

Abstract
Huntington's disease (HD), caused by an expanded triplet repeat in the huntingtin (Htt) gene, results in extensive neuropathology, but study of the Htt gene in CNS development through gene knockout is problematic as the knockout leads to embryonic lethality in mice. Here, we report that the knockdown of Htt expression in neuroepithelial cells of neocortex results in disturbed cell migration, reduced proliferation, and increased cell death that is relatively specific to early neural development. In the cerebellum, however, Htt knockdown results in cell death but not perturbed migration. The cell death phenotype in cortex can be partially reversed with co-knockdown of Casp9, indicating that mitochondria-mediated cell apoptotic processes are involved in the neuronal death. The timing of knockdown during early development is also an important variable. These results indicate a spatial and temporal requirement for Htt expression in neural development. Although it is uncertain whether the loss of wild-type huntingtin function contributes to pathogenesis in Huntington's disease, these results clearly contraindicate the use of nonspecific knockdown of Htt as a therapeutic measure in HD, particularly in utero.
Introduction
An expanded CAG repeat in the huntingtin (Htt) gene, which is ubiquitously expressed, is causal for marked neurodegeneration in the striatum and cortex and elsewhere in the brain of mid-aged humans, and leads to death in an uncompromising fashion. The role that the protein derived from the normal Htt allele plays in neural development has not been delineated. Using Htt knock-out mice, it has been found that Htt is required for early embryonic development (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995). Embryonic stem cells with the Htt deletion, however, when injected into the blastocyst, can participate in embryonic development, and chimeric mice with null and wild-type cells survive (Dragatsis et al., 1998). We found that Htt−/− cells in such chimeras can readily survive in certain regions of the brain, but are greatly underrepresented in cortical and striatal areas, the major targets of Huntington's disease (HD) (Reiner et al., 2001). In embryonic day 12.5 (E12.5) blastocyst injection chimeras, we found that Htt−/− neuroblasts are as abundant in cortex, striatum, and thalamus as they are in mid-brain and hindbrain regions (Reiner et al., 2003) However, the mutant cells show evidence of pathology (i.e., apparent cell death) at this early stage in development. These studies suggested that Htt is needed specifically for development of cortical and striatal neurons.
In this study, to determine the temporal and spatial requirement of Htt in neuronal development, we used shRNA to knock down its expression at different time points early in development. We examined two regions of the brain, one where our previous data indicated a critical role for Htt (i.e., cortex), and one where we found that Htt-null cells seemed to have uncompromised survival (i.e., cerebellum). Our results revealed that Htt plays a critical role for neuron survival, proliferation, and neuronal migration in early cortical development; and for survival but not migration in cerebellar development.
Materials and Methods
Mice and in utero transfection
Htt shRNA and EGFP (the shRNA experimental condition) or unrelated control shRNA and EGFP plasmids (the control condition) were cointroduced into the neural epithelium of the lateral or fourth ventricle of timed-pregnant ICR mice (originally from Charles River) using an in utero electroporation method described previously (Tabata and Nakajima, 2001). The use of mice was in accord with Society for Neuroscience guidelines and Institutional Animal Care and Use Committee-approved protocols.
Design of shRNA
The mU6pro vector with a mU6 promoter was a kind gift from Drs. Jenn-Yah Yu and Dave Turner (Yu et al., 2002). Four sequences of the mouse Htt and three sequences of the mouse Casp9 were designed by using GenScript (www.genscript.com/ssl-bin/app/sirna, Htt-204: 5′-gaaggaactctcagccaccaa-3′; Htt-2502: 5′-gcctctgaagaacagctccta-3′; Htt-4908: 5′-gttcatcactccaagcacaat-3′; Htt-5596: 5′-ggaatgtgcaatagagaaata-3′; Casp9-124: 5′-gatattcagcaggcaggatct-3′; Casp9-469: 5′-gaggttctcagaccagaaaca-3′; Casp9-554: 5′-gacatgcagatatggcataca-3′). An unrelated shRNA made from the Mcm5 gene (Mcm5-86: 5′-gcaactgcagaggcgattcaa-3′) that is expressed in the neuroepithelium of cortex and cerebellum was used as a control.
