

Abstract
In 1976, Sporn has defined chemoprevention as “the use of pharmacologic or natural agents that inhibit the development of invasive breast cancer either by blocking the DNA damage that initiates carcinogenesis, or by arresting or reversing the progression of premalignant cells in which such damage has already occurred.” Although the precise mechanism or mechanisms that promote a breast cancer are not completely established, the success of several recent clinical trials in preventive settings in selected high-risk populations suggests that chemoprevention is a rational and an appealing strategy. Breast cancer chemoprevention has focused heavily on endocrine intervention using selective estrogen receptor modulators (SERMs) and aromatase inhibitors (AIs). Achieving much success in this particular setting and new approaches as low-dose administration are actually under investigations in several topics. Unfortunately, these drugs are active in prevention of endocrine responsive lesions only and have no effect in reducing the risk of estrogen-negative breast cancer. Thus, recently new pathways, biomarkers, and agents likely are to be effective in this subgroup of cancers and were put under investigation. Moreover, the identification of new potential molecular targets and the development of agents aimed at these targets within cancer have already had a significant impact on advanced cancer therapy and provide a wealth of opportunities for chemoprevention. This paper will highlight current clinical research in both ER-positive and ER-negative breast cancer chemoprevention, explaining the biologic effect of the various agents on carcinogenesis and precancerous lesions, and finally presenting an excursus on the state-of-the-art about new molecular targets under investigations in breast cancer settings.
1. Introduction
While decreases in both breast cancer incidence and mortality have been apparent in recent years, the societal and economic impact of this malignancy continues to be enormous [1]. Breast cancer remains the most commonly diagnosed malignancy among females [2]. The idea of preventing breast cancer dates back to history (Figure 1). Positive associations between environmental and individual factors and increased risk of breast cancer development have been alleged for at least a century. Several progresses were made in understanding the underlying mechanisms of cancer development and some drugs were recently approved for the preventive approach of this disease. Thus, the current thinking is that prevention is a highly feasible approach to breast cancer control. Despite several factors which increase the woman' risk (gender, age, and family history) are not changeable, other modified risk factors such as alcohol intake, dietary fat, obesity in postmenopausal age, and hormonal stimulations have been identified and for these reasons interest in strategies to prevent breast cancer remains strong and intriguing [3]. Cancer chemoprevention is defined as the use of natural, synthetic, or biochemical agents to reverse, suppress or prevent carcinogenic process to neoplastic disease [4]. The epithelial carcinogenesis is a multistep, multipath, and multiyear disease of progressive genetic and associated tissue damage (Figure 2) [5]. In detail, the carcinogenetic process starts with unspecified accumulations of genetics events which lead to a progressive dysplastic cellular appearance with genotypic and phenotypic alterations, deregulated cell growth, and finally cancer [6]. This process is similar in every epithelial cancer, and the ability to arrest one or the several of these steps may impede or delay the development of cancer.
Figure 1.
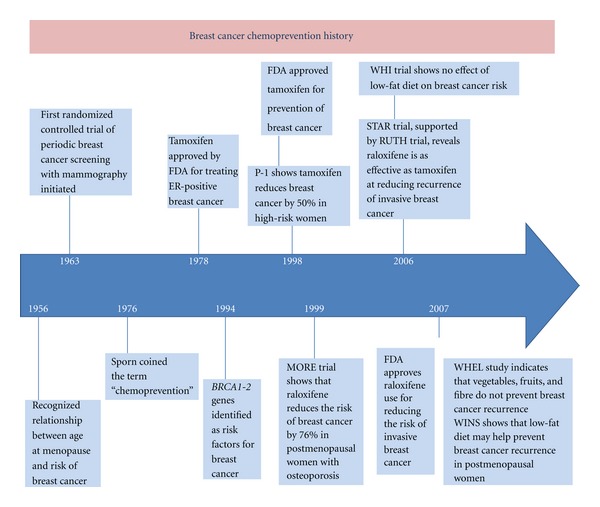
Breast cancer chemoprevention history.
Figure 2.
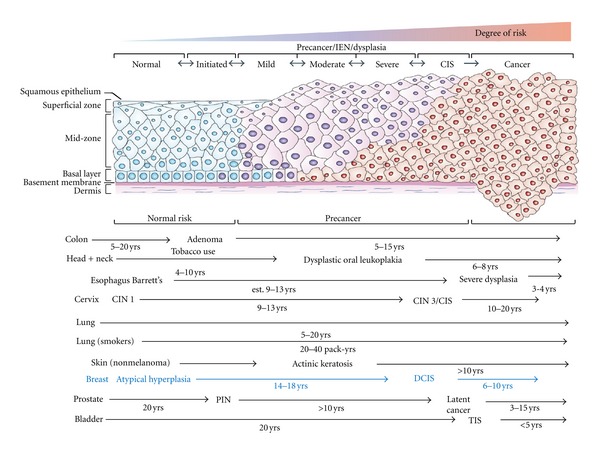
Model of human carcinogenesis.
2. ER-Positive Breast Cancer Prevention
Although the precise mechanism that causes breast cancer is not fully established itis recognized that hormones play a significant role in almost 70% of cases [7] and current chemopreventive strategies have targeted hormonally responsive breast cancers.
Estrogen is well established as a promoter of cell division in the breast, where it causes proliferation of both normal and malignant cells [8]. The two major classes of antiestrogenic drugs, the selective estrogen receptor modulators (SERMs) and the aromatase inhibitors (AIs), have been recently used for their activity in breast cancer prevention.
3. SERMs
3.1. Tamoxifen
This class of drugs includes in particular Tamoxifen (TAM) and Raloxifene, acting as both estrogen agonist and antagonists. Tamoxifen citrate is the first generation of SERMs that competes with circulating estrogen for binding the estrogen receptor (ER) [9]. Like tamoxifen, also raloxifene, a second generation of SERMs, has both estrogen agonist and antagonist properties. It differs from tamoxifen principally by its lack of stimulation of endometrium [10].
TAM has been in clinical use for breast cancer treatment for more than 30 years to reduce the risk of both recurrence and contralateral neoplasia, 42% and 47%, respectively [11]. These data lead to choose TAM as a potential chemopreventive agent, and several studies were conducted in last decades in this particular setting.
The BCPT NSABP-1 [12] was a placebo-controlled trial of TAM in more than 13000 women at high risk of breast cancer. Women were randomized to receive tamoxifen 20 mg/die or placebo for 5 years. This trial was closed early after the interim analysis showed a 49% reduction in incidence of invasive breast cancer in the treatment arm. Moreover, the highest level of benefits was observed in patients with precancerous lesions as LCIS (relative risk = 0.44) and atypical ductal hyperplasia (relative risk = 0.14). Tamoxifen appeared able to reduce breast cancer incidence also in healthy BRCA2 carriers by 62% but not in BRCA1 [13]. The study showed also an increased risk of endometrial cancer and thrombotic events, and these conclusions suggested that despite its extraordinary preventive efficacy the utilization of TAM in this particular setting should be extremely individualized. The results of this study were the first to show the benefit of TAM in breast cancer prevention.
In addition, 3 European tamoxifen prevention trials have been completed and have reported long-term follow-up data of the effect of this agent in BC incidence: The Italian Tamoxifen Prevention Study, The Royal Marsden Hospital tamoxifen randomized chemoprevention trial, and the International Breast Cancer Intervention Study (IBIS)-1 [14]. Although they differ in many details in study design and other, these trials were similar enough to be evaluated together in an overview of their main outcomes [15]. Their combined data indicate an overall 30 to 40% reduction in breast cancer ER-positive incidence following 5 years of TAM versus placebo (Figures 3 and 4), and these effects remains also after more than ten years of followup. The serious adverse events that occurred with TAM were as anticipated from previous adjuvant trials, an increase of endometrial cancers and venous thromboembolic events (VTEs). Other expected TAM-associated toxicities were observed as cataracts, hot flushes, and vaginal discharge.
Figure 3.
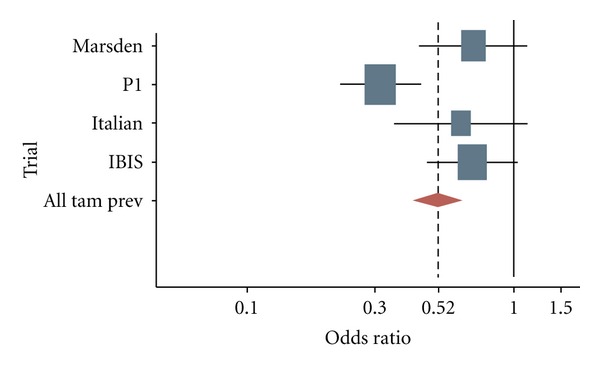
ER+: odds ratios for developing an estrogen receptor-positive invasive breast cancer among women involved in tamoxifen prevention trials.
