmembrane G-protein-coupled receptor termed IP [6]. IP is coupled to a guanosine nucleotide-binding α-stimulatory protein (Gα s). When activated by PGI2, IP stimulates adenyl cyclase leading to increased intracellular cyclic AMP (cAMP) (see Figure 1). Increased cAMP then leads to activation of protein kinase A (PKA) and further phosphorylation of key proteins [14–16]. These actions culminate in relaxation of smooth muscle, reduced cell proliferation, and other inhibitory mechanisms. IP is found on a variety of cell types and exhibits broad physiological effects. Mouse IP receptors have been identified on neurons, smooth muscle cells of the aorta, coronary arteries, pulmonary arteries, and megakaryocytes. Human IP receptors are present on multiple cell types including platelets, medullary thymocytes, neutrophils, dendritic cells, eosinophils, T regulatory cells, and activated T cells [4, 17]. IP receptors are also found on many cell types in the lung such as macrophages, pneumocytes, smooth muscle cells, and fibroblasts [17]. In addition to the single known membrane IP receptor, a peroxisome proliferator-activated nuclear receptor (PPAR) functions as a transcription factor after activation by PGI2 [3, 15]. There are three PPAR isoforms, α, δ (β), and γ [15]. PPARγ, expressed in adipose tissue, spleen, and large intestines predominately, is thought to be downstream of the activated IP membrane receptor and can be stimulated via stable PGI2 analogues. PPARs are responsive to not only PGI2 but also a broad range of ligands [15].
Figure 1.
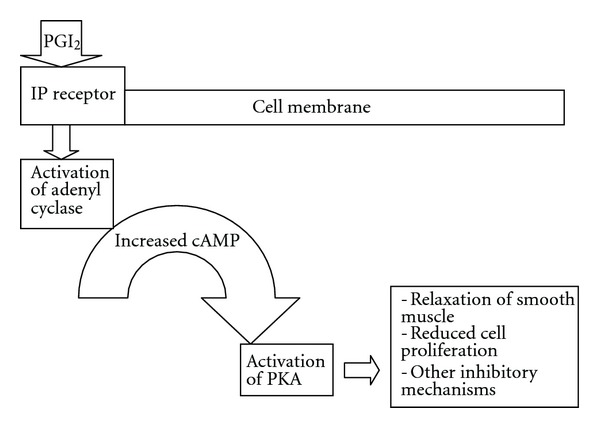
PGI2 receptor signaling.
3. PGI2 in Specific Disease States
3.1. Allergic Inflammation
One of the areas that PGI2 has been extensively studied is allergic inflammation. PGI2 is produced in the human lung during allergic reactions, suggesting that it may have a regulatory role in allergen-induced inflammation [18, 19]. The in vivo role of PGI2 in mediating allergic inflammation has been investigated both in IP-deficient mice, which examines the role of endogenous PGI2 signaling, as well as exogenous administration of PGI2. First, we will review studies that use IP-deficient mice in determining how endogenous PGI2 modulates allergen-induced lung disease.
In an acute model of allergic inflammation induced by ovalbumin (OVA), IP-deficient mice had significantly greater airway inflammatory responses consisting of increased plasma extravasation, leukocyte accumulation, and both IL-4 and IL-5 production in the airways after sensitization and exposure to inhaled antigen [20]. In addition, the IP-deficient mice had elevated total serum IgE and antigen-specific IgE when compared to wild-type mice [20]. These findings support endogenous PGI2 as an important suppressor of acute allergic inflammation. Similar findings were reported in a chronic model of OVA-induced lung inflammation. In this IP-deficient mouse model, there was a significant increase in the level of inflammatory leukocytes in the airway, serum OVA-specific IgE, and lung expression of T helper type 2 (Th2) cytokines such as IL-4, IL-5, and IL-13. The IP-deficient mice had more subepithelial fibrosis compared to wild-type mice, possibly related to the upregulation of collagen synthesis [21]. Therefore, inability to signal through IP led to enhanced acute and chronic allergic inflammation and airway remodeling.
In addition to IP-deficient mice, the role of COX-2 inhibitors contributes to the knowledge of how endogenous PGI2 regulates allergic inflammation. After OVA inhalation in a DO11.10 transgenic mousemodel of T-cell-mediated airway inflammation, there was an increased level of PGI2 [4]. Blocking PGI2 via COX-2 inhibition resulted in a marked increase in Th2 mediated lung inflammation in response to OVA challenge. COX-2 inhibition and the prevention of PGI2 formation was associated with increased bronchial airway hyperresponsiveness, elevated lung expression of Th2 cytokines, and decreased IL-10 production [4]. These results highlight a possible risk enhanced airway inflammation with use of specific COX-2 inhibitors in allergic asthmatics by blocking production of an immunoinhibitory prostanoid such as PGI2.
