

Abstract
Background
Unloading a failing heart with a left ventricular assist device (LVAD) can improve ejection fraction (EF) and left ventricular (LV) size; however, recovery with LVAD explantation is rare. We hypothesized that evaluation of myocyte contractility and biochemistry at the sarcomere level before and after LVAD may explain organ level changes.
Methods and Results
Paired LV tissue samples were frozen from 8 patients with nonischemic cardiomyopathy at LVAD implantation (Before LVAD) and prior to transplant (After LVAD). These were compared to 8 nonfailing hearts. Isolated skinned myocytes were purified, attached to a force transducer, and dimensions, maximal calcium saturated force (Fmax), calcium sensitivity, and myofilament cooperativity were assessed. Relative isoform abundance and phosphorylation levels of sarcomeric contractile proteins were measured. With LVAD support, the unloaded EF improved (10.0±1.0 to 25.6±11.0%, p=0.007), LV size decreased (LVIDd 7.6±1.2 to 4.9±1.4cm, p<0.001), and myocyte dimensions decreased (cross-sectional area 1247±346 to 638±254μm2, p=0.001). Fmax improved after LVAD (3.6±0.9 to 7.3±1.8mN/mm2, p<0.001), but was still lower than nonfailing (7.3±1.8 vs. 17.6±1.8mN/mm2, p<0.001). An increase in troponin I (TnI) phosphorylation after LVAD was noted, but protein kinase C phosphorylation of TnI decreased. Biochemical changes of other sarcomeric proteins were not observed after LVAD.
Conclusions
There is significant improvement in LV and myocyte size with LVAD, but there is only partial recovery of EF and myocyte contractility. LVAD support was only associated with biochemical changes in TnI. This suggests that alternate mechanisms might contribute to contractile changes after LVAD and that additional interventions may be needed to alter biochemical remodeling of the sarcomere to further enhance myofilament and organ level recovery.
Keywords: mechanical circulatory support, remodeling heart failure, mechanical unloading, heart failure
The left ventricular assist device (LVAD) is a well established therapy for patients with end-stage heart failure (HF). The mechanical unloading of the left ventricle with the LVAD and the subsequent restoration of cardiac output results in improvements in HF symptoms, functional status, quality of life, and end-organ perfusion. In addition to these systemic effects, some patients undergoing LVAD support demonstrate improved function of the native left ventricle (LV), termed reverse remodeling. Such organ level improvements include decreased LV chamber size, decreased LV mass, and improved LV ejection fraction (EF)1 and have been accompanied by systemic effects such as normalization of catecholamine levels,2 natriuretic peptides levels,3 and circulating cytokines like TNF-α.1,4
Some of these clinical markers of reverse remodeling at the organ level have also been demonstrated at the cellular and molecular level. Previous studies have noted reduction in myocyte hypertrophy5,6 as well as improvement in overall cardiac histology7 after LVAD support. Furthermore, in vitro studies have demonstrated improvements in myocardial contractile properties, restoration of adrenergic receptor density and responsiveness, and improved calcium handling with LVAD support.8-11
As both clinical and cellular evidence suggested the plausibility of reversing end-stage disease, the concept arose that LVADs may be used as a bridge to recovery in HF patients. Indeed, this was met with great enthusiasm after a single center’s report of the successful explantation of LVADs in 11 of 15 patients with nonischemic cardiomyopathy treated with a combination of unloading with a LVAD followed by conventional HF medical therapy and the selective β2 adrenergic receptor agonist clenbuterol.12 However, such high rates of recovery were not observed in the multi-institutional LVAD Working Group study where only 6 of 67 patients (9%) underwent LVAD explantation for recovery.1
At this time, the mechanisms of reverse remodeling remain poorly understood, and it appears that restoring myocardial contractility may be more complex than what had been initially considered. Therefore, we postulated that evaluation of myocyte contractility and biochemistry at the most fundamental contractile level of the heart, the sarcomere, before and after LVAD placement might reveal an explanation for this phenomenon. Our first goal was to assess sarcomeric contractile properties by measuring direct isometric forces on skinned isolated cell preparations. Second, since the cardiac sarcomeric proteins myosin binding protein C (MyBPC), troponin T (TnT), troponin I (TnI), tropomyosin (Tm), and regulatory myosin light chain (MLC-2) are known to effect contractility,13 we assessed whether post-translational modifications and/or other alterations in these proteins were affected with LVAD support.
Methods
Patient Selection and Tissue Acquisition
Paired LV tissue samples were collected and flash frozen from 8 patients with end-stage nonischemic cardiomyopathy at the time of LVAD implantation (Before LVAD) and prior to cardiac transplantation (After LVAD). Nonfailing control samples were obtained from 8 donor hearts that were harvested for transplant but then unused for non-cardiac reasons. The tissue obtained at the time of LVAD implantation consisted of a core sample from the LV apex – at the in-flow cannula of the LVAD. The tissue collected at the time of the cardiac transplant consisted of an analogously sized fragment excised from the explanted heart. All tissue samples were immediately flash frozen in liquid nitrogen in the operating room (without the use of any cardioplegia solutions) and transported to a -80 °C freezer where they were stored until use. This storage method has been shown to preserve sarcomeric contractile protein phosphorylation state and also allow for the accurate assessment of the myocyte contractile parameters that are described below.14-16 The ischemia time from removal of LV tissue from the patient to freezing in liquid nitrogen for the Before LVAD samples was less than 10 seconds and for the After LVAD and Nonfailing samples was less than 10 minutes. The Colorado Multicenter Institutional Review Board approved the protocol for the collection, storage, and analysis of human tissue.