Figure 4.
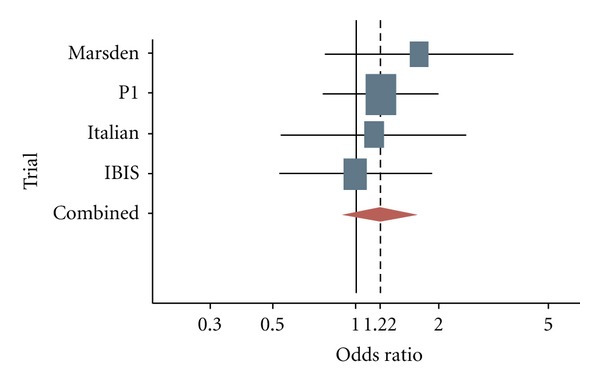
ER−: odds ratios for developing an estrogen receptor-negative invasive breast cancer among women involved in tamoxifen prevention trials.
The data from the NSABP P-1 trial, which showed a reduction in both invasive and noninvasive breast cancers, led to the 1998 US Food and Drug Administration (FDA) approval of TAM for reduction of breast cancer incidence in high-risk women. However, the adverse effects of this drug have hampered its uptake by women at increased risk. When TAM's benefits are balanced against its major toxicities, younger women at very high risk and possibly hysterectomized postmenopausal women appear to be the best candidates for preventive TAM.
3.2. Tamoxifen at Lower Dose
A simple and economic approach to retain tamoxifen efficacy while reducing the risks may be a dose reduction. In a study conducted by our group, standard dose of tamoxifen (20 mg/die) and two differ lower doses (10 mg/die and 10 mg on alternate days) were administered for 2 months in a cohort of more than 120 healthy women [16], and changes in serum biomarkers regulated by the ER were evaluated. No evidence for a concentration-response relationship was observed for most of these biomarkers. The concept of dose reduction was further supported by the observation of tamoxifen as very high tissue distribution (5–60 times its blood concentrations) [17] and a prolonged half-life [18]. Moreover, the low-dose concept has been confirmed in a preoperative trial in which 120 breast cancer women were treated with either 20, 5, or 1 mg/die of TAM for 4 weeks before surgery [19]. The effects of these different doses of Tam on proliferation were analyzed using the Ki67 expression as the main surrogate endpoint marker. Interestingly, the change in Ki67 expression induced by lower doses of tamoxifen was comparable to that achieved with the standard dose, implying that low-dose TAM retains its antiproliferative activity. Moreover, several blood biomarkers of TAM estrogenicity associated with the risk of breast cancer, cardiovascular disease and bone fracture showed a dose-response relationship, and suggesting that this approach may be associated with reduced, positive and negative oestrogenic effects of TAM.
These fundamental data provide a strong rationale for the formal assessment of low-dose TAM in preventive setting, and for these reasons we started two phase III randomized placebo trials (actually ongoing in our institute) in order to assess the efficacy of 5 mg/die of TAM in high-risk women as current HRT (HOT study) and with breast intraepithelial neoplasia (IEN).
3.3. Raloxifene
Raloxifene, a second generation of SERMs has reduced the incidence of breast cancer in preclinical models and several clinical trials evaluated it for the prevention of osteoporosis and heart disease [20–22]. Raloxifene, which has a well-established and favourable effect on bone metabolism, was in fact initially approved (by FDA) for the prevention and treatment of osteoporosis in postmenopausal women. In the Multiple Outcomes of Raloxifene Evaluation (MORE) trial [23], raloxifene (60 mg or 120 mg compared to placebo) shows a 30% reduction in the risk of vertebral fracture [24] in postmenopausal women with osteoporosis. One of the secondary endpoints of the study was the incidence of breast cancer in this target of population, and in raloxifene-treated group the risk of invasive breast cancer was significantly reduced by 72% (RR = 28; 95% CI 0.17–0.46) that becomes 62% after 4 years of followup in the Continuing Outcomes Relevant to Evista (CORE) trial [25]. As was noted in the tamoxifen trials, the benefits appeared to be specific only to receptor-positive invasive breast cancer. The adverse events were different in tamoxifen. An increasing risk in thromboembolic events included DVT (deep venous thrombosis) and pulmonary embolism as observed in a raloxifene study (RR = 3.1; 95% CI 1.5–6.2), but unlike what occurs in tamoxifen there was no difference in the incidence of endometrial carcinoma compared with placebo arm [23, 24].
Results of MORE and CORE trials led researchers to conduct a comparative, randomized phase III study of raloxifene versus tamoxifen in more than 19000 postmenopausal women at increased risk for breast cancer [26]. The Study of Tamoxifen and Raloxifene (STAR) trial or NSABP-P2 compared 20 mg of tamoxifen daily to 60 mg of raloxifene daily for 5 years with the incidence of breast cancer as a primary endpoint. The secondary end points included noninvasive breast cancer, uterine malignancies, thromboembolic events, fractures, cataracts, quality of life, and death from any cause. Interestingly, although no untreated control group was included, there was no difference in the incidence of the disease between the two groups (RR: 1.02; 95% CI: 0.82–81.28). Furthermore, while there was a difference between the two treatment groups for the rate of in situ(ductal and lobular) breast cancer, this was not shown to be statistically significant (RR: 1.40; 95% CI: 0.98–92.00). The numbers of invasive breast cancers in both groups of women were statistically equivalent. Conclusive results based on the risk reduction seen in the BCPT for tamoxifen show that both drugs reduced the risk of developing invasive breast cancer by about 50%. While tamoxifen reduced the incidence of LCIS and DCIS, raloxifene did not have an effect on these diagnoses.
A mechanism to explain the difference in noninvasive breast cancer incidence is unknown, but long-term follow-up results for the STAR trial may result in additional information regarding this issue. Regarding the side effects, more uterine malignancies occurred in the Tamoxifen arm and no statistically significant differences were noted between the 2 groups relative to the incidence of any cardiovascular events.
More recently, results from the Raloxifene Use for the Heart (RUTH) study affirmed the benefit of raloxifene with regard to reduced risk of breast cancer. This trial, designed to focus on heart disease, randomized more than 10,000 postmenopausal women with coronary health disease or multiple coronary health disease risk factors to receive either raloxifene 60 mg per day or placebo [27].
Data from the STAR trial and the other raloxifene/placebo trial resulted in the approval of raloxifene by the US Food and Drug Administration for a reduction in the risk of invasive breast cancer in postmenopausal women with osteoporosis and reduction in the risk of invasive breast cancer in postmenopausal women at high risk of invasive breast cancer. No data are currently available on the use of raloxifene in patients with BRCA1 or BRCA2 mutations, nor was raloxifene approved for women with a previous invasive breast cancer or for the treatment of invasive breast cancer. However, the approval of raloxifene gives an important new option to postmenopausal women beyond that of tamoxifen, one that avoids an excess of endometrial cancers and reduces the risk of thromboembolic events.
4. Aromatase Inhibitors (AIs)
High circulatory estrogen levels, as well as high aromatase levels in breast tissue, have been known to increase breast cancer risk. Thus, inhibition of aromatase would be expected to decrease estrogen production and ultimately estrogen-related breast carcinogenesis.
In adjuvant setting, third generation of AIs (anastrozole, letrozole, and exemestane) has been found to superior to tamoxifen and be able to reduce the incidence of contralateral breast cancers by 37 to 55% [28–33]. These agents have resulted in improved disease-free survival and are associated with fewer life-threatening side effects than SERMs [34]. The principle toxicity of AIs is accelerated bone resorption. AIs are generally welltolerated with the primary side effects being musculoskeletal and joint discomfort. Thus, the third-generation aromatase inhibitors (AIs) have been introduced into the treatment of breast cancer, and their greater efficacy compared to tamoxifen, along with a more favorable side-effect profile, makes them attractive agents for use in breast cancer prevention [35, 36].
The International Breast Cancer Intervention (IBIS)-II prevention trial [37], direct consequence of ATAC trial, is actually ongoing and is comparing anastrazole (ANA) to placebo in 6000 postmenopausal women at increased risk to breast cancer. A second complimentary study (IBIS-II) will look at the role of ANA in affected postmenopausal women who underwent a locally excised (or mastectomy) hormone receptor-positive intraductal neoplasia with clear margins. In this second group, ANA is compared to tamoxifen. Both arms address the ability of ANA to reduce the incidence of first primary invasive breast cancers.