Animal models of exogenous PGI2 administration further supports that this prostanoid inhibits allergic inflammation. In an OVA sensitized mouse model using adoptive transfer of DO11.10 Th2 cells pretreated with PGI2, there was significantly decreased pulmonary inflammation and airway hyperreactivity [22]. A protective effect of PGI2 on acute airway function is further suggested by a null effect on ozone induced airway inflammation and hyperresponsiveness in mPGES-1 deficient mice. PGI2 metabolites in the BAL of these mice were increased, while changes in other prostanoids favored deleterious effects on lung function [23]. PGI2 suppressed Th2 infiltration of the lung via strong inhibition of CCL17-induced chemotaxis. IP deficient Th2 cells were unaffected and migrated normally [22]. Lastly, in a mouse model of asthma, inhaled iloprost, a PGI2 analogue, decreased the cardinal features of asthma such as Th2 cytokine production, eosinophilic airway inflammation, goblet cell hyperplasia, and bronchial airway hyperresponsiveness [24]. Iloprost inhibited the maturation and migration of antigen presenting lung myeloid dendritic cells, decreased costimulatory molecules, and decreased the induction of allergen-specific Th2 response [24]. These findings suggest a role for inhaled iloprost in the treatment of asthma.
3.2. Inflammation-Induced Anorexia
We have shown that PGI2 signaling via the IP receptor reduces allergic inflammation. The effect of decreased appetite in acute inflammation, such as in the setting of IL-1β and lipopolysaccharide (LPS) administration, has been suggested to be PG dependent. In a mouse model, PGI2 signaling decreased the level of circulating ghrelin, a peptide produced predominantly by the stomach, which has potent stimulatory effects on appetite [25]. This finding was very similar to the effect of LPS on circulating levels of ghrelin and suggests a role for PGI2 in the acute sickness behavior of anorexia. IL-1β induced ghrelin expressing cells to produce PGI2. Nonspecific inhibition of PG production via NSAIDs reversed the decrease in circulating ghrelin caused by the acute inflammatory stimulation of LPS specifically [25]. These results suggest that PGI2 may suppress appetite in certain acute inflammatory disease states.
3.3. Liver Injury
In a mouse model of a concanavalin-A (ConA-) induced immune-mediated liver injury mimicking hepatic inflammation, beraprost, a PGI2 analogue, decreased tissue damage [7]. COX-2 deficient mice developed more severe ConA-induced liver damage compared to wild-type mice or COX-1 deficient mice. Treatment with beraprost/ConA had a protective effect with a more than 10-fold decrease in serum alanine aminotransferase (ALT) levels compared to those treated with vehicle/ConA alone. Hepatic mRNA levels and expression of both TNF-α and IFN-γ by natural killer T cells (NKT) and T cells, key in the development of ConA induced liver disease, were decreased in the COX-2 deficient mice after treatment with beraprost when compared to vehicle/ConA alone treated mice. The protection provided by beraprost is postulated to stem from the maintenance of hepatic blood flow via beraprost's vasodilatory effects [7]. These findings suggest that PGI2 analogues may be of benefit to patients suffering from inflammatory liver disease such as hepatitis due to viral infection, autoimmune conditions, use of certain drugs, or alcohol ingestion.
3.4. Cardiovascular Disease
It is hypothesized that early depletion of PGI2 from endothelial tissue could lead to the pathogenesis of atherosclerosis by causing deposition of adipocyte lipid in smooth muscle cells [26]. Cellular micro RNA (miRNA) is an important known negative regulator of gene expression. PGI2 regulated miRNA expression in a mouse adipose tissue-derived cell line leading to diminished deposition of lipid in cells [15]. The importance of this lies in the possibility of a relationship between loss of normal PGI2 production in the setting of obesity and atherosclerosis.
Iloprost was investigated for its effects on leukocyte adherence in intestinal venules and subsequent microvascular blood flow in a rat endotoxemia model. Iloprost attenuated leukocyte adherence in both postcapillary and collecting intestinal venules and improved intestinal microvascular blood flow without affecting mean arterial pressure or heart rate [27]. These findings suggest a role for iloprost therapy to reduce endotoxin-induced intestinal injury.
Retinoic acid induces PGIS and therefore synthesis of PGI2 in human umbilical vein endothelial cells [2]. Retinoic acid is important in the development of the cardiovascular system during embryonic development, angiogenesis and has antithrombotic and antiatherogenic qualities. 13-cis-retinoic acid (13-cis-RA) a molecule with anti-inflammatory, antitumor, and immunomodulatory effects elevated PGI2 levels as measured by 6-oxo-PGF1α. Consistently, arachidonic acid induced platelet aggregation was inhibited by 13-cis-RA. Treatment with the inflammatory cytokine IL-1β alone rapidly inactivated PGIS in these same cells followed by a diminution of PGI2. These findings support a role for 13-cis-RA as a possible selective treatment for patients with inflammatory cardiac disease. When IL-1β was given in combination with 13-cis-RA, 13-cis-RA was able to overcome the inhibitory effects of IL-1β, leading to increased PGIS expression and PGI2 levels [2]. Similarly, in human vascular smooth muscle cells exposed to IL-1β, hypoxia overcame the inhibitory effects of IL-1β and increased PGI2 production [5]. Hypoxia alone in these same cells elevated PGIS expression and PGI2 levels. These findings suggest that hypoxia could drive an adaptive response in vascular cells and plays a role in protecting vascular cells when inflammation is present [5].
3.5. Emphysema
PGIS expression was lower in arteriolar endothelium of human emphysema lung tissue compared with normal lung [28]. Cigarette smoke extract suppressed PGIS gene expression suggesting that its decrease could lead to deleterious effects on lung vasculature in this setting. Mice exhibiting overexpression of PGIS in the pulmonary vasculature had decreased endothelial cell apoptosis after chronic tobacco smoke exposure [28]. Cigarette smoke extract may bind to CpG sites in DNA leading to disruption of transcriptional regulation suggesting a mechanism for the minimization of PGIS gene expression.