Processing of embryos for histology
Embryos were killed 2–3 d following electroporation, and the fetal brains were immersion fixed in 4% paraformaldehyde in 0.1 m PBS for 6 h at 4°C. To study cell proliferation, BrdU (50 μg/g body weight, Sigma) was injected intraperitoneally 1 h before embryos were killed. Brains were cryoprotected, sectioned (14 μm), and mounted on slides. To demonstrate cell bodies, propidium iodine or Toto3 (Invitrogen) was used. To detect the HTT protein, the sections were preincubated with 1× PBS containing 0.2% Triton-100 (PBS-T) and 5% normal goat serum for 2 h at room temperature, and incubated with the anti-HTT antibody (1:400; MAB2166, Millipore Bioscience Research Reagents) overnight. The rinsed sections were then incubated with goat-anti-mouse antibody conjugated with Alexa Fluor 595 for 1.5 h at room temperature. Tissue was coverslipped and fluorescence preserved with FluorSave (Calbiochem). For BrdU staining, sections were treated with 1 m HCl for 30 min at 37°C to denature DNA, rinsed three times with 0.1 m PBS, and immunostained as described above with the BrdU antibody (1:200; catalog #ab6326, Abcam). For caspase immunostaining, sections were antigen retrieved in 0.01 m NaOAC buffer, pH 5.8, in a microwave oven for 15 min and washed in PBS-T buffer three times. The sections were incubated with anti-cleaved CASP3 (anti-cCASP3, 1:400; catalog #9664, Cell Signaling Technology) or CASP9 antibody (anti-CASP9, 1:1000; catalog #M054-3, MBL).
Quantitative analyses
Effect of shRNA on protein expression.
Immunofluorescent images obtained with a confocal microscope (60×) were analyzed using “Analysis” 3.0 software (Soft Imaging System Corp.). An elliptical window was extended to cover each identified transfected or control cell, and optical density was measured. Data were obtained from four fetuses with a minimum of 40 cells tallied in each group.
Assessing cellular phenotypes of migration and death.
As plasmid injection and transfection are inherently variable, only samples that showed robust labeling were processed for quantification. Furthermore, as the quantification was made from digital images, only sections that yielded high-quality images were used for analysis. At least two digital images were counted for each embryo. Three to 10 fetuses were analyzed for each manipulation and time point. To assess cellular migration, the number of EGFP-positive cell bodies was counted in the cortical plate (CP) compared with the combined number of cells in the intermediate zone (IZ), subventricular zone (SVZ), and ventricular zone (VZ). To examine cell death or proliferation, EGFP+ cells immunopositive for anti-cleaved CASP3 or anti-BrdU were counted, and the percentage of apoptotic or proliferating cells (mean ± SEM) was determined by calculating the ratio of double-positive cells over all transfected cells in the confocal images.
Statistical analysis
For statistical analysis, Student's t test was performed for pairwise comparison between Htt shRNA knockdown and a control group, or Htt shRNA and Htt shRNA+Casp9 shRNA group.
Results
Htt shRNA abolishes normal cell migration from the VZ to the CP of the developing cerebral cortex
The E12.5 time point was assessed in control and Htt-shRNA-transfected embryos as this was the time when we observed apoptotic Htt-null cells in cortex (Reiner et al., 2003). In control E12.5 embryos, with 72 h of survival after EGFP plasmid was injected into the lateral ventricle, 82% of all EGFP-labeled cells had migrated to the CP, and many axons were present in the IZ (Fig. 1A). Four Htt shRNA constructs were separately injected to determine their effect upon cortical development. In all experimental cases, transfected cells were able to leave the VZ/SVZ region but then demonstrated variable pathologies in progressing toward the CP. Construct Htt-5596 had the most profound effect on cell migration, resulting in 90 ± 1% of the transfected cells remaining deep in the IZ and SVZ (p < 0.001; Fig. 1B). Even the few cells that entered the CP could only migrate into the deep CP region. As the Htt-5596 construct yielded the greatest developmental defect, this construct was used in all subsequent experiments.
