

Abstract
Only two steps?
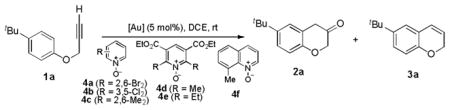
Chroman-3-ones are important intermediates for organic synthesis and medicinal chemistry. However, their syntheses require multiple steps and are not efficient. By using gold-catalyzed alkyne oxidation, this versatile heterocycle can be prepared in only two steps from readily available phenols and with mostly high efficiencies.
Keywords: gold, carbene, chroman-3-one, oxidation, catalysis
Chroman-3-one is an important structural motif, and its derivatives have served as key intermediates in the total synthesis of natural product such as miroestrol,[1] (+)-myristinin A,[2] and afzelechin[3] and in accessing a range of bioactive chromans for managing various diseases including hypertension,[4] HIV,[3] sexual dysfunction,[5] and melanoma.[6] In his 1991 review,[7] Kirkiacharian concluded that there was a lack of efficient synthetic methods for this structure. Since then, little progress[8] has been made. To date, the 20-year old statement still stays largely true. Invariably, these compounds are prepared via multiple-step routes[7, 9] from basic chemicals. For example, a typical route starts from basic condensation of salicylaldehyde with acrylonitrile to form 2H-chromene-3-carbonitrile under refluxing conditions, followed by sequential basic hydrolysis, acidification, formation of acyl azide, Curtius rearrangement, and acidic hydrolysis of the resulting vinyl isocyanate.[1, 9–10] While the overall yield could be as high as 60%,[1] the step[11] and atom[12] economy is low. An alternative approach via toxic α-diazo-α′-phenoxy acetones still requires four steps starting from phenols (Scheme 1),[13] and no application of this method has been reported in literature due to a limited scope[14] and clear drawback associated with using dangerous diazomethane. Herein, we report an efficient synthesis of chroman-3-ones via gold-catalyzed oxidation of propargyl aryl ethers, which in turn can be easily prepared in one-step from readily available phenols.
Scheme 1.

Access to chroman-3-ones via metal carbene intermediates?
We have in the past two years developed several gold-catalyzed intermolecular oxidations of alkynes using pyridine/quinoline N-oxides as oxidants.[15] The common intermediates generated in these reactions were most likely α-oxo gold carbenes, which appeared to be highly reactive and underwent facile O-H,[15b, 15c] N-H,[15a] or 1,2-C-H insertions[15d] or reaction with nitriles en route to oxazoles.[15e] Several recent works by other research groups also corroborated the intermediacy of gold carbenes.[16] Since metal carbenes in general can be generated via dediazotization,[17] this strategy permits to replace toxic, potentially explosive and sometimes difficult-to-access α-diazo ketones with benign and readily available alkynes in gold carbene chemistry.[18] In the context of chroman-3-one synthesis, we envisioned that this strategy would allow replacing α-diazo-α′-phenoxy acetones by readily available propargyl aryl ethers (Scheme 1), thereby establishing a succinct, safe and potentially highly efficient route to this class of heterocycles.
At the outset, 4-tert-butylphenyl propargyl ether 1a was chosen as the substrate for condition optimization. Some of the results are listed in Table 1. When either Ph3PAuNTf2[19] (entry 1) or the more electrophilic phosphite-coordinated L1AuNTf2[20] [L1 = tris(2,4-di-tert-butylphenyl)phosphite, entry 2] was employed as the catalyst and 2,6-dibromopyridine N-oxide as the oxidant, both reactions were sluggish, and the desired chroman-3-one 2a was formed in <10% yield.
Table 1.