3.6. Cytokine-Mediated Inflammation
IL-1β, TGF-β, and bradykinin, in human pulmonary artery smooth muscle, decreased the production of cAMP in response to subsequent administration of PGI2 analogues, providing data that inflammation can impair the actions of PGI2 analogues in pulmonary hypertension treatment [14]. IL-1β, TGF-β, and bradykinin also decreased adenylyl cyclase mRNA and increased G-α inhibitory (Gα i) protein levels with subsequent reduction in IP mRNA expression [14]. Importantly, this could be a rational for the development of tolerance to PGI2 analogue medications.
Cytokine toxicity is mediated in many cell types through the arachidonic acid metabolism pathway via inducible COX-2 production [3]. Proinflammatory cytokines are major effectors of programmed cell death in the development of type 1 diabetes mellitus. Using a model of human insulin producing pancreatic β cells, PGIS overexpression protected against cytokine toxicity via decreased activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and subsequent prevention of inducible nitric oxide synthase (iNOS) and decreased cytokine-induced caspase-3 activation. Therefore, low-endogenous PGIS expression may have a pathogenic part in the development of pancreatic β cell death [3].
3.7. Fibrosis
PGI2 has regulatory affects on fibroblast proliferation. For example, in a bleomycin-induced pulmonary fibrosis mouse model, PGI2 functioned as an antiproliferative molecule preventing an increase in fibroblasts and providing protection against loss of lung function [17]. Similarly, in an additional mouse model of bleomycin-induced pulmonary fibrosis, mice treated with iloprost had a significant reduction in airway and pulmonary parenchyma inflammatory cells and deposition of collagen with improvement in lung static compliance, tissue elastance, and overall survival [29]. The proposed mechanism for these changes is the upregulation of antifibrotic mediators such as interferon-γ (IFN-γ), the chemokine CXCL10, and the downregulation of proinflammatory and profibrotic cytokines such as TNF-α, IL-6, and TGF-β [29].
Using a mouse model of PGIS overexpression specifically in lung epithelium, mortality related to bleomycin-induced acute lung injury was decreased [30]. In addition to decreased mortality, there was a reduction in parenchymal consolidation, apoptosis of lung tissue and weight loss. These findings were explained by in vitro and in vivo PGI2 induced expression of NADP(H) : quinone oxide reductase 1 (Nqo1), an enzyme known to inhibit the generation of reactive oxygen species and therefore protective against oxidative stress. The PGIS-overexpressing mice had elevated levels of this antioxidant prior to administration of bleomycin and afterwards [30]. Taken together, these three studies suggest that PGI2 may provide protection against bleomycin-induced lung injury.
3.8. Viral Infection
Signaling through IP had protective effects in the setting of respiratory syncytial virus (RSV) infection in a mouse model and one study suggests it may be beneficial in human disease. During RSV infection, in a mouse model of overexpressed PGIS in bronchial epithelium, weight loss, viral replication, and IFN-γ production were all decreased compared to controls [31]. Histopathology results showed decreased pulmonary edema. In contrast, IP-deficient mice had longer more severe illness with prolonged viral replication [31].
RSV infection also elevated a urinary metabolite of PGI2 in human infants [32] suggesting PGI2 may modulate virally-induced illness in people. Low numbers of a genetic polymorphism in the PGIS gene with subsequent decreased urinary PGI2 metabolite levels, described as a genetic 9-base variable-number tandem repeat (VNTR), were correlated with more severe RSV infection. Infants with greater VNTR had less severe RSV-induced infections. Therefore, an association between lower numbers of PGIS VNTR repeats and increased severity of RSV infection identifies a host genetic factor associated with RSV disease severity [32]. Both of these studies support a protective role for PGI2 in RSV-induced illness and suggest a possible therapy for acute RSV infection.
3.9. Rheumatoid Arthritis
In contrast to the seemingly anti-inflammatory properties of PGI2, there is still debate about its effect in the setting of specific conditions [16, 33, 34]. PGI2 and PGE2 have been questioned as causative factors in inflammation as levels are increased in inflammatory tissues. The synovial fluid of rheumatoid arthritis (RA) patients has rich quantities of PGI2 but the exact role of PGI2 in RA is not fully understood [33, 34]. In a mouse model of chronic RA, IP-deficient mice were subjected to collagen-induced arthritis and had significantly decreased clinical and histologic arthritic scores despite anticollagen antibodies and complement activation similar to wild-type mice [34]. These mice were noted to have decreased levels of IL-6 in their arthritic paws. Administration of an IP agonist elevated the inflammatory cytokine IL-6 and amplified arthritis related genes such as IL-11, VEGF, FGF-2, and RANKL, increasing inflammation in the joint. Elevated IL-6 led to proliferation of the synovium, maturation of B cells, and formation of osteoclasts, all important in the pathogenesis of RA [34]. Importantly, signaling through the IP receptor required the presence of inflammatory IL-1β for many of its effects in RA. One mechanism proposed for the effects of increased PGI2 was the enhanced production of IL-6 via activated synovial fibroblasts in the setting of IL-1β. Lack of elevated IL-6 due to IL-1β explains why patients receiving PGI2 agonists, for example, due to PAH, do not exhibit an increase in arthritic symptoms as a side effect. The results of this study are intriguing because PGI2 was traditionally thought to play a role in the mediation of acute inflammation, but its role in chronic inflammation has been less studied [34].