Another prevention trial with AIs (MAP3) is actually underway with exemestane (EXE). Authors are comparing placebo or EXE, or EXE plus celecoxib for 5 years in more than 5000 high-risk postmenopausal women. In September 2004 the disclosure of an excess of adverse cardiovascular events in the COX-2 inhibitor arm has recommended authors to revised the study design. They modified it in two different arms (exemestane vs placebo) and a new simple size of 4.560. Despite this, the MAP3 study was reopened to accrual in March 2005 with a revised sample size of 4,560 and two arms, EXE 25 mg/d alone and placebo. The primary endpoint is the incidence of breast cancer specifically to determine if EXE is able to reduce invasive breast cancer by 65% compared to placebo. Secondary endpoints regard also safety and incidence of noninvasive breast cancer.
These data obtained by adjuvant trial provide a rational for exploring AIs in prevention setting. They are superior to tamoxifen, and we hypothesized that the major of ER-positive breast cancer (but not for ER negative) can be prevented by these drugs. Moreover, they are also well tolerated than tamoxifen without uterine and thrombotic effects, but they do lead to bone mineral loss. These effects should be contrasted by the use of bisphosphonates.
5. ER-Negative Breast Cancer Prevention
Although a number of antiestrogenic agents are being extensively tested in clinical trials, all these agents affect the endocrine pathway and suppress only the development, of estrogen receptor (ER)-positive breast cancer. They have no effect in reducing the risk of ER-negative breast cancer, which accounts for 20–30% of breast cancers and has a poor prognosis [38].
Thus, it is worth identifying new pathways, biomarkers, and agents that are effective in the treatment and prevention of these subtypes. With the accumulating knowledge in understanding the biology of cancer development several classes of a new generation of chemopreventive agents modulating the nonendocrine biochemical pathways have been developed and many of these are still currently under investigation.
These agents include retinoids, epidermal growth factor receptor (EGFR), tyrosine kinase inhibitors (TKIs), cyclooxygenase-2 (COX-2) inhibitors, bisphosphonates, vitamin D receptor (VDR), statins, peroxisome proliferator- activated receptor (PPAR), and others. A complete summary of involved agents, with their specific pathways, is shown in Table 1 and a brief state of the art of the more compounds involved are analyzed below.
Table 1.
Class, specific pathways, and agents actually involved in the treatment and prevention of ER-breast cancer.
| Class | Targets | Drugs or agents |
|---|---|---|
| Nuclear receptors | Retinoid acid receptor RXR | Fenretinide (4-HPR) 9 cis-retinoic acid (Targretin) |
| VDR | VIT D3 analogues | |
| PPARγ | Troglitazone, rosiglitazone, pioglitazone | |
|
| ||
| Membrane receptors and signal transduction | HMG-CoA | Statins |
| Tyrosine kinase | Gefitinib (Iressa) | |
| HER-1, HER-2 | Trastuzumab (Herceptin), lapatinib, gefitinib, erlotinib | |
| IGF-R, IGF-1, IGFBP3 | Metformin | |
|
| ||
| Anti-inflammatory and antioxidant | COX-2 | celecoxib, rofecoxib, NSAIDs |
|
| ||
| Angiogenesis | VEGF | Bevacizumab |
|
| ||
| DNA modulation | BRCA1-BRCA2 | PARP inhibitors |
4-HPR: N-(4-hydroxyphenyl) retinamide; COX: cyclooxygenase; ER: oestrogen receptor; HMGCoA: 3 hydroxy-3 methylglutaryl coenzyme A; NSAIDs: nonsteroidal anti-inflammatory drugs; PARP: poly (ADP-ribose) polymerases; PPAR: peroxisome proliferator-activated receptor; RXr: retinoid X receptors, VDR: vitamin D receptor.
5.1. Retinoids
Retinoids are natural and synthetic derivative of Vitamin A (Retinol) that have profound effects on development, metabolism, differentiation, and cell growth.
The retinoid, the most widely studied in chemoprevention clinical trials, is the synthetic amide of retinoic acid N-(4-hydroxyphenyl) retinamide (4-HPR), or fenretinide. Fenretinide has been found to exert significant chemopreventive activity in various in vitro and in vivo studies [39–42]. A phase III clinical trial, using 4-HPR to reduce the incidence of secondary breast cancer in almost 3000 patients, was published in 1999 and showed no difference in contralateral and ipsilateral breast cancers; however, a posthoc analysis suggested a significant treatment interaction with menopausal status. In particular, it showed a 35% reduction in premenopausal women and an opposite trend in postmenopausal women [43]. Moreover, the 15-year follow-up of this trial substantially confirmed these results, and the risk reduction is of the order of 50% in women aged 40 years or younger and persists for 10 years after retinoid cessation [44]. Moreover, 4-HPR was observed to reduce secondary tumours in premenopausal women irrespective of the hormone receptor status of the primary cancer, suggesting that retinoids have a potential chemopreventive effect on ER-negative and ER-positive breast cancers.
Recently, also a new RXR-selective retinoid, commonly named as rexinoids, has been studied as cancer preventative agent. Preclinical studies in fact have demonstrated that this compound is able to maintain the chemopreventive efficacy of the retinoids, also in ER-negative setting, but with substantial minor toxicity [45, 46]. For this reason this agent is actually considered particularly attractive in prevention setting.
5.2. EGFR-Tyrosine Kinase Inhibitors
The EGFR is one of a family of four closely related receptors (EGFR or erbB-1, HER-2/neu or erbB-2, HER-3 or erbB-3, and HER-4 or erbB-4) that uses tyrosine kinase activity and contributes to a large number of processes involved in tumour survival and growth, including cell proliferation and inhibition of apoptosis, angiogenesis, and metastasis [47], thus making it an attractive target for cancer prevention and treatment, because agents that are able to block the erbB-signaling pathways are promising in the treatment and prevention of breast cancer.
In particular, the researchers focused their attention to EGFR-HER-1 and HER-2 pathways, because the mechanism of resistance to antioestrogen therapy is usually associated with an increased expression of HER-1 and HER-2 receptors. Inhibition of tyrosine kinase activity, with TKIs, involved in the EGFR signaling cascade could be the right pathway for the treatment and prevention of ER-independent breast.
There are two different and concomitant strategies able to inhibit erbB activity. One involves blockade of this activity with monoclonal antibodies (trastuzumab), whereas the second involves the TKIs. The two strategies differ in several pharmacological properties [48].
Amplification of the HER2 gene and overexpression of it's related protein have been found in almost 30% of human breast cancer and it is generally correlated with poorer outcomes compared with tumors HER2 negative [49, 50]. Moreover, there is substantial evidence of an inverse correlation between HER2 expression and hormone receptor [51]. In an effort to improve the prognosis of these HER2+ cancers, research has focused therapies directly against this pathway and in particular included the monoclonal antibodies trastuzumab (Herceptin).
Trastuzumab has largely showed its benefit in adjuvant therapy; in particular, it is able to increase the clinical benefit of first-line chemotherapy in metastatic HER-2 breast cancer [52], and this benefit seems to be irrespective of the ER status [53]. Important results were also obtained in early breast cancer setting [54–56]. The drug is generally well tolerated, but its possible cardiotoxicity and its route of administration (intravenously) make it difficult to propose it to healthy women as chemoprevention.
Apart from the monoclonal antibodies directed against the extracellular receptor domain of HER-2, there is another way to contest erbB activity. As previously explained, the use of small molecules inhibit intracellular tyrosine kinase activity, named TKIs. TKIs have several advantages over monoclonal antibodies such as oral bioavailability, potentially less toxicity, and ability to inhibit truncated forms of EGF receptors [57].
Lapatinib (Tykerb) is a reversible small-molecule TKI that targets both HER-2 and EGFR tyrosine kinase. It is able to interrupt signal transduction from both EGFR and HER-2 receptors, and because of its dual-receptor activity it has been evaluated in several phase II and III trials in various forms of breast cancer [58–60]. Moreover, in the prevention setting, it showed a significant delay in the ER-negative mammary tumors development [61]. This preventive action was seen in premalignant mammary lesions, and this suggests a drug efficacy also in initiation and progression of breast carcinogenesis.
Gefitinib (Iressa), another EGFR tyrosine kinase inhibitor that suppressed ER-negative mammary tumor formation in MMTV-ErbB2 transgenic mice [47]. Its mechanism of action is complex and involves cell cycle, angiogenesis, and growth factors [62, 63]. Moreover, results of preclinical and clinical studies about breast cancer treatment remain controversial [64, 65], but in preventive setting, the ability of gefitinib to inhibit the proliferation at the early stages of breast cancer and also in the normal adjacent epithelium [66] could be the rationale for using this compound in prevention trial.