Figure 1.
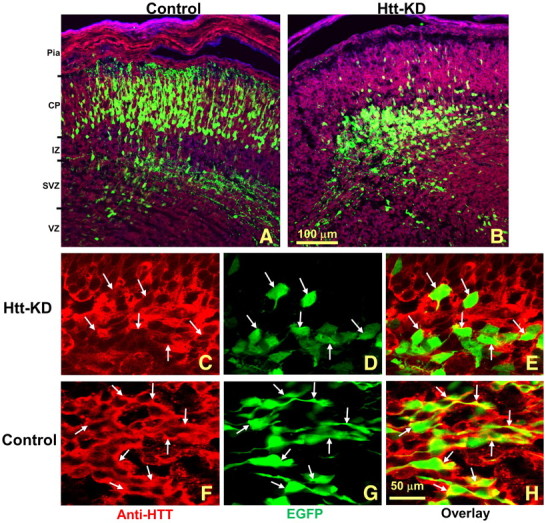
Htt shRNA disrupts the migration of neurons in the developing cortex. A, The normal development of cortical neurons is shown at E15.5 following transfection with EGFP at E12.5. More than 80% of the EGFP-labeled cells have migrated from the VZ to the CP. B, Transfection of Htt shRNA inhibited the number of cells that successfully migrated to the CP. Pia, Pia mater. C–H, Htt shRNA significantly reduced Htt immunostaining in transfected cells (C–E); anti-HTT immunostaining is not changed in the control (F–H). Note that yellow, indicating costaining with EGFP and HTT, is apparent in the control (H) but is not observed in the Htt shRNA-transfected group (E); white arrows indicate individual transfected cells. Scale bars: A, B, 200 μm; C–H, 50 μm.
Brains transfected with EGFP at E12.5 and harvested 48 h later contained cells in cortex with moderate to strong anti-HTT immunostaining in the cytosol and processes (Fig. 1F). There was no obvious change in the HTT immunofluorescent signal between EGFP-labeled and unlabeled cells in control embryos (Fig. 1F–H). In the shRNA-transfected cortex (Htt-5596), EGFP-negative cells had similar HTT expression to that from control embryos. However, adjacent EGFP-positive cells were only weakly immunostained for HTT (Fig. 1C). The intensity of the fluorescent staining revealed an average 51% reduction in immunofluorescent signal in transfected cells for the whole cell body (n = 43 cells, p < 0.001). If only cytosol expression (whole cell body minus nucleus area) was considered, where HTT was primarily localized, the average reduction was 75% of control HTT levels (n = 43 cells, p < 0.001), with only a limited overlap between immunofluorescent HTT and EGFP signals (Fig. 1C–E).
Knockdown of Htt leads to a time-dependent defect in cell migration and apoptosis in the developing cortex
When there is a prolific movement of cortical neuroblasts from the ventricular zone to the cortical plate, the temporal window is limited (Takahashi et al., 1996). In control tissue, this age-dependent process was clearly visible (Fig. 2A,C,E). When the control shRNA construct was introduced into E12.5 fetal neuroepithelia, 85 ± 1% of the labeled cells migrated into the CP at 72 h of survival (n = 8; Fig. 3A). In contrast, only 24 ± 3.5% of the transfected cells migrated into the CP when fetuses were injected on E14.5 with 72 h of survival (n = 3; Fig. 2C), and migration was further reduced with control injection at E16.5 (survival for 72 h; n = 4), with only 4 ± 1% of EGFP-labeled cells reaching the CP (Fig. 2E).
Figure 2.