Similarly, in an IP-deficient mouse model of collagen-antibody induced chronic inflammatory arthritis, the reduction in arthritis scores was 91% compared to a wild-type control group [33]. When a highly selective IP antagonist was used in a RA mouse model, it decreased pain similar to use of NSAIDs [33]. Therefore, it appears that signaling through the IP receptor, in the setting of RA, may increase inflammation in the joint. Currently, it is believed that both PGI2 and PGE2 are the primary prostaglandins involved in the inflammatory pain response in RA and this provides a rationale for the empiric use of NSAIDS and COX inhibitors for treatment of chronic arthritis.
4. PGI2 Regulation of the Immune System
4.1. PGI2 Regulation of the Innate Immune System
PGI2 regulates both innate and adaptive immunity and its effects are, for the most part, anti-inflammatory or immunosuppressive in nature. PGI2 modulates the function of dendritic cells, macrophages, monocytes, endothelial cells and eosinophils [35–41]. We will now review this data.
Dendritic cells are an important bridge between the innate and the adaptive immune system. PGI2 analogues decreased mouse bone marrow derived dendritic cell (BMDC) maturation, function, and proinflammatory cytokine production after LPS stimulation. PGI2 analogues also increased production of anti-inflammatory IL-10 and diminished chemokine production in mice suggesting an overall anti-inflammatory effect [42]. In a dose-dependent fashion, PGI2 analogues, decreased secretion of TNF-α, IL-1α, IL-6, and IL-12 in vitro. After LPS stimulation, both iloprost and cicaprost decreased levels of costimulatory molecules CD86, CD40, and MHC class II molecules on BMDC. The BMDC had a diminished ability to activate antigen-specific CD4 T cells of DO11.10 mice and led to reduced IL-4 and IL-5 production by the CD4 cells after iloprost and cicaprost treatment [42]. When the biological activity of iloprost on human monocytes-derived dendritic cells was examined, similar findings were identified. In a dose dependent fashion, iloprost inhibited the secretion of TNF-α, IL-6, IL-8, and IL-12 by monocytes-derived dendritic cells and increase secretion of IL-10 [39].
PGI2 analogues produced a differential pattern of regulation on alveolar versus peritoneal rat macrophages suggesting variation in immunomodulatory effects of these agents on these specific cell types. Using a well characterized FcR-mediated model of phagocytosis, activation of the IP receptor inhibited phagocytosis of IgG-opsonized targets in peritoneal macrophages to a much greater extent compared to alveolar macrophages [35]. In addition, under conditions of LPS treatment, peritoneal macrophages increased production of IL-6 after administration of iloprost or carbaprostacyclin, but alveolar macrophages did not increase production to the same degree. Iloprost and carbaprostacyclin also significantly inhibited peritoneal macrophage of bacterial killing when compared to alveolar macrophages. A postulated reason for the differences is the differential expression of IP receptors on the two macrophage cell types and different receptor binding properties of PGI2 analogues [35].
In addition to its effects on mouse innate immune cells, PGI2 also has important regulatory effects on human dendritic cells. Iloprost increased IL-10 expression but inhibited toll-like receptor-mediated expression of TNF-α and IFN-α in human plasmacytoid dendritic cells suggesting an overall anti-inflammatory role [36]. Iloprost may, therefore, increase the tolerogenic ability of plasmacytoid dendritic cells and could potentially be useful in asthma treatment. Human follicular dendritic cells, found in germinal centers of secondary lymphoid follicles, strongly express PGIS and are able to produce PGI2. Application of beraprost significantly reduced T cell proliferation stimulated by anti-CD3 antibody and, therefore, production of PGI2 in the germinal centers may be a mechanism for controlling T cell numbers [38] suggesting a reason why T cells constitute a smaller population when compared to B cells in the germinal centers. Iloprost reduced IFN-γ and IL-6 induced MCP-1, IL-8, RANTES, and TNF-α production in human monocytes [40]. STAT1 activation, critical in cardiovascular inflammation, was reduced with iloprost administration and led to decreased IFN-γ induced MCP-1 expression and therefore iloprost had an overall anti-inflammatory effect [40].
The evaluation of PGI2 analogues on the expression of Th1 and Th2 related chemokines has also been ongoing as chemokines are known to play a role in the development of asthma [43]. Monocytes are main contributors of chemokines. Human monocytes were pretreated with iloprost and treprostinil prior to LPS stimulation and then evaluated for production of Th1-related chemokines interferon-γ-inducible protein-10 (IP-10/CXCL10) and Th2-related chemokine macrophage-derived chemokine (MDC/CCL22). The PGI2 analogues decreased IP-10 production, but enhanced MDC. The enhancement of MDC suggests that use of PGI2 analogues could potentially increase Th2 inflammation [43].