5.3. COX-2 Inhibitors
The inducible isoenzyme COX-2 is expressed in invasive and in situ breast cancers cells [67], and several epidemiological studies have shown the inverse relationship between nonsteroidal, anti-inflammatory drugs (NSAIDs) and cancer incidence [68, 69]. COX-2 is the main target of NSAIDs, and despite the mechanism by which it contributes to tumor formation is not fully understood, it is possible to hypothesize an involvement of a multidisciplinary process which involves proliferative stimulation, mutagen production, and apoptosis inhibition. The COX-1 and COX-2 pathway, which converts arachidonic acid to prostaglandin, is involved in the development and growth of several different neoplastic lesions [70], and it is frequently overexpressed not only in invasive breast cancer but also in adjacent intraductal neoplasia; therefore, it might be an early event in mammary tumorigenesis [71]. A meta-analysis, published in 2001, demonstrated that NSAIDs were associated with a 20% reduction of breast cancer risk, and the same results were confirmed in a more recent publication [72, 73]. These data suggested the chemopreventive potential (including breast cancer) of anti-inflammatory drugs.
Celecoxib, a selective COX-2 inhibitor, reduced the incidence and multiplicity of DMBA-induced mammary tumors in rat models by 68 and 86%, respectively [74]. Nimesulide, another selective COX-2 inhibitor, significantly reduced the incidence and multiplicity of PhIP and NMU-induced rat mammary tumors [75]. Similar effects were observed with aspirin, but the level of evidence for both of agents on breast cancer incidence is, at present, too small to justify their use solely as a preventive therapy and insufficient to make any recommendations.
5.4. Bisphosphonates
Bisphosphonates, the drugs of choice for the treatment of osteoporosis, act on the mevalonate pathway [76] and for this reason are currently of considerable interest in the treatment and prevention of breast cancer. Their mechanism of action involved osteoclasts, and in particular, they are able to inhibit their activity [77]. Thus they have proven efficacy in control of breast cancer bone metastases and also in bone loss induced by other treatment as AIs [78]. Previous studies showed the antiangiogenic, antiproliferative, and proapoptotic effect of these drugs [79]. Moreover, interestingly recent two cohort studies showed a reduction of 30% in breast cancer incidence in bisphosphonates users [80, 81], irrespective of hormonal status. Bisphosphonates are generally well tolerated, but randomised prevention trials with composite endpoints in women with osteopenia and increased risk of a new breast cancer are required to fully investigate the risk-benefit profile of these drugs.
5.5. Poly(ADP-Ribose) Polymerases Inhibitors
Poly(ADP-ribose) polymerases (PARPs) are a family of enzymes that play a key role in the repair of DNA damage [82]. In particular, the most important seems to be PARP enzymes (PARP-1 and PARP-2) [83, 84]. Their role was recently considerable of interest in oncology treatment and prevention.
A key role for PARP-1 and PARP-2 is maintaining genomic integrity, in particular, repair of single strand DNA lesions and breaks using the base excision repair (BER) pathway. The inhibition of these enzymes leads to accumulation of DNA single-strand breaks, which can lead to DNA double-strand breaks at replication forks [85, 86]. Normally, these breaks are repaired and a key component of this mechanism comprises the tumour-suppressor proteins BRCA1 and BRCA2. In BRCA mutated cells, this DNA repair ability is lost and the aberrations drive to carcinogenesis. Consequently, the requirement for a BRCA mutation to be present for a PARP inhibitor to be effective constitutes a synthetic lethal strategy selectively affecting mutant tumour cells comprising BRCA1-BRCA2, which are 1000-fold more sensitive than others [87–89]. Recent preclinical studies have shown encouraging results, and at present PARP inhibitors are usually studied in combination with other cytotoxic agents [90, 91]. The only published study with a PARP inhibitor as a single-agent treatment is a phase I trial with olaparib in patient with BRCA-associated cancer, which showed a good efficacy to inhibit PARP activity and that it has few side effects compared conventional chemotherapy [92].
The efficacy of a particular risk group as the mutation carriers and the relative good tolerability make these agents well suited for cancer prevention. Further investigations should be proposed in BRCA mutation carriers to assess the ability of this class of agents to prevent cancer, evaluate the safety profile, and reduce the incidence of breast cancer.
5.6. Metformin
There is increasing evidence that presence of hyperinsulinemia and insulin resistance increased breast cancer risk, worsen, the prognosised and partly explained the obesity-breast cancer risk association in postmenopausal women [93–98]. Several epidemiological and observational studies have confirmed the relationship between insulin levels and cancer induction [99, 100]. Insulin may promote tumorigenesis via a direct effect on epithelial tissues, or indirectly by affecting other modulators, such as insulin-like growth factors, sex hormones, and adipokines [101, 102]. Thus, there is a great interest in exploring the possibility that antidiabetic therapies, which lower insulin levels, could decrease breast cancer incidence or its related mortality.
Metformin, a biguanide derivative, is the most commonly used drug worldwide to treat type II diabetes. It is generally well tolerated with low toxicity and a very low cost. Epidemiological studies have shown a significant risk reduction in cancer incidence and mortality in diabetic patients on metformin, relative to other antidiabetic drugs, including positive results specifically in breast cancer [103–105]. It may impact cancer through a direct (insulin-independent) activation of AMPK-mTOR pathway mechanism or indirect effect (insulin-dependent) reducing hepatic gluconeogenesis obtaining lower circulating insulin levels with inhibition of proliferation cells and protein synthesis over increase apoptosis (Figure 5).
Figure 5.
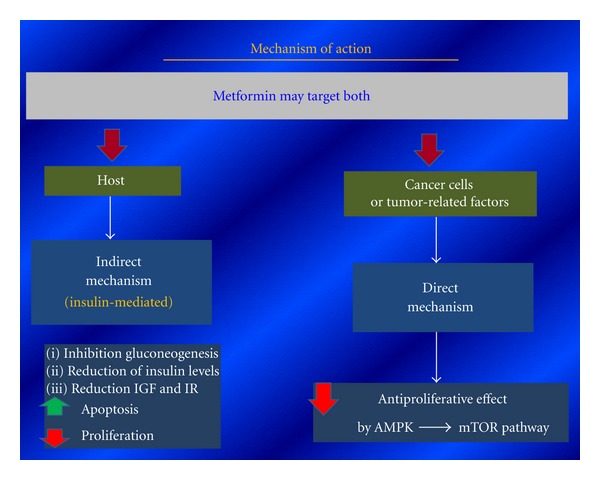
Metformin anticancer mechanism of action.
Several preclinical studies have confirmed these effects of metformin in vitro and in vivo and showed a significant reduction of both breast epithelial cell proliferation and protein synthesis [106, 107]. [108] In particular, it is confirmed that metformin produces a significant repression of cell proliferation, and moreover, they also found that this effect was different in human breast cancer cell lines if related to either positive or negative ERs. They in fact detected a complete cell growth repression in ER-positive cell lines, although only a partial inhibition was detected in ER-negative phenotypes. These data suggest that, although ER-negative cells are not as sensitive as ER-positive ones, both of them show a reduction in cell growth under metformin treatment. These data make metformin an intriguing compound for treatment and prevention of breast cancer. Several important phase II and III studies are actually ongoing in the world in order to confirm and clarify these promising settings [109].
6. New Molecular Targets for Breast Cancer Chemoprevention
Many molecularly pathways and the correlated targeted drugs are actually in development for advanced cancer therapy, and they have potential activity and tolerability also in cancer chemoprevention setting. The identification of new potential molecular targets and the development of agents aimed at these targets within cancer have already had a significant impact on advanced cancer therapy and provide a wealth of opportunities for chemoprevention.
6.1. Microenvironment and Its Molecular Targets
There is substantial evidence that together with the epithelial cells alterations the microenvironment dysfunction is crucial for carcinogenesis, and this makes the microenvironment an interesting target for breast cancer chemoprevention. In particular, there are many excellent publications which consider microenvironment as a good target for cancer therapy, but the application of chemoprevention to control the tumour microenvironment during the early stages of carcinogenesis is not yet adequately analyzed. We will briefly explain a recent progress that indicates that the effects of chemopreventive agents on the microenvironment are an important aspect of their preventive action and that many classes of agents, which showed to have significant chemopreventive actions on epithelia, also have similar useful actions on the microenvironment.
Many molecular targets inside the microenvironment with the correlated drugs are summarized in the Table 2.
Table 2.
Molecular targets and chemopreventive agents in the microenvironment.
| Molecular targets | Chemopreventive agents |
|---|---|
| Oestrogen receptors | Tamoxifen; raloxifene; aroxifene |
| Akt and NFκB | Curcumin; N-acetyl cysteine; silibinin; xanthohumol; deguelin; EGCG; resveratrol |
| NRF2-KEAP1 | Sulphoraphane; oltipraz |
| COX2 | Rofecoxib; celecoxib; EGCG |
| COX1/2 | Aspirin and other NSAIDs |
| Histone deacetylases | Sulphoraphane |
| TGFβ pathway | CDDO-Imidazolide |
| HIF1α | WGCG; resveratrol; apigenin; sulphoraphane |
| STATs | CDDO-Imidazolide |
| VEGF | Sulphoraphane; EGCG; fenretinide |
Some of the specific targets in the microenvironment and specific agents that interact with these targets: CDDO: 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid; COX2: cyclooxygenase 2; EGCG: epigallocatechin-3-gallate; HIF1α: hypoxia-inducible factor 1α; KEAP1: kelch-like ECH-associated protein 1; NFκB: nuclear factor κB; NSAIDs: nonsteroidal anti-inflammatory drugs; STATs: signal transducers and activators of transcription; TGFβ: transforming growth factor-β; VEGF: vascular endothelial growth factor.