The effect of Htt shRNA on cell migration and survival is time dependent in the developing cerebral cortex. A–O, Htt shRNA transfection was performed at E12.5 (A, B, G–I), E14.5 (C, D, J–L), and E16.5 (E, F, M–O) with 72 h survival. For E12.5 transfection, while many EGFP-labeled control cells migrate to the CP (A), Htt shRNA-transfected cells remain in the IZ (B). For E14.5 and E16.5 transfection, control transfected cells have left the VZ and largely reside in the IZ, with some cells present in the CP (C, E). Following Htt shRNA at these time points, most EGFP-labeled cells still remain in the VZ or the nearby SVZ (D, F). Pia, Pia mater. G–O, Higher-magnification confocal images show that cleaved CASP3-positive cells were also Htt shRNA positive and were easily detectible in the E12.5 condition (G–I, arrows). The apoptotic cells were greatly reduced at E14.5 (J–L, arrows), and only a few were observed at E16.5 transfection (M–O). Red, Anti-cleaved CASP3 immunostaining (anti-cCASP3); green, EGFP. Scale bars: (in E) A–F, 200 μm; (in O) G–O, 50 μm.
Figure 3.

Htt shRNA does not impact cell migration but leads to cell death in the developing cerebellum. Htt shRNA was introduced into the cerebellar neuroepithelium at E12.5 and examined 48 h later. A, As in controls (data not shown), abundant numbers of transfected cells have left the neuroepithelium and traveled toward the region of the differentiating zone. EGFP-labeled cells have well formed processes and surround the cerebellar nuclei. B–D, Higher-magnification confocal images of anti-cleaved CASP3 immunostaining (B), EGFP (C), and costaining (D) that show cell death in transfected neurons. CN, Cerebellar nuclei; ITN, inferior tectal neuroepithelium; NE, cerebellum neuroepithelium; DZ, differentiating zone. Scale bars: A, 100 μm; B–D, 50 μm.
When the Htt shRNA was introduced into neuroepithelial cells of E12.5 embryos and examined 72 h later (n = 9), cell migration was greatly reduced in the transfected cells, with only 10 ± 1% making their way to the CP compared with 85 ± 1% in the control (p < 0.001; Fig. 2B). With Htt shRNA introduced into E14.5 and 16.5 fetuses and examined 72 h later (n = 11 and 4, respectively), the percentage of neurons that successfully migrated was also diminished [9 ± 1% and 1% compared with 24 ± 3.5% (p < 0.05) and 4 ± 1% (p < 0.05) in the control groups, respectively; Fig. 2D,F]. In addition to its disruptive effect on cell migration into the developing CP, many Htt-5596-transfected cells along the migratory pathway from the VZ to the IZ were also seen with condensed chromatin at 48 h (n = 14) or 72 h (n = 37). Using an anti-cleaved CASP3 antibody to mark cells that have entered the cell death pathway, we found numerous Htt-5596 shRNA-transfected cells that were also cleaved CASP3 positive (Fig. 2G–I, arrows); 38.2 ± 1.4% of the E12.5 transfected cells were costained with the antibody to cleaved CASP3 compared with 4.4 ± 1% in the control (p < 0.001). At E14.5 and E16.5 transfections, 35.7 ± 1.2% and 6.6 ± 0.5% of Htt shRNA-transfected cells were immunopositive for cleaved CASP3, whereas in the control group there were very few anti-cleaved CASP3-positive cells (1 and 0.5% respectively, p < 0.001 for both Htt shRNA vs control; Fig. 2J–L,M–O).
Dissociation of cell death and arrested migratory phenotypes: examination of the developing cerebellum
In the Htt-null chimera, Htt-null cells are well represented in the cerebellum, suggesting that Htt is not required for cells to develop in this region of the brain (Reiner et al., 2001). Thus, we transfected cells in the cerebellar anlagen with Htt shRNA at E12.5, and allowed 48 h survival (n = 3). Migration appeared normal in both the migrating cells of the neuroepithelium (Fig. 3A) as well as in the external germinal layer in the E14.5 transfected cerebellum (data not shown). As in the control, there were many transfected cells with long leading processes characteristic of migrating cells in the cerebellum. However, when cleaved CASP3 was examined in these cerebella, immunostained cells were found in the VZ of the developing cerebellum (Fig. 3B–D, arrows). Thus, while knockdown of Htt in the cerebellum did not affect migration, it did result in abundant cell death (39 ± 3.5% of transfected cells compared with 5 ± 1% in the control group, p < 0.05).