Lastly, in a model using human lung microvascular endothelial cells, eosinophils and the chemoattractants eotaxin and C5a, PGI2 inhibited eosinophilic migration through the endothelial barrier by affecting their chemotaxis, adhesion, and transmigration and by strengthening the endothelial barrier [37]. Chemotaxis was limited by direct stimulation of the IP receptor on eosinophils as PGI2 caused upregulation of cAMP leading to an increase is adenylyl cyclase despite use of chemoattractants such as eotaxin and C5a. Rapid upregulation of eotaxin-induced CD11b adhesion molecule was diminished by PGI2 leading to decreased adhesion to fibronectin. PGI2 also prevented eosinophil transmigration by strengthening endothelial barrier function. These findings were reversed by exposing eosinophils and endothelium to an IP antagonist [37]. These studies are important because they suggest a role for PGI2 in the maintenance of the endothelial barrier and the prevention of allergic disease.
4.2. PGI2 Regulation of the Adaptive Immune System
Currently, it is widely believed that signaling through the IP receptor has immunosuppressive properties. PGI2 analogues play a key role via inhibition of Th1 and Th2 cytokine production from CD4 T cells [21, 42] although the specifics remain controversial as we will see in the discussion below. B cells are also influenced by analogues of PGI2 and are discussed at the end of this section.
In a mouse study using PGI2 analogues, cicaprost and iloprost, IFN-γ from Th1 cells and IL-4, L-10, and IL-13 from Th2 cells were diminished in a dose dependent manner [41]. Elevated cAMP levels and downregulation of NF-κB correlated with the inhibition of these cytokines. These findings suggest an immunosuppressive capability of PGI2 analogues [41]. In IP-deficient mice subjected to a chronic contact hypersensitivity model showed a significantly decreased contact hypersensitivity response [44]. In contrast to the study above, iloprost promoted Th1 cells differentiation suggesting that PGI2-IP-signaling promotes contact hypersensitivity. Promotion of Th1 differentiation was decreased by a PKA inhibitor suggesting mediation through the cAMP-PKA pathway [44]. These findings suggest a role for PGI2 in the promotion of Th1 pathology, which is in opposition to findings suggesting that it has more immunosuppressive effects.
In addition to immunomodulatory effects on T cells, PGI2 regulates B cell function. In activated B cells, beraprost increased the costimulatory molecule CD86 via the IP receptor with subsequent elevation in cAMP [45]. CD86 is important as it acts as a costimulatory molecule required for T cell activation specifically Th2 activation. Its expression is elevated on B cells in the light zone of germinal center where some Th2 cells are found. Increased T cell proliferation was noted after exposure to beraprost treated B cells [45] and suggests a role for beraprost as an adjuvant in vaccine development.
5. Therapeutic Use of PGI2
PGI2 has a long history of use for the treatment of pulmonary arterial hypertension (PAH) [14, 16, 28]. Regrettably, it efficacy has been less than expected possibly due to the development of tolerance. A postulated mechanism for the development of tolerance is the impaired production of PGI2 in the setting of increased thromboxane synthesis, which is also thought to be an initial step in the pathogenesis of PAH. Nonetheless, it is one of the main therapies in severe idiopathic PAH and has been shown to improve overall survival.
Epoprostenol, treprostinil, and iloprost are approved for the treatment of PAH [46] (see Table 1). Their use in PAH has been investigated since 1980 and their initial use in long-term therapy began in 1984 in the research setting. Intravenous formulations are nonselective and can cause both pulmonary and systemic hypotension via vasodilation. Side effects shared by these medications include headache, flushing, hypotension, jaw pain with initial mastication, diarrhea, nausea, musculoskeletal pain of the legs and feet, and an erythematous blotchy skin rash [46].
Table 1.
Therapeutic use of PGI2—approved agents.
| Agent | Pharmacology | Indications |
|---|---|---|
| Epoprostenol | Synthetic salt of PGI2 | PAH, transplantation, renal dialysis, and extracorporeal circulation systems |
| Treprostinil | IP receptor agonist | PAH |
| Iloprost | IP receptor agonist | PAH |
Epoprostenol (Flolan), a stable freeze dried synthetic salt of PGI2, is also used in transplantation, renal dialysis, and extracorporeal circulation systems as it is an effective inhibitor of platelet aggregation. It was the first prostanoid approved for use in the treatment in PAH by the FDA in 1995 and is currently the most commonly used PGI2 analogue [46]. It has a very short half life of 6 minutes in vivo. Any interruption of its infusion can cause severe rebound pulmonary hypertension and even death. Tolerance may develop over time. Due to its unstable nature requiring a continuous intravascular infusion, its use as an antithrombotic in the general population has been limited.
Treprostinil (Remodulin) was approved for use by the FDA in 2002 and is a potent IP receptor agonist. It is administered as a continuous subcutaneous (SQ) or IV infusion or an inhaled formulation and provides a long-term survival benefit in patients with idiopathic PAH. Pain at the site of administration when given SQ has limited this form of administration. The inhaled form given 4 times per day was approved for use in 2009. Currently, an oral form of treprostinil is being investigated. Overall, treprostinil has fewer side effects when compared to epoprostenol [46] and a longer half life of about 4 hours.
Iloprost (Ventavis), a PGI2 analogue and potent IP receptor agonist, received FDA approval in 2004. It is administered via nebulization 6–9 times per day requiring 10–15 minutes per administration. Its half life is about 20–30 minutes. Administration via inhalation limits side effects such as systemic hypotension. Inhaled iloprost can cause the development of reactive airway obstruction therefore limiting its use [46].
Cicaprost is a synthetic PGI2 analogue which is metabolically stable and bioavailable after oral administration. It is currently used in the research setting [46].