Medical records were retrospectively reviewed by a trained physician to obtain demographic and clinical data. Echocardiographic and hemodynamic data were obtained from the medical record at the time closest to before LVAD implantation (to ensure conditions reflecting the time the Before LVAD tissue was obtained) followed by the time closest to cardiac transplantation (to ensure conditions reflecting the time the After LVAD tissue was obtained). The data obtained from patients being supported with a LVAD reflect the device settings that were clinically indicated at the time for the patients, and represent the combined effects of native LV function as well as LVAD related unloading.
Myocyte Contractility Measurements
Details of the myocyte isolation and experimentation protocols have been previously described.14,17-19 Briefly, myocytes were purified from the frozen LV samples by mechanical homogenization and subsequently permeabilized with 0.3% Triton X-100. The resulting suspension was placed on the stage of an inverted microscope, and in order to ensure adequate tissue quality, myocyte fragments with an organized myofibril pattern with clear sarcomeric striations were chosen for the contractility experiments.15 These isolated skinned myocyte fragments were then attached to a force transducer and motor and mechanical experiments were conducted. A representative photomicrograph is shown in Figure 1. The length and width of the skinned myocyte fragment were measured when it was attached to the force transducer in relaxing solution and a side view mirror was used to measure the thickness. Cross-sectional area was calculated using an elliptical approximation. The isolated myocytes were stored on ice and used within 20 hours of isolation. All experiments were performed at 15 °C and since sarcomere length can effect force measurements, the sarcomere length was set to 2.1 um for all experiments.18
Figure 1.
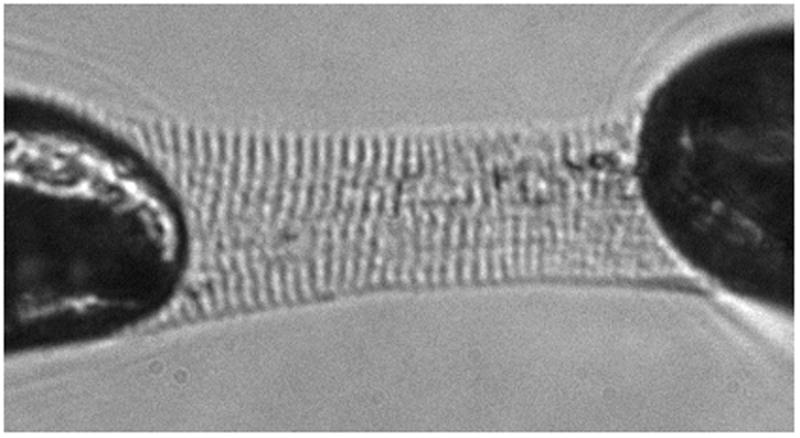
Photomicrograph example of an isolated skinned myocyte fragment attached to the force transducer and motor in relaxing solution for mechanical contractile experiments. The visible sarcomere striations were set at 2.1μm for all myocyte experiments.
Myocytes were exposed in random order to six different activation solutions containing varying concentrations of calcium (pCa 9.0 to 4.5) and the developed force for each of the calcium concentrations was recorded as shown in Figure 2a. The developed force at the maximal calcium concentration was measured before and after obtaining the measurements of force at the other 6 calcium concentrations. If there was >15% decline in force between the first and last force measurement at the maximal calcium concentration, we considered this as evidence of cell fragment degradation and this data was excluded from the analysis. Using these measurements, a force-pCa curve was plotted (Figure 2b) and the Hill equation was fit to derive the following contractile parameters: Fmax (the maximal calcium saturated developed force normalized to the cross-sectional area of the myocyte), pCa50 (the calcium concentration at which the force is half maximal, a measure of calcium sensitivity), and Hill coefficient (the slope of the calcium-force relation, an index of myofilament cooperative activation). In order to minimize measurement variations for any single isolated myocyte, the individual patient contractile data reflect the average of measurement recordings for 3 to 5 myocytes per patient.
Figure 2.
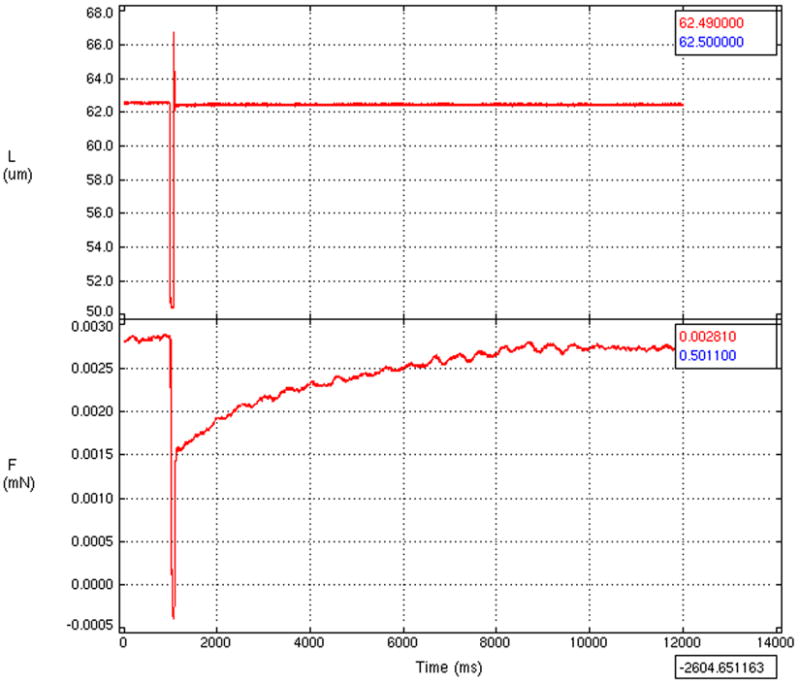

(a) Representative tracing of force measurements from the experimental protocol. After the myocyte had achieved a steady force in the calcium containing activation solution, it was rapidly step shortened to break cross-bridges (top tracing) and the maximum force for the particular activation solution was then recorded (bottom tracing).