These transcription factors and their associated regulatory proteins are an ideal target of chemoprevention, and in particular three attractive pathways as the nuclear factor κB (NFκB), hypoxia-inducible factor 1α (HIF-1α), and PI3κ-mTOR are analyzed in this section.
Nuclear factor-κB pathway plays important roles in the control of cell proliferation, differentiation, apoptosis, inflammation, stress response, cell signaling transduction, and other physiological processes, but it is also critically involved in the processes of development and progression of cancers [110–113]. NF-kappaB is critically involved in the processes of oxidative stress response. Oxidative stress is defined as an increase in intracellular reactive oxygen species (ROS) such as H2O2, superoxide, and hydroxyl radical and. ROS in cells are increased in response to agents that also activate NFkappaB. These findings suggest that oxidative stress activates NF-kappaB activity in the cells [114, 115]. Moreover, NFκB is activated not only by the ROS but also by various carcinogen and tumor promoters [116], and these are the reasons why NFκB is overexpressed and activated in various cancers, especially in the poorly differentiated.
Experimental studies have shown that natural antioxidant compounds including isoflavones, indole-3-carbinol (I3C), 3,3′-diindolylmethane (DIM), curcumin, epigallocatechin-3-gallate (EGCG), rosveratrol, curcumin and others seems to be able to inhibit the activity of NF-kappaB and the growth of cancer cells and also to induce apoptosis, suggesting that NF-kappaB could be a target for cancer prevention [117–121].
Similarly, HIF-1—a master regulator in the control of tissue homeostasis, crucial in adaptive responses to tissue oxygenation including energy status, glucose, and iron metabolism as well as growth factor signaling [122, 123]—is a key target for the prevention and treatment of cancer.
Recent experimental evidence in fact suggests that HIF-1 is a key player in carcinogenesis. Interest in the role of HIF-1 in cancer has grown exponentially over the last two decades, as this factor activates the transcription of many genes that code for proteins involved in several pathways intimately related to cancer [124–126].
Tumors are invariably less well oxygenated than the normal tissues from which they arise. Hypoxia-inducible factor-1 (HIF-1) plays a central role in the adaptation of tumor cells to hypoxia by activating the transcription of genes, which regulate several biological processes.
For these reasons, HIF-1 is considered a potential target for cancer therapy, and, recently, many efforts to develop new HIF-1-targeting agents have been made [127–130]. Interestingly, they are recently identified by increased HIF-1 expression (relative to adjacent normal tissue) in 13 tumor types, including lung, prostate, breast, and colon carcinoma. Moreover, HIF-1 was also overexpressed in preneoplastic and premalignant lesions, such as colonic adenoma, breast ductal carcinoma in situ, and prostate intraepithelial neoplasia. These data show that overexpression of HIF-1 may occur very early in carcinogenesis, before histologic evidence of angiogenesis or invasion [131], and suggest that HIF-1 might be a biomarker of carcinogenesis and a suitable target for cancer chemoprevention. Because HIF-1 seems to have an important function in carcinogenesis, HIF-1 inhibitors may be considered a source of potential cancer chemopreventive agents. Several, approved anticancer drugs (e.g., topotecan, imatinib mesylate, trastuzumab, NS398, celecoxib, and ibuprofen) inhibit HIF-1 activity [127]. Moreover, also several natural products (e.g., resveratrol, genistein, apigenin, and berberin) have also been found to inhibit the activity of this transcription [129]. In this setting it is important to say that: however, the use of HIF-1 inhibitors in cancer chemoprevention might be associated with toxicity. An excessive inhibition of HIF-1 may produce adverse effects, as HIF-1 regulates many cellular processes under physiologic conditions [125, 132]. Therefore, although HIF-1 inhibitors may represent a useful source of chemopreventive agents, the potential toxicity associated with these agents should be considered carefully, especially when chemopreventive interventions are aimed at preventing cancer in healthy populations.
The mammalian target of rapamycin (mTOR) is a signaling kinase of the phosphatidylinositol 3-kinase/protein kinase B or PI3K pathway that mediates cell growth and metabolism and coordinates cell cycle progression in response to genetic, epigenetic, and environmental conditions. Pathways involved in mTOR signaling are dysregulated in precancerous human tissues, including breast cancer, and is associated with the development of resistance to endocrine therapy [133–135] and to the anti-human epidermal growth factor receptor-2 (HER2) monoclonal antibody trastuzumab [136, 137]. Rapamycin and the rapalogues have been used in clinical trials for many cancer types. Phase I trials have demonstrated that mTOR inhibitors are fairly well tolerated with the most frequent drug-related toxic effects being acne-like maculopapular rash, mucositis, and stomatitis, all of which were reversible on discontinuation of treatment [138].
Rapamycin and its analogues, the “rapalogues,” decrease tumor growth in many xenograft models, including those with breast cancer cell lines [139, 140]. Thus, preclinical data have confirmed the antitumor activity of rapamycin and the rapalogues and have suggested that patients with breast cancer may especially respond to mTOR inhibitors. Phase I-II clinical trials have demonstrated that everolimus (RAD-001), an mTOR inhibitor with demonstrated preclinical activity against breast cancer cell lines, has been shown to reverse Akt-induced resistance to hormonal therapy and trastuzumab. It has promising clinical activity in women with HER2-positive, HER2-negative, and estrogen receptor-positive breast cancer when combined with HER2-targeted therapy, cytotoxic chemotherapy, and hormonal therapy, respectively.
The involvement of mTOR pathways in precancerous lesions makes the mTOR signaling an intriguing target for chemopreventive intervention. Thus, several recent preclinical studies explored also the possibility of a chemopreventive action through the mTOR inhibition. In one of this, rapamycin showed chemopreventive activity against mammary gland tumors in transgenic mice, bearing activated ErbB2 (HER-2/neu) receptor either alone (NeuYD) or with VEGF expression [141], where it dramatically inhibited tumor formation in NeuYD mice.
These results seem to suggest the mTOR inhibition as a possible chemopreventive strategy against metachronous tumors or recurrence in high-risk patients, whose primary tumors overexpressed ErbB-2, or in patients showing dysregulation of the PI3K/AKT/mTOR signaling pathway.
Another recent preclinical study evaluated chemopreventive effects of rapamycin in a transgenic mouse model of human breast carcinogenesis [142], where it significantly inhibited growth of mammary intraepithelial neoplasia outgrowths, invasive tumor incidence, and tumor burden.
Finally, some natural products, such as epigallocatechin gallate (EGCG), caffeine, curcumin, and resveratrol, have been found to inhibit mTOR as well and are actually under investigations in this setting.
7. Conclusions
In conclusion, the success of chemopreventive approach depends on a tumor-specific risk model for identifying high-risk subjects, increasing preclinical drug test over the development novel and more safety chemopreventative agents, and identifying new surrogate endpoint using molecular pathways and new targets of drugs activity.
Safety is a very important point to take into account, because several large randomized prevention trials in several cancers have shown that major adverse events can prevent widespread public acceptance of active chemoprevention agents.
Despite the success of action showed for example in endocrine intervention is a promising starting point in order to continue to evolve with the rapid integration of molecular approaches into research and clinical practice. It is urgent to find active agents in other fields as nonhormone-responsive lesions. The personalized approaches in advanced cancer therapy and the evolution of molecularly targeted will streamline chemoprevention research and facilitate the development of rational, effective, and safe preventive drugs, involving different pathways and with the ability to modify carcinogenesis in early phases.