Beraprost is a stable, orally active PGI2 analogue, used experimentally in the treatment of primary pulmonary hypertension, peripheral occlusive disease, ischemia, reperfusion injury, and in the vascular complication of diabetes mellitus [7]. Beraprost has a high-affinity binding to the human IP receptor and a longer half life of about 1 hour [45]. Benefits of beraprost have been shown in short-term trials but it appears to have attenuated effects in longer treatment courses [46].
5.1. Potential New Therapeutic Options
ONO-1301 is a novel nonprostanoid long-acting PGI2 agonist that is currently being used in the research setting. It has both PGI2 activity and thromboxane synthase inhibitory activity [47]. It is unlike PGI2 as it does not contain a five-membered ring and allylic alcohol. This property adds to its stability and allows the drug to be given two times per day as a subcutaneous injection. In mouse studies, it diminished pulmonary fibrosis associated with bleomycin intratracheal injection and improved survival rates. In addition, it decreased the total cell count, neutrophil count, thromboxane B2, and total protein level in BAL fluid and inhibit ICAM-1 and VCAM-1 adhesion molecule expression in the lung tissue [47]. Therefore, it may be a prospective candidate in the treatment of idiopathic pulmonary fibrosis in the future.
Taken together, PGI2 functions mostly as an immunoinhibitor molecule through multiple cell types such as dendritic cells, macrophages, and T-cells.
Acknowledgments
This work was supported by National Institutes of Health Grants U19 AI095227, R01 HL 090664, R01 AI 070672, R01 AI 059108, R21 HL106446, and K12HD043483-08 and Veteran Affairs Grant 1I01BX000624.
Abbreviations
- ALT:
Alanine aminotransferase
- BMDC:
Bone marrow dendritic cells
- CCL17:
Chemokine (C-C motif) ligand 17
- 13-cis-RA:
13-cis-retinoic acid
- CD:
Clusters of differentiation
- ConA:
Concanavalin A
- CXCL10:
C-X-C motif chemokine 10
- cAMP:
Cyclic adenosine monophosphate
- COX:
Cyclooxygenase
- FGF-2:
Fibroblast growth factor
- FDA:
Food and Drug Administration
- Gαi:
Guanosine nucleotide-binding-α-inhibitory protein
- Gαs:
Guanosine nucleotide-binding α-stimulatory protein
- iNOS:
Inducible nitric oxidesynthase
- IgE:
Immunoglobulin-E
- IFN-γ:
Interferon-γ
- IL:
Interleukin
- LPS:
Lipopolysaccharide
- MDC:
Macrophage derived chemokine
- MHC:
Major histocompatibility complex
- mRNA:
Messenger RNA
- miRNA:
Micro-RNA
- MCP-1:
Monocyte chemotactic protein-1
- mPGES-1:
Membrane-associated PGE synthase-1
- Nqo 1:
NADP(H) : Quinone oxide reductase
- NKT:
Natural killer T cells
- NSAIDS:
Nonsteroidal anti-inflammatory
- NF-κB:
Nuclear factor kappa-light-chain-enhancer of activated B cells
- OA:
Osteoarthritis
- OVA:
Ovalbumin
- PPAR:
Peroxisome proliferators activated receptor
- IP:
Prostacyclin receptor
- PGIS:
Prostacyclin synthase
- PGI2:
Prostacyclin
- PG:
Prostaglandin
- PKA:
Protein kinase A
- PAH:
Pulmonary arterial hypertension
- RANKL:
Receptor activator of nuclear factor kappa-B ligand
- RANTES:
Regulated upon activation, normal Tcell expressed, and secreted protein
- RSV:
Respiratory syncytial virus
- RA:
Rheumatoid arthritis
- STAT:
Signal transducers and activators of transcription
- SQ:
Subcutaneous
- Th1:
T helper type 1 T cells
- Th2:
T helper type 2 T cells
- TGF-β:
Transforming growth factor-β
- TNF-α:
Tumor necrosis factor-α
- VNTR:
Variable-number tandem repeat
- VEGF:
Vascular endothelial growth factor.