(b) Representative plot of the force-calcium relation. Force is shown plotted against the inverse log of the calcium concentration (pCa). The Fmax (the maximal calcium saturated developed force normalized to the cross-sectional area of the myocyte), pCa50 (the calcium concentration at which the force is half maximal, a measure of calcium sensitivity), and Hill coefficient (the slope of the calcium-force relation, an index of myofilament cooperative activation) for each myocyte were derived from these force-pCa curves.
Sarcomeric Protein Phosphorylation Assessment
The remaining myocyte preparations not used for mechanical study were acetone precipitated to clamp their phosphorylation state and homogenized in 8M urea, 2.5M thiourea, 4% CHAPS, 10 mM EDTA, and a mixture of protease and phosphatase inhibitors as previously described.20 Protein concentration of these samples was measured using a BCA Protein Assay Kit (Thermo Scientific).
Phosphorylation levels of the cardiac sarcomeric proteins were determined by separating the proteins with 12% 1D-SDS-PAGE and fixing and staining with a phosphoprotein stain (ProQ Diamond Phosphoprotein Gel Stain, Invitrogen). After imaging, the same gels were rinsed and stained with a total protein stain (BioSafe Coomassie Blue, BioRad). All gels were imaged using a Typhoon 9410 Gel Imager (GE Lifesciences) and protein optical densities were measured using ImageJ (version 1.42 NIH). In order to adjust for subtle differences in protein loading, the phosphorylation levels were calculated by dividing the optical density of the each sarcomeric protein on the ProQ diamond gel by the optical density of the same protein on the Coomassie gel. In the case of MyBPC, the total protein stain optical density merges with the abundant protein myosin, so the phosphorylation level of MyBPC on the phosphoprotein stain was normalized to essential myosin light chain (MLC-1) as prior studies have reported.20
Additional Assessment of Sarcomeric Proteins
Site-specific phosphorylation of troponin I was assessed by Western blots. Proteins were separated with 12% 1D-SDS-PAGE as above and transferred to PVDF membranes. After the membranes were blocked in 5% BSA and rinsed with TBST, they were incubated overnight at 4 °C with a phosphospecific primary antibody to TnI phosphorylated at the putative protein kinase A (PKA) site, Serine 22,23 (Cell Signaling 1:1000) or to troponin I phosphorylated at the putative protein kinase C (PKC) site Serine 43 using an epitope specific phosphoserine antibody (Abcam 1:1000) as previously described.20 The blots were then washed and incubated with secondary antibody (anti-mouse from Sigma, 1:10000) for one hour at room temperature, washed, and visualized using enhanced chemiluminescence. Membranes were stripped and subsequently incubated in primary total cardiac TnI antibody (Fitzgerald, Inc 1:2500). Site specific phosphorylation was calculated by dividing the optical density of the phosphospecific antibody blot by the density of the total protein blot.
Changes in myosin heavy chain isoforms were assessed as previously described20 by separating proteins using modified 6% 1D-SDS-PAGE and subsequent staining with BioSafe Coomassie Blue total protein stain. MLC-1 and MLC-2 phosphorylation and isoforms changes were assessed using 2D-SDS-PAGE21 and percent phosphorylated MLC-2 and percent atrial isoform of MLC-1 were calculated.
Changes in TnT isoform expression were assessed using by modified Western blots as previously described.22 The primary cardiac TnT isoform antibody used was Ab-1 (Clone 13-11, Thermo scientific 1:1000).
Statistical Analysis
Results are expressed as mean ± standard deviation (SD). The paired t-test was used to compare differences between before and after LVAD implantation. The unpaired t-test was used to compare differences between before LVAD implantation failing and nonfailing and between after LVAD implantation failing and nonfailing groups. Statistical significance was defined as a two-tailed P-value of less than 0.05. The R version 2.9.1 (Vienna, Austria) statistical program was used for all analyses.
Results
Patient Characteristics
The baseline characteristics of patients with end-stage nonischemic cardiomyopathy requiring LVAD as a bridge to transplant are provided in Table 1. All patients were inotrope dependent with NYHA class IV HF at the time of LVAD implantation, and 7 of 8 patients required mechanical support with an intra-aortic balloon pump. The mean duration for the diagnosis of HF at the time of LVAD implantation was 59±22 months. Both first generation pulsatile displacement LVADs (HeartMate XVE, Thoratec, Pleasanton, CA) and second generation continuous axial flow LVADs (HeartMate II, Thoratec, Pleasanton, CA) were utilized. The mean duration of LVAD support was 143±41 days. LVAD support resulted in significant reductions in LV echocardiographic dimensions, improvement in EF, and normalization of several hemodynamic measures as summarized in Table 2.
Table 1.
Baseline Patient Characteristics
| Patient Characteristics (n=8) | |
|---|---|
| Mean age (years) | 40.5 ± 11.5 |
| Male gender | 7 (88%) |
| Caucasian | 4 (50%) |
| African American | 4 (50%) |
| Mean duration of HF prior to LVAD (months) | 56 ± 22 |
| NYHA Class IV | 8 (100%) |
| Mean duration of LVAD support (days) | 143 ± 41 |
| HeartMate XVE LVAD* | 5 (62%) |
| HeartMate II LVAD* | 3 (39%) |
| Medical Therapy or Interventions Before LVAD | |
| Intravenous inotropic agent | 8 (100%) |
| Intravenous vasodilator | 5 (62%) |
| Beta-blocker | 0 (0%) |
| ACE inhibitor/ARB | 2 (25%) |
| Aldosterone antagonist | 7 (88%) |
| Diuretic | 8 (100%) |
| Implantable cardioverter-defibrillator | 4 (50%) |
| Cardiac resynchronization therapy | 2 (25%) |
| Intraaortic balloon pump | 7 (88%) |
| Medical Therapy during LVAD Support | |
| Intravenous inotropic agent | 0 (0%) |
| β-blocker | 6 (75%) |
| ACE inhibitor/ARB | 7 (88%) |
| Aldosterone antagonist | 7 (88%) |
| Diuretic | 6 (75%) |
HF=Heart Failure, LVAD=left ventricular assist device, NYHA=New York Heart Association, ACE=angiotensin-converting enzyme, ARB=angiotensin II receptor blocker
Thoratec, Pleasanton, CA
Table 2.