Initial discovery and reaction condition screening.[a]
| ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | oxidant (1.1 equiv) | time | 1a remained[b] | yield[b] | |
| 2a | 3a[c] | |||||
| 1 | Ph3PAuNTf2 | 4a | 16 h | 41% | 6% | − (66%) |
| 2 | L1AuNTf2 | 4a | 16 h | 58% | 7% | − (73%) |
| 3 | Cy-JohnPhosAuNTf2 | 4a | 16 h | 17% | 9% | 2% (64%) |
| 4 | tBuXPhosAuNTf2 | 4a | 16 h | - | 27% | 4% (76%) |
| 5 | BrettPhosAuNTf2 | 4a | 0.5 h | - | 65% | 4% (87%) |
| 6 | Me4tBuXPhosAuNTf2 | 4a | 0.5 h | - | 78% | 8% (79%) |
| 7 | Me4tBuXPhosAuNTf2 | 4b | 15 h | 18% | 38% | <2% |
| 8 | Me4tBuXPhosAuNTf2 | 4c | 15 h | 39% | 29% | 8% |
| 9 | Me4tBuXPhosAuNTf2 | 4f | 15 h | 10% | 45% | 5% |
| 10 | Me4tBuXPhosAuNTf2 | 4d | 1 h | 6% | 76% | ~2% |
| 11 | Me4tBuXPhosAuNTf2 | 4d[d] | 1 h | - | 84%[f] | ~2% |
| 12 | Me4tBuXPhosAuNTf2 | 4e[e] | 1 h | - | 87% | ~2% |
[1a] = 0.05 M.
Estimated by 1H NMR using diethyl phthalate as the internal reference.
Yield in parenthesis is without oxidant, and the reaction time was 1 h except for BrettPhosAuNTf2 (15 min) and Me4tBuXPhosAuNTf2 (25 min).
1.3 equivalent.
1.2 equivalent.
Isolated yield: 82%.
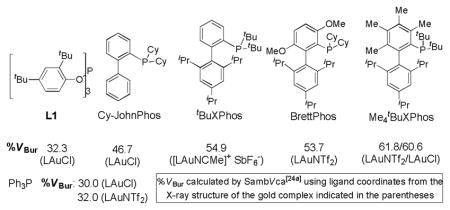
In our previous work on azetidine-3-one synthesis,[15a] we noticed that gold catalysts based on biphenylphosphine ligands[21] were increasingly effective as the biphenyl moiety became bulkier and BrettPhosAuNTf2 was the optimal catalyst. To our delight, the same trend was observed with this reaction (comparing entries 3–5), and chroman-3-one 2a was formed in 65% NMR yield in only 0.5 h with BrettPhosAuNTf2 as the catalyst (entry 5). Seeking to further improve this reaction, we tried Me4tBuXPhosAuNTf2.[22] To our delight, the yield was improved to 78% (entry 6). Single crystal X-ray diffraction studies established the structure of this effective catalyst (Figure 1A). In comparison with BrettPhosAuNTf2 (Figure 1B),[15a] the Au1-P1-C2 angle is smaller by 5.52, and the distance between Au1 and C1’ is shorter by 0.265 Å, suggesting that Me4tBuXPhos presses Au more toward the shielding 2,4,6-triisopropylphenyl group[23] and thereby increasing the crowdedness of the metal center. This is reflected by its %VBur [24][25] (61.8 based on the X-ray structure of its AuNTf2 complex and 60.6 on its AuCl complex[22c]), significantly higher than that of BrettPhos[26] (53.7%VBur based on the X-ray structure of its AuNTf2 complex[15a]). In fact, Me4tBuXPhos has a higher %VBur than any of the Au(I) ligands investigated by Nolan.[25] This bulky ligand likely prevents the corresponding gold carbene B (see Scheme 1) from deleterious intermolecular side reactions due to access difficulty, thereby leading to increased efficiency. Notably, Barriault[22c] recently reported that [Me4tBuXPhosAuNCMe]+ SbF6− is the best catalyst for promoting 6-endo-dig cyclizations selectively over typically favored 5-exo-dig ones.
Figure 1.

Ortep ellipsoid drawing of complexes Me4tBuXPhosAuNTf2 and BrettPhosAuNTf2 with 50% probability. The hydrogen atoms have been omitted for clarity.