References
- 1.Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature. 1976;261(5561):558–560. doi: 10.1038/261558a0. [DOI] [PubMed] [Google Scholar]
- 2.Camacho M, Rodríguez C, Salazar J, et al. Retinoic acid induces PGI synthase expression in human endothelial cells. Journal of Lipid Research. 2008;49(8):1707–1714. doi: 10.1194/jlr.M700559-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Gurgul-Convey E, Lenzen S. Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. The Journal of Biological Chemistry. 2010;285(15):11121–11128. doi: 10.1074/jbc.M109.054775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. Journal of Immunology. 2002;169(10):5997–6004. doi: 10.4049/jimmunol.169.10.5997. [DOI] [PubMed] [Google Scholar]
- 5.Camacho M, Rodríguez C, Guadall A, et al. Hypoxia upregulates PGI-synthase and increases PGI2 release in human vascular cells exposed to inflammatory stimuli. Journal of Lipid Research. 2011;52(4):720–731. doi: 10.1194/jlr.M011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuoka T, Narumiya S. The roles of prostanoids in infection and sickness behaviors. Journal of Infection and Chemotherapy. 2008;14(4):270–278. doi: 10.1007/s10156-008-0622-3. [DOI] [PubMed] [Google Scholar]
- 7.Yin H, Cheng L, Langenbach R, Ju C. Prostaglandin I2 and E2 mediate the protective effects of cyclooxygenase-2 in a mouse model of immune-mediated liver injury. Hepatology. 2007;45(1):159–169. doi: 10.1002/hep.21493. [DOI] [PubMed] [Google Scholar]
- 8.Egan KM, Lawson JA, Fries S, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306(5703):1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 9.Hui Y, Ricciotti E, Crichton I, et al. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010;121(24):2654–2660. doi: 10.1161/CIRCULATIONAHA.109.910687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(5):1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 11.Zou MH, Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. The FEBS Letters. 1996;382(1-2):101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
- 12.Zou M, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biological Chemistry. 1997;378(7):707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- 13.Wade MI, Voelkel NF, Fitzpatrick FA. ‘Suicide’ inactivation of prostaglandin I2 synthase: characterization of mechanism-based inactivation with isolated enzyme and endothelial cells. Archives of Biochemistry and Biophysics. 1995;321(2):453–458. doi: 10.1006/abbi.1995.1417. [DOI] [PubMed] [Google Scholar]
- 14.El-Haroun H, Clarke DL, Deacon K, et al. IL-1β, BK, and TGF-β1 attenuate PGI 2-mediated cAMP formation in human pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP kinase and PKA. American Journal of Physiology. 2008;294(3):L553–L562. doi: 10.1152/ajplung.00044.2006. [DOI] [PubMed] [Google Scholar]
- 15.Mohite A, Chillar A, So SP, Cervantes V, Ruan KH. Novel mechanism of the vascular protector prostacyclin: regulating microRNA expression. Biochemistry. 2011;50(10):1691–1699. doi: 10.1021/bi101654w. [DOI] [PubMed] [Google Scholar]
- 16.Soberman RJ, Christmas P. Revisiting prostacyclin: new directions in pulmonary fibrosis and inflammation. American Journal of Physiology. 2006;291(2):L142–L143. doi: 10.1152/ajplung.00102.2006. [DOI] [PubMed] [Google Scholar]
- 17.Lovgren AK, Jania LA, Hartney JM, et al. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. American Journal of Physiology. 2006;291(2):L144–L156. doi: 10.1152/ajplung.00492.2005. [DOI] [PubMed] [Google Scholar]
- 18.Dahlen SK, Hansson G, Hedqvist P, Bjorck T, Granstrom E, Dahlen B. Allergen challenge of lung tissue from asthmatic elicits bronchial contraction that correlates with the release of leukotrienes C4, D4, and E4. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(6):1712–1716. doi: 10.1073/pnas.80.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulman ES, Newball HH, Demers LM, Fitzpatrick FA, Adkinson NF., Jr. Anaphylactic release of thromboxane A2, Prostaglandin D2, and prostacyclin from human lung parenchyma. American Review of Respiratory Disease. 1981;124(4):402–406. doi: 10.1164/arrd.1981.124.4.402. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y, Tokuoka S, Masuda T, et al. Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. British Journal of Pharmacology. 2002;137(3):315–322. doi: 10.1038/sj.bjp.0704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagao K, Tanaka H, Komai M, Masuda T, Narumiya S, Nagai H. Role of prostaglandin I2 in airway remodeling induced by repeated allergen challenge in mice. American Journal of Respiratory Cell and Molecular Biology. 2003;29(3):314–320. doi: 10.1165/rcmb.2003-0035OC. [DOI] [PubMed] [Google Scholar]
- 22.Jaffar Z, Ferrini ME, Buford MC, FitzGerald GA, Roberts K. Prostaglandin I2-IP signaling blocks allergic pulmonary inflammation by preventing recruitment of CD4+ Th2 cells into the airways in a mouse model of asthma. Journal of Immunology. 2007;179(9):6193–6203. doi: 10.4049/jimmunol.179.9.6193. [DOI] [PubMed] [Google Scholar]
- 23.Wang M, Cooper PR, Jiang M, et al. Deletion of microsomal prostaglandin E synthase-1 does not alter ozone-induced airway hyper-responsiveness. Journal of Pharmacology and Experimental Therapeutics. 2010;334(1):63–68. doi: 10.1124/jpet.110.166678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Idzko M, Hammad H, van Nimwegen M, et al. Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. Journal of Clinical Investigation. 2007;117(2):464–472. doi: 10.1172/JCI28949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madison LD, Scarlett JM, Levasseur P, et al. Prostacyclin signaling regulates circulating ghrelin during acute inflammation. Journal of Endocrinology. 2008;196(2):263–273. doi: 10.1677/JOE-07-0478. [DOI] [PubMed] [Google Scholar]