Echocardiographic and Hemodynamic Parameters Before and After LVAD Support
| Before LVAD | After LVAD | P-value | |
|---|---|---|---|
| Ejection fraction (%) | 10.0 ± 1 | 25.6 ± 11 | 0.007 |
| Left ventricular end-systolic internal dimension (cm) | 6.8 ± 1.2 | 4.1 ± 1.3 | <0.001 |
| Left ventricular end-diastolic internal dimension (cm) | 7.6 ± 1.2 | 4.9 ± 1.4 | <0.001 |
| Heart rate (beats/min) | 96 ± 27 | 79 ± 14 | 0.13 |
| Mean arterial pressure (mmHg) | 72 ± 6 | 90 ± 12 | 0.016 |
| Right atrial pressure (mmHg) | 16.8 ± 6.9 | 1.3 ± 0.6 | <0.001 |
| Mean pulmonary artery pressure (mmHg) | 40 ± 8 | 19 ± 6 | 0.006 |
| Pulmonary capillary-wedge pressure (mmHg) | 27 ± 7 | 5 ± 3 | <0.001 |
| Cardiac output (liters/min) | 2.9 ± 0.8 | 4.7 ± 0.8 | 0.024 |
| Cardiac index (liters/min/m2) | 1.5 ± 0.4 | 2.5 ± 0.7 | 0.16 |
The 8 nonfailing patients (3 male and 5 female) were organ donors whose hearts were not utilized for transplant for non-cardiac reasons. The mean age of this group was 48 ± 8 years, the mean EF 70.6 ± 5.4%, and none of these patients had any prior cardiac history. Seven (87.5%) of these patients were on adrenergic vasopressor agents during the period of brain death and as organs were being harvested.
Myocyte Size and Contractility Parameters
Significant differences were noted for all myocyte fragment dimensions as summarized in Table 3. Myocytes obtained from patients before LVAD implant were significantly larger than myocytes from patients after LVAD implantation. However, the myocytes from patients after LVAD implantation were still larger than the myocytes obtained from nonfailing patients. Fmax doubled with LVAD support (3.6 ± 0.9 vs. 7.3 ± 1.8 mN/mm2, p<0.001), but was still less than half of nonfailing subjects (7.3 ± 1.8 vs. 17.6 ± 1.8 mN/mm2, p<0.001) as described in Table 4. The pCa50 was unchanged in all groups and the Hill coefficient was reduced in both LVAD groups relative to controls.
Table 3.
Isolated Skinned Myocyte Fragment Morphometric Data
| Before LVAD | After LVAD | Nonfailing | |
|---|---|---|---|
| Length (μm) | 105 ± 21 | 86 ± 14* | 75 ± 10† |
| Width (μm) | 41 ± 6 | 33 ± 7* | 23 ± 3† |
| Thickness (μm) | 37 ± 6 | 24 ± 5* | 17 ± 4† |
| Cross-sectional Area (μm2) | 1247 ± 346 | 638 ± 254* | 330 ± 111† |
| Volume (μm3) | 138660 ± 63460 | 56923 ± 29858* | 25435 ± 10803† |
P-value <0.05 for comparison between Before LVAD and After LVAD
P-value <0.05 for comparisons between Nonfailing vs. Before LVAD and for Nonfailing vs. After LVAD.
Table 4.
Mean contractile parameters among control patients and patients with nonischemic cardiomyopathy before and after LVAD implantation.
| Before LVAD | After LVAD | P-value* | Nonfailing | |
|---|---|---|---|---|
| Fmax (mN/mm2) | 3.6 ± 0.9 | 7.3 ± 1.8 | <0.001 | 17.6 ± 1.8† |
| pCa50 | 5.89 ± 0.08 | 6.03 ± 0.28 | 0.257 | 5.85 ± 0.14 |
| Hill coefficient | 1.64 ± 0.28 | 1.55 ± 0.45 | 0.579 | 2.30 ± 0.81‡ |
The Fmax is the maximum calcium-saturated developed force normalized to the cross-sectional area of the myocytes, pCa50 is the calcium concentration at which the force is half maximal and represents a measure of myofilament calcium sensitivity, Hill coefficient is the slope of the calcium-force relation and is an index of myofilament cooperative activation.
P-value for comparison between Before LVAD and After LVAD
P<0.001 for comparisons of Fmax between Before LVAD vs. Nonfailing and After LVAD vs. Nonfailing
P<0.05 for comparisons of Hill coefficient between Before LVAD vs. Nonfailing and After LVAD vs. Nonfailing
Sarcomeric Proteins and Phosphorylation Levels
Total TnI phosphorylation increased with LVAD support (12.8 ± 4.1 vs. 21.6 ± 9.4 OD units, p=0.030) (Table 5 and Figure 3). While there were no significant differences noted in the Serine 22,23 site specific phosphorylation of TnI, Serine 43 site specific phosphorylation of TnI decreased after LVAD (7.5 ± 2.2 vs. 5.2 ± 1.9 OD units, p=0.044) (Figure 4). Total phosphorylation levels of MyBPC were higher in both failing groups compared with nonfailing donors (Table 5). Phosphorylation levels of TnT, Tm, and MLC-2 were not significantly different before and after LVAD (Table 5, and Figure 3).