References
- 1.Smigal C, Jemal A, Ward E, et al. Trends in breast cancer by race and ethnicity: update 2006. Ca-A Cancer Journal for Clinicians. 2006;56(3):168–183. doi: 10.3322/canjclin.56.3.168. [DOI] [PubMed] [Google Scholar]
- 2.Youlden DR, Cramb SM, Dunn NAM, Muller JM, Pyke CM, Baade PD. The descriptive epidemiology of female breast cancer: an international comparison of screening, incidence, survival and mortality. Cancer Epidemiology. 2012;36(3):237–248. doi: 10.1016/j.canep.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Tirona MT, Sehgal R, Ballester O. Prevention of breast cancer (Part I): epidemiology, risk factors, and risk assessment tools. Cancer Investigation. 2010;28(7):743–750. doi: 10.3109/07357907.2010.494321. [DOI] [PubMed] [Google Scholar]
- 4.Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Research. 1976;36(7):2699–2702. [PubMed] [Google Scholar]
- 5.O’Shaughnessy JA, Kelloff GJ, Gordon GB, et al. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development: recommendations of the American association for cancer research Task force on the Treatment and Prevention of intraepithelial neoplasia. Clinical Cancer Research. 2002;8(2):314–346. [PubMed] [Google Scholar]
- 6.Braakhuis BJM, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Research. 2003;63(8):1727–1730. [PubMed] [Google Scholar]
- 7.Althuis MD, Fergenbaum JH, Garcia-Closas M, Brinton LA, Madigan MP, Sherman ME. Etiology of hormone receptor-defined breast cancer: a systematic review of the literature. Cancer Epidemiology Biomarkers and Prevention. 2004;13(10):1558–1568. [PubMed] [Google Scholar]
- 8.Pelekanou V, Leclercq G. Recent insights into the effect of natural and environmental estrogens on mammary development and carcinogenesis. International Journal of Developmental Biology. 2011;55(7-9):869–878. doi: 10.1387/ijdb.113369vp. [DOI] [PubMed] [Google Scholar]
- 9.Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Advanced Drug Delivery Reviews. 2010;62(13):1265–1276. doi: 10.1016/j.addr.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan VC. Selective estrogen receptor modulation: a personal perspective. Cancer Research. 2001;61(15):5683–5687. [PubMed] [Google Scholar]
- 11.Early Breast Cancer Trialists’ Collaborative Groups. Tamoxifen for early breast cancer: an overview of the randomized trials. The Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 12.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 study. Journal of the National Cancer Institute. 1998;90(18):1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 13.King MC, Wieand S, Hale K, et al. Tamoxifen and breast cancer incidence among women with inherited mutations in brca1 and brca2 national surgical adjuvant breast and bowel project (nsabp-p1) breast cancer prevention trial. Journal of the American Medical Association. 2001;286(18):2251–2256. doi: 10.1001/jama.286.18.2251. [DOI] [PubMed] [Google Scholar]
- 14.Cuzick J. First results from the International Breast Cancer Intervention Study (IBIS-I): a randomised prevention trial. The Lancet. 2002;360(9336):817–824. doi: 10.1016/s0140-6736(02)09962-2. [DOI] [PubMed] [Google Scholar]
- 15.Cuzick J, Powles T, Veronesi U, et al. Overview of the main outcomes in breast-cancer prevention trials. The Lancet. 2003;361(9354):296–300. doi: 10.1016/S0140-6736(03)12342-2. [DOI] [PubMed] [Google Scholar]
- 16.Decensi A, Bonanni B, Guerrieri-Gonzaga A, et al. Biologic activity of tamoxifen at low doses in healthy women. Journal of the National Cancer Institute. 1998;90(19):1461–1467. doi: 10.1093/jnci/90.19.1461. [DOI] [PubMed] [Google Scholar]
- 17.Johnston SRD, Haynes BP, Sacks NPM, et al. Effect of oestrogen receptor status and time on the intra-tumoural accumulation of tamoxifen and N-desmethyltamoxifen following short-term therapy in human primary breast cancer. Breast Cancer Research and Treatment. 1993;28(3):241–250. doi: 10.1007/BF00666585. [DOI] [PubMed] [Google Scholar]
- 18.Guerrieri-Gonzaga A, Baglietto L, Johansson H, et al. Correlation between tamoxifen elimination and biomarker recovery in a primary prevention trial. Cancer Epidemiology Biomarkers and Prevention. 2001;10(9):967–970. [PubMed] [Google Scholar]
- 19.Decensi A, Robertson C, Viale G, et al. A randomized trial of low-dose tamoxifen on breast cancer proliferation and blood estrogenic biomarkers. Journal of the National Cancer Institute. 2003;95(11):779–790. doi: 10.1093/jnci/95.11.779. [DOI] [PubMed] [Google Scholar]
- 20.Barrett-Connor E, Mosca L, Collins P, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. New England Journal of Medicine. 2006;355(2):125–137. doi: 10.1056/NEJMoa062462. [DOI] [PubMed] [Google Scholar]
- 21.Martino S, Cauley JA, Barrett-Connor E, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. Journal of the National Cancer Institute. 2004;96:1751–1761. doi: 10.1093/jnci/djh319. [DOI] [PubMed] [Google Scholar]
- 22.Sporn MB, Dowsett SA, Mershon J, Bryant HU. Role of raloxifene in breast cancer prevention in postmenopausal women: clinical evidence and potential mechanisms of action. Clinical Therapeutics. 2004;26(6):830–840. doi: 10.1016/s0149-2918(04)90127-0. [DOI] [PubMed] [Google Scholar]
- 23.Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Journal of the American Medical Association. 1999;281(23):2189–2197. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 24.Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Journal of the American Medical Association. 1999;282(7):637–645. doi: 10.1001/jama.282.7.637. [DOI] [PubMed] [Google Scholar]
- 25.Cauley JA, Norton L, Lippman ME, et al. Continued breast cancer risk reduction in postmenopausal women treated with raloxifene: 4-Year results from the MORE trial. Breast Cancer Research and Treatment. 2001;65(2):125–134. doi: 10.1023/a:1006478317173. [DOI] [PubMed] [Google Scholar]
- 26.Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. Journal of the American Medical Association. 2006;295(23):2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 27.Mosca L, Barrett-Connor E, Wenger NK, et al. Design and methods of the Raloxifene Use for The Heart (RUTH) study. American Journal of Cardiology. 2001;88(4):392–395. doi: 10.1016/s0002-9149(01)01685-x. [DOI] [PubMed] [Google Scholar]
- 28.Coates AS, Keshaviah A, Thürlimann B, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. Journal of Clinical Oncology. 2007;25(5):486–492. doi: 10.1200/JCO.2006.08.8617. [DOI] [PubMed] [Google Scholar]
- 29.Coombes R, Kilburn L, Snowdon C, et al. Survival and safety of exemestane versus tamoxifen after 2-3 years’ tamoxifen treatment (Intergroup Exemestane Study): a randomised controlled trial. The Lancet. 2007;369(9561):559–570. doi: 10.1016/S0140-6736(07)60200-1. [DOI] [PubMed] [Google Scholar]
- 30.Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. New England Journal of Medicine. 2003;349(19):1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 31.Baum M, Buzdar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. The Lancet. 2002;359(9324):2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 32.Howell A, Cuzick J, Baum M. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years' adjuvant treatment for breast cancer. The Lancet. 2005;365(9453):60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 33.Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. New England Journal of Medicine. 2004;350(11):1081–1092. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 34.Perez EA. Safety profiles of tamoxifen and the aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Annals of Oncology. 2007;18(supplement 8):viii26–viii35. doi: 10.1093/annonc/mdm263. [DOI] [PubMed] [Google Scholar]
- 35.Cuzick J. Aromatase inhibitors for breast cancer prevention. Journal of Clinical Oncology. 2005;23(8):1636–1643. doi: 10.1200/JCO.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 36.Goss PE, Strasser-Weippl K, Brown M, Santen R, Ingle J, Bissell M. Prevention strategies with aromatase inhibitors. Clinical Cancer Research. 2004;10(1):372S–379S. doi: 10.1158/1078-0432.ccr-031210. [DOI] [PubMed] [Google Scholar]
- 37.Cuzick J. IBIS II: a breast cancer prevention trial in postmenopausal women using the aromatase inhibitor anastrozole. Expert Review of Anticancer Therapy. 2008;8(9):1377–1385. doi: 10.1586/14737140.8.9.1377. [DOI] [PubMed] [Google Scholar]
- 38.Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. Journal of the American Medical Association. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 39.Moon RC, Thompson HJ, Becci PJ. N-(4-hydroxyphenyl)retinamide, a new retinoid for prevention of breast cancer in the rat. Cancer Research. 1979;39(4):1339–1346. [PubMed] [Google Scholar]
- 40.Budd GT, Adamson PC, Gupta M, et al. Phase I/II trial of all-trans retinoic acid and tamoxifen in patients with advanced breast cancer. Clinical Cancer Research. 1998;4(3):635–642. [PubMed] [Google Scholar]
- 41.Decensi A, Bonanni B, Baglietto L, et al. A two-by-two factorial trial comparing oral with transdermal estrogen therapy and fenretinide with placebo on breast cancer biomarkers. Clinical Cancer Research. 2004;10(13):4389–4397. doi: 10.1158/1078-0432.CCR-04-0087. [DOI] [PubMed] [Google Scholar]