Caspase 9 shRNA significantly reduces the Htt shRNA-caused cell apoptosis
To examine whether blockade of the apoptotic pathway can reverse the cell death caused by Htt knockdown in the cortex, we cotransfected Casp9 shRNA plasmids together with the Htt shRNA at age E12.5, and allowed a survival of 72 h, as previously done. When the Casp9 shRNA (Casp9–469) was cotransfected with Htt shRNA (n = 10), there was an average 43.6% reduction in fluorescent signal upon immunostaining for CASP9 (n = 45 cells, p < 0.001), thus showing its efficacy. The morphology of cotransfected cells was greatly improved, and 26 ± 1% of the cotransfected cells successfully migrated to the CP compared with only 10 ± 1% in the Htt shRNA-only group (p < 0.05; Fig. 4A). Anti-cleaved CASP3 immunostaining of this tissue revealed decreased numbers of apoptotic cells (28.8 ± 1% of transfected cells compared with 38.2 ± 1.4% in the Htt shRNA-only group, p < 0.001; Fig. 4C,D). Thus, the Casp9 shRNA improved cell morphology, limited the number of apoptotic cells caused by Htt knockdown, and partially rescued the migration defect in the developing cerebral cortex.
Figure 4.

Cotransfection of Casp9 shRNA partially rescues the deleterious effects of Htt shRNA on neuronal survival and proliferation. A, B, Casp9 shRNA was cotransfected with Htt shRNA at E12.5 (with 72 h survival). The cotransfection of the Casp9 shRNA greatly improved the morphology of the transfected cells (A) compared with the Htt shRNA transfection alone (see Figs. 1B, 2B). Pia, Pia mater. Scale bar, 200 μm. B, Percentage of transfected cells reaching CP and those remaining cells in VZ, SVZ, and IZ for control, Htt shRNA, and Htt and Casp9 shRNA-cotransfected groups. C, D, Anti-cleaved CASP3 immunostaining revealed that cotransfection of Casp9 shRNA with Htt shRNA decreased the percentage of transfected cells undergoing apoptosis (C). Arrows indicate the colocalization of anti-cleaved CASP3 immunostaining (red) and EGFP (green). D, Percentage of apoptotic cells over all transfected cells for control, Htt shRNA, and Htt and Casp9-cotransfected groups. E–G, Knockdown of Htt results in decreases of BrdU-positive cells, and cotransfection with Casp9 shRNA rescues the disruptive effect of Htt on mitosis. Arrows indicate the colocalization of anti-BrdU immunostaining (red) and EGFP (green). Control (E), Htt shRNA (F), and Htt and Casp9 shRNA (G) cotransfection. H, Percentage of BrdU-double-labeled cells over all transfected cells for control, Htt shRNA, and Htt and Casp9 shRNA-cotransfected groups. Scale bar, 50 μm. Error bars in B, D, and H represent SEM.
In light of the recent discovery that Htt is required for mitotic spindle orientation and neurogenesis (Godin et al., 2010), we examined the effect of shRNA knockdown of Htt on mitosis in cortical development with BrdU delivered 1 h before embryo harvesting with transfection at E12.5 with 72 h survival. The percentage of transfected cells (EGFP+) that were also BrdU+ was calculated in control, Htt shRNA-transfected, and Htt and Casp9 shRNA-cotransfected groups. Compared with 33.3 ± 1% of BrdU+-transfected cells in the control group (n = 3; Fig. 4E), only 8.9 ± 1% of the transfected cells were BrdU+ in the Htt shRNA group (n = 5, p < 0.001, Fig. 4F), and 20.9 ± 1% transfected cells were BrdU+ in the Htt and Casp9 shRNA-cotransfected group (n = 7, p < 0.001; Fig. 4G,H). Therefore, shRNA knockdown of Htt had an adverse effect on mitosis, and cotransfection with Casp9 shRNA was able to partially rescue this defect.