Table 5.
Phosphorylation levels of sarcomeric proteins in patients with nonischemic cardiomyopathy before and after LVAD implantation.
| Before LVAD | After LVAD | P-value* | Nonfailing | |
|---|---|---|---|---|
| Myosin Binding Protein C | 6.1 ± 1.2 | 6.7 ± 1.0 | 0.328 | 4.6 ± 0.6† |
| Troponin T | 19.2 ± 3.0 | 18.0 ± 3.8 | 0.562 | 20.9 ± 7.0 |
| Troponin I | 12.8 ± 4.1 | 21.6 ± 9.4 | 0.030 | 13.2 ± 1.9 |
| Tropomyosin | 7.9 ± 2.6 | 8.5 ± 2.9 | 0.431 | 8.0 ± 1.9 |
| Regulatory Myosin Light Chain | 0.5 ± 0.2 | 0.7 ± 0.3 | 0.109 | 0.6 ± 0.2 |
| Serine 22,23 phosphorylated Troponin I | 3.7 ± 2.3 | 3.4 ± 1.5 | 0.747 | 2.8 ± 0.6 |
| Serine 43 phosphorylated Troponin I | 7.5 ± 2.2 | 5.2 ± 1.9 | 0.044 | 12.0 ± 1.0‡ |
Phosphorylation levels expressed in arbitrary optical density units.
P-value for comparison between Before LVAD and After LVAD
P<0.01 for comparisons of Myosin Binding Protein C between Before LVAD vs. Nonfailing and After LVAD vs. Nonfailing
P<0.001 for comparisons of Serine 43 phosphorylated Troponin I between Before LVAD vs. Nonfailing and After LVAD vs. Nonfailing
Figure 3.

12% 1D-SDS-PAGE images stained with a total phosphoprotein stain (ProQ Diamond Phosphoprotein Gel Stain) in panel A and a total protein stain (BioSafe Coomassie Blue) in panel B demonstrate the increase in total TnI phosphorylation with LVAD support and higher MyBPC phosphorylation in both failing samples compared with nonfailing donors. Sarcomeric protein phosphorylation levels were calculated by dividing the optical density of the each sarcomeric protein on the ProQ diamond gel by the optical density of the same protein on the Coomassie gel. MyBPC was normalized to MLC-1 as noted in the text.
NF=nonfailing, MyBPC=myosin binding protein C, TnT=troponin T, Tm=tropomyosin, TnI=troponin I, MLC-1=myosin light chain 1, MLC-2=myosin light chain 2
Figure 4.

Western blots of phosphorylated troponin I (TnI) demonstrate while there were no significant differences in Serine 22,23 site specific phosphorylation of TnI, Serine 43 site specific phosphorylation of TnI decreased after LVAD. Samples were separated on 12.5% SDS-PAGE and probed with antibodies against phosphoserine 22/23, phosphoserine 43, and cardiac TnI (for total protein). NF=nonfailing.
There were no changes noted on 2D-SDS-PAGE in the percent phosphorylation of MLC-2 before and after LVAD (43±3 vs. 41±2%, p=0.341) and in the percent atrial isoform of MLC-1 before and after LVAD (23±5 vs. 21±7%, p=0.711). Furthermore, no differences were noted in myosin heavy chain isoforms before and after LVAD with only the β-isoform being expressed in both groups of patients (Figure 5). Finally there was no evidence of altered TnT isoform expression in the before LVAD, after LVAD, and nonfailing groups. Total TnT content was unchanged between these groups.
Figure 5.

6% 1D-SDS-PAGE separation of α and β myosin before and after LVAD demonstrates that only the β-isoform is expressed in both groups. The last lane is a mixture of 50% nonfailing human with 50% nonfailing mouse and illustrates adequate separation of α and β myosin isoforms.
Discussion
The main finding of this study is that in isolated skinned myocytes, there is a marked reduction in maximum developed force in patients with end-stage nonischemic cardiomyopathy compared to nonfailing donors. This marked reduction in force is only partially improved with LVAD support. Furthermore, there is strong evidence that structural changes accompany these changes in force at the organ level (reduction in LV size) as well as at the cellular level (reduction of all myocyte dimensions) and these correlate with functional changes at both the organ level (improvement in EF) and cellular level (improvement in maximum force). Despite these significant changes in structural and mechanical parameters, unloading of the LV with LVAD support did not change the biochemical properties of the sarcomere other than a change in TnI phosphorylation.
Our findings confirm that contractile dysfunction in human heart failure at least in part resides at the level of the cardiac myofilament. This is independent of cell loss or changes in the cardiac interstitium and certainly argues that therapies to treat heart failure need to acknowledge sarcomeric function. To place these changes in context, a prior report using a similar experimental protocol in human samples reported ~30% reductions in myocyte Fmax in diabetics with preserved EF compared to control patients (14.6±1.7 vs. 20.6±3.7 mN/mm2, p<0.05).14 The Fmax of the failing groups before and after LVAD from the current study are more dramatic (~60%) than seen in this prior report, consistent with the severe reductions in contractile function at the organ level as assessed by EF.