- 42.Cazzaniga M, Varricchio C, Montefrancesco C, Feroce I, Guerrieri-Gonzaga A. Fenretinide (4-HPR): A preventive chance for women at genetic and familial risk? Journal of Biomedicine and Biotechnology. 2012;2012:9 pages. doi: 10.1155/2012/172897.172897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veronesi U, De Palo G, Marubini E, et al. Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. Journal of the National Cancer Institute. 1999;91(21):1847–1856. doi: 10.1093/jnci/91.21.1847. [DOI] [PubMed] [Google Scholar]
- 44.Veronesi U, Mariani L, Decensi A, et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Annals of Oncology. 2006;17(7):1065–1071. doi: 10.1093/annonc/mdl047. [DOI] [PubMed] [Google Scholar]
- 45.Wu K, Kim HT, Rodriquez JL, et al. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiology Biomarkers and Prevention. 2002;11(5):467–474. [PubMed] [Google Scholar]
- 46.Wu K, Zhang Y, Xu XC, et al. The retinoid X receptor-selective retinoid, LGD1069, prevents the development of estrogen receptor-negative mammary tumors in transgenic mice. Cancer Research. 2002;62(22):6376–6380. [PubMed] [Google Scholar]
- 47.Lu C, Speers C, Zhang Y, et al. Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumors. Journal of the National Cancer Institute. 2003;95(24):1825–1833. doi: 10.1093/jnci/djg117. [DOI] [PubMed] [Google Scholar]
- 48.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nature Reviews Cancer. 2006;6(9):714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 49.Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. Journal of Clinical Oncology. 1993;11(10):1936–1942. doi: 10.1200/JCO.1993.11.10.1936. [DOI] [PubMed] [Google Scholar]
- 50.Tandon AK, Clark GM, Chamness GC, Ullrich A, McGuire WL. HER-2/neu oncogene protein and prognosis in breast cancer. Journal of Clinical Oncology. 1989;7(8):1120–1128. doi: 10.1200/JCO.1989.7.8.1120. [DOI] [PubMed] [Google Scholar]
- 51.Konecny G, Pauletti G, Pegram M, et al. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. Journal of the National Cancer Institute. 2003;95(2):142–153. doi: 10.1093/jnci/95.2.142. [DOI] [PubMed] [Google Scholar]
- 52.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses HER2. New England Journal of Medicine. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 53.Brufsky A, Lembersky B, Schiffman K, Lieberman G, Paton VE. Hormone receptor status does not affect the clinical benefit of trastuzumab therapy for patients with metastatic breast cancer. Clinical Breast Cancer. 2005;6(3):247–252. doi: 10.3816/CBC.2005.n.027. [DOI] [PubMed] [Google Scholar]
- 54.Buzdar AU, Valero V, Ibrahim NK, et al. Neoadjuvant therapy with paclitaxel followed by 5-fluorouracil, epirubicin, and cyclophosphamide chemotherapy and concurrent trastuzumab in human epidermal growth factor receptor 2-positive operable breast cancer: an update of the initial randomized study population and data of additional patients treated with the same regimen. Clinical Cancer Research. 2007;13(1):228–233. doi: 10.1158/1078-0432.CCR-06-1345. [DOI] [PubMed] [Google Scholar]
- 55.Gianni L, Eiermann W, Semiglazov V, et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. The Lancet. 2010;375(9712):377–384. doi: 10.1016/S0140-6736(09)61964-4. [DOI] [PubMed] [Google Scholar]
- 56.Baselga J, Perez EA, Pienkowski T, Bell R. Adjuvant trastuzumab: a milestone in the treatment of HER-2-positive early breast cancer. Oncologist. 2006;11(1):4–12. doi: 10.1634/theoncologist.11-90001-4. [DOI] [PubMed] [Google Scholar]
- 57.Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors—a review on pharmacology, metabolism and side effects. Current Drug Metabolism. 2009;10(5):470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- 58.Cameron D, Casey M, Press M, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Research and Treatment. 2008;112(3):533–543. doi: 10.1007/s10549-007-9885-0. [DOI] [PubMed] [Google Scholar]
- 59.Lin NU, Carey LA, Liu MC, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. Journal of Clinical Oncology. 2008;26(12):1993–1999. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Leo A, Gomez HL, Aziz Z, et al. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. Journal of Clinical Oncology. 2008;26(34):5544–5552. doi: 10.1200/JCO.2008.16.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strecker TE, Shen Q, Zhang Y, et al. Effect of lapatinib on the development of estrogen receptor-negative mammary tumors in mice. Journal of the National Cancer Institute. 2009;101(2):107–113. doi: 10.1093/jnci/djn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocrine-Related Cancer. 2004;11(4):689–708. doi: 10.1677/erc.1.00600. [DOI] [PubMed] [Google Scholar]
- 63.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clinical Cancer Research. 2001;7(10):2958–2970. [PubMed] [Google Scholar]
- 64.Agrawal A, Gutteridge E, Gee JMW, Nicholson RI, Robertson JFR. Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocrine-Related Cancer. 2005;12(1):S135–S144. doi: 10.1677/erc.1.01059. [DOI] [PubMed] [Google Scholar]
- 65.Dickler MN, Rugo HS, Eberle C, et al. A phase II trial of erlotinib in combination with bevacizumab in patients with metastatic breast cancer. Clinical Cancer Research. 2008;14(23):7878–7883. doi: 10.1158/1078-0432.CCR-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chan KC, Knox WF, Gee JM, et al. Effect of epidermal growth factor receptor tyrosine kinase inhibition on epithelial proliferation in normal and premalignant breast. Cancer Research. 2002;62(1):122–128. [PubMed] [Google Scholar]
- 67.Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Research. 2002;62(6):1676–1681. [PubMed] [Google Scholar]
- 68.Thun MJ, Jane Henley S, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. Journal of the National Cancer Institute. 2002;94(4):252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 69.Cuzick J, Otto F, Baron JA, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. The Lancet Oncology. 2009;10(5):501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 70.Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. Ca-A Cancer Journal for Clinicians. 2006;56(2):69–83. doi: 10.3322/canjclin.56.2.69. [DOI] [PubMed] [Google Scholar]
- 71.Shim V, Gauthier ML, Sudilovsky D, et al. Cyclooxygenase-2 expression is related to nuclear grade in ductal carcinoma in situ and is increased in its normal adjacent epithelium. Cancer Research. 2003;63(10):2347–2350. [PubMed] [Google Scholar]
- 72.Khuder SA, Mutgi AB. Breast cancer and NSAID use: a meta-analysis. British Journal of Cancer. 2001;84(9):1188–1192. doi: 10.1054/bjoc.2000.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takkouche B, Regueira-Méndez C, Etminan M. Breast cancer and use of nonsteroidal anti-inflammatory drugs: a meta-analysis. Journal of the National Cancer Institute. 2008;100(20):1439–1447. doi: 10.1093/jnci/djn324. [DOI] [PubMed] [Google Scholar]
- 74.Harris RE, Alshafie GA, Abou-Issa H, Seibert K. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Research. 2000;60(8):2101–2103. [PubMed] [Google Scholar]
- 75.Nakatsugi S, Ohta T, Kawamori T, et al. Chemoprevention by nimesulide, a selective cyclooxygenase-2 inhibitor, of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary gland carcinogenesis in rats. Japanese Journal of Cancer Research. 2000;91(9):886–892. doi: 10.1111/j.1349-7006.2000.tb01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Russell RGG. Bisphosphonates: the first 40 years. Bone. 2011;49(1):2–19. doi: 10.1016/j.bone.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 77.Rodan GA, Fleisch HA. Bisphosphonates: mechanisms of action. Journal of Clinical Investigation. 1996;97(12):2692–2696. doi: 10.1172/JCI118722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brufsky A, Harker WG, Beck JT, et al. Zoledronic acid inhibits adjuvant letrozole-induced bone loss in postmenopausal women with early breast cancer. Journal of Clinical Oncology. 2007;25(7):829–836. doi: 10.1200/JCO.2005.05.3744. [DOI] [PubMed] [Google Scholar]
- 79.Hall TJ, Schaueblin M. A pharmacological assessment of the mammalian osteoclast vacuolar H+-ATPase. Bone and Mineral. 1994;27(2):159–166. doi: 10.1016/s0169-6009(08)80217-6. [DOI] [PubMed] [Google Scholar]
- 80.Rennert G, Pinchev M, Rennert HS. Use of bisphosphonates and risk of postmenopausal breast cancer. Journal of Clinical Oncology. 2010;28(22):3577–3581. doi: 10.1200/JCO.2010.28.1113. [DOI] [PubMed] [Google Scholar]
- 81.Chlebowski RT, Chen Z, Cauley JA, et al. Oral bisphosphonate use and breast cancer incidence in postmenopausal women. Journal of Clinical Oncology. 2010;28(22):3582–3590. doi: 10.1200/JCO.2010.28.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Amé JC, Spenlehauer C, De Murcia G. The PARP superfamily. BioEssays. 2004;26(8):882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 83.Dantzer F, Amé JC, Schreiber V, Nakamura J, Ménissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods in Enzymology. 2006;409:493–510. doi: 10.1016/S0076-6879(05)09029-4. [DOI] [PubMed] [Google Scholar]
- 84.Amé JC, Rolli V, Schreiber V, et al. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. Journal of Biological Chemistry. 1999;274(25):17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 85.Peralta-Leal A, Rodríguez-Vargas JM, Aguilar-Quesada R, et al. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radical Biology and Medicine. 2009;47(1):13–26. doi: 10.1016/j.freeradbiomed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 86.Calvert H, Azzariti A. The clinical development of inhibitors of poly(ADP-ribose) polymerase. Annals of Oncology. 2011;22(1):i53–i59. doi: 10.1093/annonc/mdq667. [DOI] [PubMed] [Google Scholar]