- 26.Vane J, Corin RE. Prostacyclin: a vascular mediator. European Journal of Vascular and Endovascular Surgery. 2003;26(6):571–578. doi: 10.1016/s1078-5884(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann C, König JP, Dettmann J, Birnbaum J, Kox WJ. Effects of iloprost, a stable prostacyclin analog, on intestinal leukocyte adherence and microvascular blood flow in rat experimental endotoxemia. Critical Care Medicine. 2001;29(7):1412–1416. doi: 10.1097/00003246-200107000-00019. [DOI] [PubMed] [Google Scholar]
- 28.Nana-Sinkam SP, Jong DL, Sotto-Santiago S, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. American Journal of Respiratory and Critical Care Medicine. 2007;175(7):676–685. doi: 10.1164/rccm.200605-724OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Liu Y, Zhou W, et al. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respiratory Research. 2010;11, article 34 doi: 10.1186/1465-9921-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W, Dowell DR, Geraci MW, et al. PGI synthase overexpression protects against bleomycin-induced mortality and is associated with increased Nqo 1 expression. American Journal of Physiology. 2011;301(4):L615–L622. doi: 10.1152/ajplung.00224.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto K, Graham BS, Geraci MW, et al. Signaling through the prostaglandin I2 receptor IP protects against respiratory syncytial virus-induced illness. Journal of Virology. 2004;78(19):10303–10309. doi: 10.1128/JVI.78.19.10303-10309.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto K, Ishibashi K, Gebretsadik T, et al. Functional polymorphism of the promoter region of the prostacyclin synthase gene and severity of RSV infection in hospitalized children. Journal of Medical Virology. 2008;80(11):2015–2022. doi: 10.1002/jmv.21318. [DOI] [PubMed] [Google Scholar]
- 33.Pulichino AM, Rowland S, Wu T, et al. Prostacyclin antagonism reduces pain and inflammation in rodent models of hyperalgesia and chronic arthritis. Journal of Pharmacology and Experimental Therapeutics. 2006;319(3):1043–1050. doi: 10.1124/jpet.106.110387. [DOI] [PubMed] [Google Scholar]
- 34.Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. Journal of Experimental Medicine. 2006;203(2):325–335. doi: 10.1084/jem.20051310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aronoff DM, Peres CM, Serezani CH, et al. Synthetic prostacyclin analogs differentially regulate macrophage function via distinct analog-receptor binding specificities. Journal of Immunology. 2007;178(3):1628–1634. doi: 10.4049/jimmunol.178.3.1628. [DOI] [PubMed] [Google Scholar]
- 36.Hung CH, Chu YT, Suen JL, et al. Regulation of cytokine expression in human plasmacytoid dendritic cells by prostaglandin I2 analogues. European Respiratory Journal. 2009;33(2):405–410. doi: 10.1183/09031936.00070008. [DOI] [PubMed] [Google Scholar]
- 37.Konya V, Sturm EM, Schratl P, et al. Endothelium-derived prostaglandin I2 controls the migration of eosinophils. Journal of Allergy and Clinical Immunology. 2010;125(5):1105–1113. doi: 10.1016/j.jaci.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Lee IY, Ko EM, Kim SH, Jeoung DI, Choe J. Human follicular dendritic cells express prostacyclin synthase: a novel mechanism to control T cell numbers in the germinal center. Journal of Immunology. 2005;175(3):1658–1664. doi: 10.4049/jimmunol.175.3.1658. [DOI] [PubMed] [Google Scholar]
- 39.Müller T, Dürk T, Blumenthal B, et al. Iloprost has potent anti-inflammatory properties on human monocyte-derived dendritic cells. Clinical and Experimental Allergy. 2010;40(8):1214–1221. doi: 10.1111/j.1365-2222.2010.03558.x. [DOI] [PubMed] [Google Scholar]
- 40.Strassheim D, Riddle SR, Burke DL, Geraci MW, Stenmark KR. Prostacyclin inhibits IFN-γ-stimulated cytokine expression by reduced recruitment of CBP/p300 to STAT1 in a SOCS-1-independent manner. Journal of Immunology. 2009;183(11):6981–6988. doi: 10.4049/jimmunol.0901045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou W, Blackwell TS, Goleniewska K, et al. Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. Journal of Leukocyte Biology. 2007;81(3):809–817. doi: 10.1189/jlb.0606375. [DOI] [PubMed] [Google Scholar]
- 42.Zhou W, Hashimoto K, Goleniewska K, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. Journal of Immunology. 2007;178(2):702–710. doi: 10.4049/jimmunol.178.2.702. [DOI] [PubMed] [Google Scholar]
- 43.Kuo CH, Ko YC, Yang SN, et al. Effects of PGI2 analogues on Th1- and Th2-related chemokines in monocytes via epigenetic regulation. Journal of Molecular Medicine. 2011;89(1):29–41. doi: 10.1007/s00109-010-0694-2. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima S, Honda T, Sakata D, et al. Prostaglandin I2-IP signaling promotes Th1 differentiation in a mouse model of contact hypersensitivity. Journal of Immunology. 2010;184(10):5595–5603. doi: 10.4049/jimmunol.0903260. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Park CS, Park CH, Jeoung DI, Kim YM, Choe J. Beraprost enhances the APC function of B cells by upregulating CD86 expression levels. Journal of Immunology. 2011;186(7):3866–3873. doi: 10.4049/jimmunol.1002170. [DOI] [PubMed] [Google Scholar]
- 46.Ivy DD. Prostacyclin in the intensive care setting. Pediatric Critical Care Medicine. 2010;11(2):S41–S45. doi: 10.1097/PCC.0b013e3181d10845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murakami S, Nagaya N, Itoh T, et al. Prostacyclin agonist with thromboxane synthase inhibitory activity (ONO-1301) attenuates bleomycin-induced pulmonary fibrosis in mice. American Journal of Physiology. 2006;290(1):L59–L65. doi: 10.1152/ajplung.00042.2005. [DOI] [PubMed] [Google Scholar]