There are a number of cellular and molecular changes in the heart that occur with LVAD support and these encompass the most basic aspects of genetic regulation and involve a complex, interconnected cascade of changes.23 While there have been two prior studies that have assessed for changes in myocyte contractility with LVAD, this is the first study to directly measure isometric force of skinned myocytes from paired samples before and after LVAD support. The two prior studies (one of which used paired pre/post LVAD samples) involved patients with both ischemic and nonischemic/idiopathic cardiomyopathies. Using in vitro motility and unloaded cell shortening assays, the authors reported improvements in contractility in myocytes obtained from patients with HF vs. patients with HF supported by LVAD.8,9 Our assessment of myocyte contractility also differs from the two prior reports in that we limited our analysis to only those patients with nonischemic cardiomyopathy (a global pathologic process that involves the entire LV) as opposed to ischemic cardiomyopathy (a regional pathologic process based on areas of infarct/ischemia from coronary artery disease). The prior reports included data from patients with ischemic cardiomyopathy, and the before LVAD tissue (from the LV apex) would be inherently different than the after LVAD tissue depending on the regions of LV infarction making comparisons difficult. The results of our study confirm that among patients with nonischemic cardiomyopathy, improvement in contractility at the sarcomeric level is possible with LVAD support; however normalization of forces to levels seen in nonfailing subjects does not occur using standard LVAD protocols. This is concordant with organ level assessment of contractility where EF improved from 10.0 to 25.6% with LVAD support; which is still half that seen in a nonfailing control hearts. This may in part explain clinically why the majority of LVADs cannot be successfully explanted.
The cellular mechanisms by which contractility is depressed in these HF samples and why there is only partial reversal with mechanical unloading is not unequivocally answered in this study, although there is suggestive evidence that points towards both structural and biochemical mechanisms. The fact that the Hill coefficient, a marker of protein cooperativity, was reduced in both HF groups, coupled with the changes in MyBPC phosphorylation suggest that functional interactions between actin and myosin might have been compromised in both HF groups.24 This hypothesis is supported by the cell morphology data which suggest that sarcomere cross-bridge (and perhaps lattice) spacing is tightly coupled with contractile performance both in the two HF groups and also in the donor heart specimens, a finding which reflects previous studies demonstrating that contractile parameters in isolated muscle were influenced in a step-wise fashion by osmotic determinants that influenced lattice architecture.25,26
The mechanism for the partial recovery of contractile function following LVAD support might be in part reflective of contractile protein phosphorylation changes. Total TnI phosphorylation was lower before LVAD support compared to after LVAD and nonfailing samples which may be due to down-regulation of β-adrenergic receptors in inotrope dependent patients with end-stage HF. The increase in TnI phosphorylation after LVAD might be reflective of normalization of β-adrenergic receptor function. Despite this, there was an increase in TnI phosphorylation at serine 43—a putative PKC site—in the pre LVAD samples which decreased following LVAD support. This finding is consistent with a substantial literature that suggests that heart failure, both in humans and in experimental animals, is associated with an increase in PKC activation,27,28 and that PKC dependent phosphorylation of TnI is associated with depressed myofilament force development.29-31 The finding also provides a plausible explanation as to why phosphatase treatment of myofilament preparations from HF patients improves contractile performance despite overall low levels of overall contractile protein phosphorylation.9 The fact that there was no change seen in TnI phosphorylation at the putative PKA site, serine 22,23 despite postulated changes in adrenergic receptor activity before and after LVAD, may reflect the somewhat promiscuous nature of this site which can be targeted both by PKA and PKC,29 the former likely increasing after LVAD and the later declining.
Not seen in this study were changes in phosphorylation of other contractile proteins or isoform shifts before and after LVAD support. In particular, and in contrast to studies in experimental animals, we did not see a shift at the protein level in the α/β myosin heavy chain isoform ratio or in troponin T isoform distribution. This is particularly important to note as mechanical unloading of the normal LV in experimental animals does result in atrophy associated with a recapitulation of the fetal gene program32 whereas in this study, unloading was associated with a normalization of ventricular size without isoform changes.
Limitations
We acknowledge a number of limitations to this study. The overall number of patients in the analysis is small and all were from a single center’s LVAD program. There were no differences in the results between patients supported with pulsatile-flow HeartMate XVE pumps versus continuous-flow HeartMate II pumps, although our small numbers preclude accurate subgroup analysis to definitively test for differences in the type of mechanical unloading. In addition, the echocardiographic measures of LV size and EF during LVAD support were obtained from retrospective review and were thus obtained on the LVAD settings that were clinically indicated for the patient. While these assessments do partially reflect the influence of the LVAD, some component of native LV function also contributes as on pump measures of LV size and EF are often used in the initial screening assessments to determine whether patients should have further testing to assess for recovery.33,34 Myocyte dimensions were measured from the isolated skinned myocyte fragments that were used for mechanical force measurements rather than a specific histological stain of intact tissue. Despite this different methodology, the cross-sectional areas obtained in this study were very similar to those reported recently using such traditional histological techniques6 and the overall trends are well in line with other published reports.5
The nonfailing tissue was obtained from organ donors, and the majority of these patients were receiving adrenergic agents prior to and during harvest, so adrenergic mediated pathways were likely activated in these patients. Furthermore, concomitant medical therapy during LVAD support, such as β-blockade, varied and may have influenced some of the sarcomeric mechanical properties as has been previously demonstrated.35 In addition, while we believe the comparison between LVAD (before and after) and nonfailing donor hearts are illuminating, it is worth noting that the tissue procurement strategies were by necessity somewhat different in that the nonfailing and after LVAD hearts were excised en bloc whereas the before LVAD specimens were taken from small tissue cores obtained from beating hearts. Finally, the before and after LVAD samples were obtained in the operating room after the patient was induced with general anesthesia and placed on cardiopulmonary bypass. These procedures may have affected the sarcomeric protein phosphorylation background. Despite these limitations, the current study remains the most comprehensive examination of the effects of LVAD support on the sarcomere and thus merits attention.