Discussion
A host of developmental problems has been identified with the early deletion of Htt, with no clear resolution as to the role of Htt in neural development. Evidence for its importance in neural development, especially in the forebrain, has come from several lines of research. First, Htt has been found to be expressed very early in the human (19–21 week fetus) and mouse forebrain (as early as E10) (Dure et al., 1994; Bhide et al., 1996). Second, White et al. (1997) made and analyzed a hypomorphic knock-in allele of the Htt gene (HttneoQ50) that resulted in profound early neural defects. The neo insert produced a greatly attenuated expression of Htt, and mice homozygous for this allele showed perinatal lethality with ectopic masses of proliferating cells in the SVZ of the forebrain. Additionally, there were major disturbances in overall brain organization. Third, when the embryonic lethal effect of the Htt −/− allele was bypassed using blastocyst injection chimeras, we found that there was a striking deletion of neurons in forebrain regions (Reiner et al., 2001) and that this absence of neurons was likely due to an early apoptosis of Htt-null cells in the E12.5 embryo (Reiner et al., 2003). The chimeric setting offers one way, albeit random, to look at the role of the Htt gene in development. Another approach is to use in utero transfection of Htt shRNA in the developing brain with resolution in both time and space. In this manner, we were able to identify three elements of neuronal development that are dependent upon normal Htt expression: cell migration, cell proliferation, and survival.
Htt knockdown disrupts normal cell migration in a region-specific manner
The migration of cells from the neuroepithelium to a final destination is a critical step in CNS development. In this report, we show that Htt knockdown leads to region-specific disruption of neuronal migration. Knockdown of Htt in neurons of the neuroepithelium at E12.5 by shRNA impairs normal cell migration in cerebral cortex, but not in cerebellum. We observed the accumulation of transfected cells in the IZ of developing cortex as early as 48 h after transfection. As far as we know, this is the first report of Htt's involvement in neuronal migration, and the mechanism of regional specificity is currently unknown. The recent report by Godin et al. (2010) found that siRNA knockdown of Htt had no effect on neuronal migration. There are two main differences between the current report and the work of Godin et al. (2010) that could explain these discordant results. First, the time of transfection is different. While our data include knockdown at three time points, E12.5, E14.5 and E16.5, Godin et al. (2010) only examined knockdown at E14.5. We find that the disrupted migratory phenotype with Htt shRNA knockdown is most obvious at E12.5. By contrast, at E14.5 the normal egress of cells from the neuroepithelium has diminished such that it would have been difficult to observe the relatively minor abnormality in migration that we observed at that time point. Second, shRNA target sequences are different. We tested four constructs, and used the one that gave the most prominent phenotype (Fig. 1), while Godin et al. (2010) reported the use of only two Htt siRNA constructs that may have been less efficient than the optimal construct we identified in yielding deficits in cell migration.
Neuroprotective activity of Htt and its implication for therapeutic use of Htt RNAi
Wild-type Htt has been documented to provide a positive effect on cell survival and can mitigate the effects of the mutant Htt (Rigamonti et al., 2000; Reiner et al., 2003). The absence of the HTT protein can have profound consequences on the ability of neurons to successfully colonize cortex and striatum. We previously reported that chimeric mice composed of a mixture of wild-type and Htt−/− cells (Htt+/+ and Htt−/−) showed that forebrain neurons needed Htt for normal cortical and striatal development, and neurons throughout the brain needed Htt for the long-term health of the animal (Reiner et al., 2001). Furthermore, Dragatsis et al. (2000), using conditional inactivation of Htt, have shown that loss of Htt late in development or shortly after birth can lead to subsequent degeneration of cortical and striatal neurons. Htt −/− neurons in culture have been shown to be especially vulnerable to the effects of serum deprivation (Rigamonti et al., 2000), which suggests the possibility that Htt plays a role in enabling at least some types of neurons to withstand stressful events. This neuroprotective role of Htt may be more important for cortical and striatal neurons, which are most vulnerable in HD.