- 87.Rehman FL, Lord CJ, Ashworth A. Synthetic lethal approaches to breast cancer therapy. Nature Reviews Clinical Oncology. 2010;7(12):718–724. doi: 10.1038/nrclinonc.2010.172. [DOI] [PubMed] [Google Scholar]
- 88.Iglehart JD, Silver DP. Synthetic lethality—a new direction in cancer-drug development. New England Journal of Medicine. 2009;361(2):189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 89.Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clinical Cancer Research. 2008;14(12):3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 90.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 91.O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. New England Journal of Medicine. 2011;364(3):205–214. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 92.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 93.Xue F, Michels KB. Diabetes, metabolic syndrome, and breast cancer: a review of the current evidence. The American Journal of Clinical Nutrition. 2007;86(3):s823–835. doi: 10.1093/ajcn/86.3.823S. [DOI] [PubMed] [Google Scholar]
- 94.Hsu IR, Kim SP, Kabir M, Bergman RN. Metabolic syndrome, hyperinsulinemia, and cancer. The American Journal of Clinical Nutrition. 2007;86(3):s867–871. doi: 10.1093/ajcn/86.3.867S. [DOI] [PubMed] [Google Scholar]
- 95.Goodwin PJ, Ennis M, Pritchard KI, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. Journal of Clinical Oncology. 2002;20(1):42–51. doi: 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- 96.Pasanisi P, Berrino F, De Petris M, Venturelli E, Mastroianni A, Panico S. Metabolic syndrome as a prognostic factor for breast cancer recurrences. International Journal of Cancer. 2006;119(1):236–238. doi: 10.1002/ijc.21812. [DOI] [PubMed] [Google Scholar]
- 97.Irwin ML, McTiernan A, Bernstein L, et al. Relationship of obesity and physical activity with C-peptide, leptin, and insulin-like growth factors in breast cancer survivors. Cancer Epidemiology Biomarkers and Prevention. 2005;14(12):2881–2888. doi: 10.1158/1055-9965.EPI-05-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gunter MJ, Hoover DR, Yu H, et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. Journal of the National Cancer Institute. 2009;101(1):48–60. doi: 10.1093/jnci/djn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nature Reviews Cancer. 2004;4(7):505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 100.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Reviews. 2009;30(6):586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 101.Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. The Lancet Oncology. 2005;6(2):103–111. doi: 10.1016/S1470-2045(05)01736-5. [DOI] [PubMed] [Google Scholar]
- 102.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature Reviews Cancer. 2008;8(12):915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 103.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. British Medical Journal. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 105.DeCensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prevention Research. 2010;3(11):1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 106.Sahra IB, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 107.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Research. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 108.Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERα negative MDA-MB-435 breast cancer model. Breast Cancer Research and Treatment. 2009;113(1):101–111. doi: 10.1007/s10549-008-9916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cazzaniga M, Bonanni B, Guerrieri-Gonzaga A, Decensi A. Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiology Biomarkers and Prevention. 2009;18(3):701–705. doi: 10.1158/1055-9965.EPI-08-0871. [DOI] [PubMed] [Google Scholar]
- 110.Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-κB in cell growth regulation. American Journal of Pathology. 2001;159(2):387–397. doi: 10.1016/s0002-9440(10)61708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lenardo MJ, Baltimore D. NK-κB: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58(2):227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 112.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 113.Karin M. NF-κB and cancer: mechanisms and targets. Molecular Carcinogenesis. 2006;45(6):355–361. doi: 10.1002/mc.20217. [DOI] [PubMed] [Google Scholar]
- 114.Bowie A, O’Neill LAJ. Oxidative stress and nuclear factor-κB activation: a reassessment of the evidence in the light of recent discoveries. Biochemical Pharmacology. 2000;59(1):13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 115.Toledano MB, Leonard WJ. Modulation of transcription factor NF-κB binding activity by oxidation-reduction in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(10):4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 117.Ahmad N, Gupta S, Mukhtar H. Green tea polyphenol epigallocatechin-3-gallate differentially modulates nuclear factor κB in cancer cells versus normal cells. Archives of Biochemistry and Biophysics. 2000;376(2):338–346. doi: 10.1006/abbi.2000.1742. [DOI] [PubMed] [Google Scholar]
- 118.Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101(3):1053–1062. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 119.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20(23):2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 120.Li Y, Sarkar FH. Inhibition of nuclear factor κB activation in PC3 cells by genistein is mediated via Akt signaling pathway. Clinical Cancer Research. 2002;8(7):2369–2377. [PubMed] [Google Scholar]
- 121.Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by Akt and NF-kappaB pathways in prostate cancer cells. Frontiers in Bioscience. 2005;10(1):236–243. doi: 10.2741/1523. [DOI] [PubMed] [Google Scholar]
- 122.Brahimi-Horn MC, Pouysségur J. The hypoxia-inducible factor and tumor progression along the angiogenic pathway. International Review of Cytology. 2004;242:157–213. doi: 10.1016/S0074-7696(04)42004-X. [DOI] [PubMed] [Google Scholar]
- 123.Esteban MA, Maxwell PH. HIF, a missing link between metabolism and cancer. Nature Medicine. 2005;11(10):1047–1048. doi: 10.1038/nm1005-1047. [DOI] [PubMed] [Google Scholar]
- 124.Semenza GL. Targeting HIF-1 for cancer therapy. Nature Reviews Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 125.Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nature Reviews Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 126.López-Lázaro M. HIF-1: hypoxia-inducible factor or dysoxia-inducible factor? FASEB Journal. 2006;20(7):828–832. doi: 10.1096/fj.05-5168hyp. [DOI] [PubMed] [Google Scholar]
- 127.Semenza GL. Development of novel therapeutic strategies that target HIF-1. Expert Opinion on Therapeutic Targets. 2006;10(2):267–280. doi: 10.1517/14728222.10.2.267. [DOI] [PubMed] [Google Scholar]
- 128.Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nature Reviews Drug Discovery. 2003;2(10):803–811. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- 129.Nagle DG, Zhou YD. Natural product-based inhibitors of hypoxia-inducible factor-1 (HIF-1) Current Drug Targets. 2006;7(3):355–369. doi: 10.2174/138945006776054979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yeo EJ, Chun YS, Park JW. New anticancer strategies targeting HIF-1. Biochemical Pharmacology. 2004;68(6):1061–1069. doi: 10.1016/j.bcp.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 131.Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Research. 1999;59(22):5830–5835. [PubMed] [Google Scholar]
- 132.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nature Reviews Molecular Cell Biology. 2004;5(5):343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 133.Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. Journal of the National Cancer Institute. 2004;96(12):926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 134.Mondesire WH, Jian W, Zhang H, et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clinical Cancer Research. 2004;10(20):7031–7042. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 135.Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22(21):3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 136.Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Research. 2006;8(6, article 215) doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 138.Raymond E, Alexandre J, Faivre S, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. Journal of Clinical Oncology. 2004;22(12):2336–2347. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 139.Yu K, Toral-Barza L, Discafani C, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocrine-Related Cancer. 2001;8(3):249–258. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 140.Li H, Lin J, Wang X, et al. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. doi: 10.1007/s10549-012-2036-2. Breast Cancer Research and Treatment. In press. [DOI] [PubMed] [Google Scholar]
- 141.Liu M, Howes A, Lesperance J, et al. Antitumor activity of rapamycin in a transgenic mouse model of ErbB2-dependent human breast cancer. Cancer Research. 2005;65(12):5325–5336. doi: 10.1158/0008-5472.CAN-04-4589. [DOI] [PubMed] [Google Scholar]
- 142.Namba R, Young LJT, Abbey CK, et al. Rapamycin inhibits growth of premalignant and malignant mammary lesions in a mouse model of ductal carcinoma in situ. Clinical Cancer Research. 2006;12(8):2613–2621. doi: 10.1158/1078-0432.CCR-05-2170. [DOI] [PubMed] [Google Scholar]