Conclusion
There is significant improvement in LV and myocyte size with LVAD support, but there is only partial recovery of EF and myocyte contractility. The architectural changes of LVAD support were associated with a limited spectrum of biochemical changes which suggest that both lattice spacing and PKC activation might contribute to contractile dysfunction. These data further suggest that additional interventions beyond mechanical unloading may be needed to alter biochemical remodeling of the sarcomere to further enhance myofilament and organ level recovery.
Clinical Summary.
Sustained recovery of the failing left ventricle (LV) during pressure/volume unloading with a left ventricular assist device (LVAD) is rare and may be related to incomplete recovery of sarcomeric contractility. In this study, we evaluated contractility and biochemistry at the most fundamental contractile level of the heart–the sarcomere. Force development in muscle is the result of actin and myosin interactions and crossbridge cycling, processes regulated by modifications of the sarcomeric contractile proteins. Sarcomeric contractility was assessed by measuring isometric forces on skinned LV myocytes from patients with nonischemic cardiomyopathy before and after LVAD placement. We found that contractile dysfunction at the level of the sarcomere was present in failing hearts and paralleled organ level contractile dysfunction as assessed by ejection fraction. Furthermore, there were improvements in LV and myocyte size with partial recovery of sarcomeric force after LVAD, but that LVAD supported myocyte forces were still half of that seen in nonfailing hearts. The persistence of sarcomeric contractile dysfunction may be one of the reasons most LVADs are unable to be explanted in clinical practice. In assessing for biochemical alterations of sarcomeric proteins after LVAD, there were changes in TnI phosphorylation that may account for some of the improvement in sarcomeric force, but the other sarcomeric contractile proteins revealed minimal biochemical changes. This would suggest that additional interventions (in addition to mechanical unloading with a LVAD) may be needed to optimize TnI phosphorylation and/or modify other sarcomeric protein biochemistry to further enhance sarcomeric and organ level recovery.
Acknowledgments
Sources of Funding
Dr. Ambardekar is supported by a Research Fellowship Award from the Heart Failure Society of America (St. Paul, MN). Additional work on this review was supported by grants from the National Institutes of Health (HL077195 and HL101435).
Abbreviations
- LVAD
left ventricular assist device
- EF
ejection fraction
- LV
left ventricle
- LVIDd
left ventricular internal dimension end-diastole
- TnI
troponin I
- HF
heart failure
- MyBPC
myosin binding protein C
- TnT
troponin T
- Tm
tropomyosin
- MLC-2
regulatory myosin light chain
- MLC-1
essential myosin light chain
- PKA
protein kinase A
- PKC
protein kinase C
- SD
standard deviation
Footnotes
Disclosures
None.
References
- Maybaum S, Mancini D, Xydas S, Starling RC, Aaronson K, Pagani FD, Miller LW, Margulies K, McRee S, Frazier OH, Torre-Amione G. Cardiac improvement during mechanical circulatory support: A prospective multicenter study of the LVAD working group. Circulation. 2007;115:2497–2505. doi: 10.1161/CIRCULATIONAHA.106.633180. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, Klotz S, Mancini DM. LVAD-induced reverse remodeling: Basic and clinical implications for myocardial recovery. J of Card Failure. 2006;12:227–239. doi: 10.1016/j.cardfail.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Bruggink AH, de Jonge N, van Oosterhout MF, Van Wichen DF, de Koning E, Lahpor JR, Kemperman H, Gmelig-Meyling FH, de Weger RA. Brain natriuretic peptide is produced both by cardiomyocytes and cells infiltrating the heart in patients with severe heart failure supported by a left ventricular assist device. J Heart Lung Transplant. 2006;25:174–180. doi: 10.1016/j.healun.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Torre-Amione G, Stetson SJ, Youker KA, Durand JB, Radovancevic B, Delgado RM, Frazier OH, Entman ML, Noon GP. Decreased expression of tumor necrosis factor-α in failing human myocardium after mechanical circulatory support: A potential mechanism for cardiac recovery. Circulation. 1999;100:1189–1193. doi: 10.1161/01.cir.100.11.1189. [DOI] [PubMed] [Google Scholar]
- Zafeiridis A, Jeevanandam V, Houser SR, Margulies KB. Regression of cellular hypertrophy after left ventricular assist device support. Circulation. 1998;98:656–662. doi: 10.1161/01.cir.98.7.656. [DOI] [PubMed] [Google Scholar]
- Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, Rasmusson BY, Whitehead KJ, Salama ME, Selzman CH, Stehlik J, Clayson SE, Bristow MR, Renlund DG, Li DY. Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol. 2010;56:382–391. doi: 10.1016/j.jacc.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AG, Park SJ. Pathology in patients with ventricular assist devices: A study of 21 autopsies, 24 ventricular apical core biopsies and 24 explanted hearts. Cardiovascular Pathology. 2005;14:19–23. doi: 10.1016/j.carpath.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Dipla K, Mattiello JA, Jeevanandam V, Houser SR, Margulies KB. Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation. 1998;97:2316–2322. doi: 10.1161/01.cir.97.23.2316. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Hunlich M, Camp PC, Begin KJ, El-Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Thin filament-based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–987. doi: 10.1161/01.CIR.0000139334.43109.F9. [DOI] [PubMed] [Google Scholar]