In this report, with shRNA knockdown we demonstrate that the Htt gene product is a critical protein in early cortical and cerebellar development for cell survival. Knockdown of Htt expression leads to caspase-mediated cell apoptosis both in cortex and cerebellum. The application of Casp9 shRNA to block the mitochondria-mediated apoptotic pathway reduced cell apoptosis, improved cell morphology and mitosis, and rescued the migration of some cells in cortex. Interestingly, much of the cell apoptosis was still present in the Casp9 shRNA-cotransfected tissues. This can be due to two possibilities. First, the Casp9 shRNA may not have completely knocked down the Casp9 message in all transfected neurons, resulting in some apoptotic cells and incomplete rescue of migration defect in the Casp9 shRNA-cotransfected fetus tissues. The second possibility is that the Casp9 shRNA in our experiment may target only one of multiple apoptotic pathways engaged by Htt knockdown (Graham et al., 2006).
A link between abnormal neurogenesis and Htt knockdown is suggested by the findings of White et al. (1997) that reduced expression of Htt was associated with reduced neurogenesis and aberrant brain development. In this report, we show that Htt shRNA knockdown reduced the number of cells undergoing mitosis in the SVZ/VZ and cotransfection with Casp9 shRNA partially reversed the disruption. This is also in line with the recent report by Godin et al. (2010), who showed that loss of Htt expression increased neuronal differentiation, and decreased proliferation and maintenance of cortical progenitors in the VZ.
Finally, we show that there is a restricted temporal window when Htt is critical to cortical development. The deleterious effect of Htt shRNA is most pronounced at E12.5 and is attenuated during later development, pointing to a critical time when Htt is needed in developing neurons. Furthermore, our results point toward a cell-autonomous need for Htt expression in cortical development. The phenotypic abnormalities of impaired migration and cell death were only found in transfected cells, while neighboring nontransfected cells showed no signs of a defect in cell migration or apoptosis, which is consistent with our previous work on wild-type and Htt−/− chimeric mice (Reiner et al., 2001). These findings are important for understanding the role of Htt in normal neural development, and for strategies for Htt gene knockdown or elimination as a cure for HD.
Notes
Supplemental material for this article is available at http://grits.dglab.org/Downloads/Tong_2011_Supplemental_Figures_and_Tables.pdf. The PDF file contains two supplemental figures regarding Htt shRNA and Casp9 shRNA constructs and eight supplemental tables that summarize the results. This material has not been peer reviewed.
Footnotes
This work is supported by NIH Grant HD 52472, Hereditary Disease Foundation HDF, HighQ Foundation, the Methodist Hospitals Foundation, and the Centre for Molecular Medicine and Therapeutics and the Child and Family Research Institute.
References
- Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J Neurosci. 1996;16:5523–5535. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Efstratiadis A, Zeitlin S. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development. 1998;125:1529–1539. doi: 10.1242/dev.125.8.1529. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Dure LS, 4th, Landwehrmeyer GB, Golden J, McNeil SM, Ge P, Aizawa H, Huang Q, Ambrose CM, Duyao MP, Bird ED. IT15 gene expression in fetal human brain. Brain Res. 1994;659:33–41. doi: 10.1016/0006-8993(94)90860-5. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Reiner A, Del Mar N, Meade CA, Yang H, Dragatsis I, Zeitlin S, Goldowitz D. Neurons lacking huntingtin differentially colonize brain and survive in chimeric mice. J Neurosci. 2001;21:7608–7619. doi: 10.1523/JNEUROSCI.21-19-07608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28:259–276. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Interkinetic and migratory behavior of a cohort of neocortical neurons arising in the early embryonic murine cerebral wall. J Neurosci. 1996;16:5762–5776. doi: 10.1523/JNEUROSCI.16-18-05762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]