- Klotz S, Barbone A, Reiken S, Holmes JW, Naka Y, Oz MC, Marks AR, Burkhoff D. Left ventricular assist device support normalizes left and right ventricular beta-adrenergic pathway properties. J Am Coll Cardiol. 2005;45:668–76. doi: 10.1016/j.jacc.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Chaudhary KW, Rossman EI, Piacentino V, Kenessey A, Weber C, Gaughan JP, Ojamaa K, Klein I, Bers DM, Houser SR, Margulies KB. Altered myocardial Ca2+ cycling after left ventricular assist device support in the failing human heart. J Am Coll Cardiol. 2004;44:837–45. doi: 10.1016/j.jacc.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Birks EJ, Tansley PD, Hardy J, George RS, Bowles CT, Burke M, Banner NR, Khaghani A, Yacoub MH. Left ventricular assist device and drug therapy for the reversal of heart failure. N Engl J Med. 2006;355:1873–84. doi: 10.1056/NEJMoa053063. [DOI] [PubMed] [Google Scholar]
- Jin W, Brown AT, Murphy AM. Cardiac myofilaments: From proteome to pathophysiology. Proteomics Clin Appl. 2008;2:800–810. doi: 10.1002/prca.200780075. [DOI] [PubMed] [Google Scholar]
- Jweied EE, McKinney RD, Walker LA, Brodsky I, Geha AS, Massad MG, Buttrick PM, de Tombe PP. Depressed cardiac myofilament function in human diabetes mellitus. Am J Physiol Heart Circ Physiol. 2005;289:H2478–H2483. doi: 10.1152/ajpheart.00638.2005. [DOI] [PubMed] [Google Scholar]
- Jweied E, de Tombe P, Buttrick PM. The use of human cardiac tissue in biophysical research: The risks of translation. J Mol Cell Cardiol. 2007;42:722–726. doi: 10.1016/j.yjmcc.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LA, Medway AM, Walker JS, Cleveland JC, Buttrick PM. Tissue procurement strategies affect the protein biochemistry of human heart samples. J Muscle Res Cell Motil. 2011;31:309–314. doi: 10.1007/s10974-010-9233-6. [DOI] [PubMed] [Google Scholar]
- Fan D, Wannenburg T, de Tombe PP. Decreased myocyte tension development and calcium responsiveness in rat right ventricular pressure overload. Circulation. 1997;95:1077–1083. doi: 10.1161/01.cir.95.9.2312. [DOI] [PubMed] [Google Scholar]
- Dobesh DP, Konhilas JP, de Tombe PP. Cooperative activation in cardiac muscle: Impact of sarcomere length. Am J Physiol Heart Circ Physiol. 2002;282:H1055–H1062. doi: 10.1152/ajpheart.00667.2001. [DOI] [PubMed] [Google Scholar]
- Walker JS, Li X, Buttick PM. Analysing force-pCa curves. J Muscle Res Cell Motil. 2010;31:59–69. doi: 10.1007/s10974-010-9208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LA, Walker JS, Ambler SK, Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol. 2010;48:1180–1186. doi: 10.1016/j.yjmcc.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in Situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2009;284:5097–5106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson PAW, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Troponin T isoform expression in humans: A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ Res. 1991;69:1226–1233. doi: 10.1161/01.res.69.5.1226. [DOI] [PubMed] [Google Scholar]
- Ambardekar AV, Buttrick PM. Reverse remodeling with left ventricular assist devices: A review of clinical, cellular, and molecular effects. Circ Heart Failure. 2011;4:224–233. doi: 10.1161/CIRCHEARTFAILURE.110.959684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Irving TC, de Tombe PP. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: Role of interfilament spacing. Circ Res. 2002;90:59–65. doi: 10.1161/hh0102.102269. [DOI] [PubMed] [Google Scholar]
- Farman GP, Walker JS, de Tombe PP, Irving TC. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H1847–H1855. doi: 10.1152/ajpheart.01237.2005. [DOI] [PubMed] [Google Scholar]
- Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, Solaro RJ, Buttrick PM. Protein kinase Cε overexpression alters myofilament properties and composition during the progression of heart failure. Circ Res. 2004;95:424–432. doi: 10.1161/01.RES.0000138299.85648.92. [DOI] [PubMed] [Google Scholar]
- Roman BB, Goldspink PH, Spaite E, Urboniene D, McKinney R, Geenen DL, Solaro RJ, Buttrick PM. Inhibition of PKC phosphorylation of cTnI improves cardiac performance in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H2089–H2095. doi: 10.1152/ajpheart.00582.2003. [DOI] [PubMed] [Google Scholar]
- Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase C-α-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- Liu Q, Chen X, MacDonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase Cα, but not PKCβ or PKCγ, regulate contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Ying J, Razeghi P, Stepkowski S, Taegtmeyer H. Atrophic remodeling of the transplanted rat heart. Cardiology. 2006;105:128–136. doi: 10.1159/000090550. [DOI] [PubMed] [Google Scholar]
- Dandel M, Weng Y, Siniawski H, Potapov E, Drews T, Lehmkuhl HB, Knosalla C, Hetzer R. Prediction of cardiac stability after weaning from left ventricular assist devices in patients with idiopathic dilated cardiomyopathy. Circulation. 2008;118(suppl 1):S94–S105. doi: 10.1161/CIRCULATIONAHA.107.755983. [DOI] [PubMed] [Google Scholar]
- Formica P, Murthy S, Edwards P, Goldstein D, Maybaum S. A structured 3-step approach to evaluate cardiac recovery with continuous flow circulatory support. J Heart Lung Transplant. 2010;29:1440–1442. doi: 10.1016/j.healun.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Hamdani N, Paulus WJ, van Heerebeek L, Borbely A, Boontje NM, Zuidwijk MJ, Bronzwaer JGF, Simonides WS, Niessen HWM, Stienen GJM, van der Velden J. Distinct myocardial effects of beta-blocker therapy in heart failure with normal and reduced left ventricular ejection fraction. Eur Heart J. 2009;30:1863–1872. doi: 10.1093/eurheartj/ehp189. [DOI] [PubMed] [Google Scholar]