In these reactions, only small amounts of benzopyran 3a were formed via a competitive gold-catalyzed 6-endo-dig cyclization. However, in the absence of oxidant 4a, this cyclization was facile and generally efficient in the presence of a range of gold catalysts (see yields in parenthesis in entries 1–6, Table 1).[27] These results suggested that the N-oxide, besides being the oxidant, played the requisite role of retarding this side reaction, most likely due to its coordination to the gold catalyst.
 |
(1) |
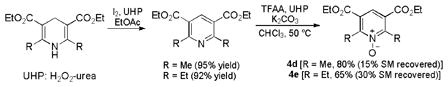
Other oxidants that we previously used [15] did not improve the reaction (entries 7–9). However, it was clear that hindered and electron-deficient N-oxides fared better. Consequently, we prepared pyridine N-oxides 4d and 4e by double oxidation of the corresponding Hantzsch esters (Eq. 1), which in turn can be easily prepared.[28] To the best of our knowledge, these hindered and electron-deficient N-oxides have never been used as oxidants before. Gratifyingly, with 1.3 equivalent of 4d, the reaction yield was improved to 84% (entry 11); a better yield was obtained with even more hindered 4e (entry 12).
With Me4tBuXPhosAuNTf2 as the catalyst and 4d or 4e as the oxidant, we explored the reaction scope, which is shown in Table 2. For the parent phenyl propargyl ether, unsubstituted chroman-3-one 2b was obtained in 76% yield (entry 1). An ortho-Me substitution on the benzene ring were inconsequential (entry 2). The cyclization in the case of a meta-Me (entry 3) was highly regioselective, favoring the position para to the Me group; an even higher regioselectivity was achieved with a tBu substrate (entry 4). A second methyl substitution on the benzene ring did not affect much the reaction yield (entry 5). Substrates with differently substituted methoxy groups reacted smoothly, affording corresponding products in good yields (entries 6–8). Importantly, the regioselectivity in the case of a meta-MeO group (entry 8) could be improved to 11:1 when the reaction was run at 0 °C. Notably, these chroman-3-ones, i.e., 2g–2i, were previously prepared from more costly MeO-substituted salicylaldehydes following afore-mentioned multiple step sequence, and the overall yields were mostly less than 41%.[9] The functional group tolerance of this chemistry is good as substrates with a TBS ether (entry 9), a benzyl ether (entry 10), an acetate (entry 11), and a secondary amide (entry 12) all reacted well. With electron-withdrawing groups including halides (entries 13 and 14) and methoxycarbonyl (entry 15), the reactions were less efficient albeit still serviceable considering the short sequences. This chemistry also worked well with naphthalene substrates (entries 16 and 17), showing excellent regioselectivities.
Table 2.
The reaction scope studies.[a]

| ||
|---|---|---|
1
 2b (76%, B) |
2
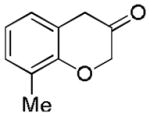 2c (78%, B) |
3
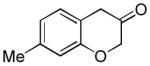 2d (67%, p:o = 12:1, A) |
4
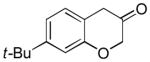 2e (74%, p:o = 16:1, A) |
5
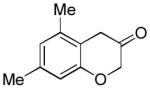 2f (70%, A) |
6
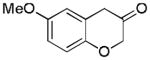 2g (83%, B) |
7
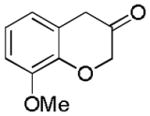 2h (70%, B) |
8[b]
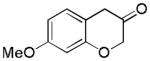 2i (75%, p:o = 11:1, B) |
9
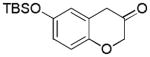 2j (70%, A) |
10
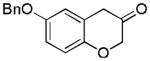 2k (86%, B) |
11
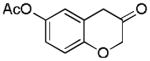 2l (63%, B) |
12
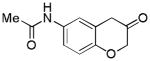 2m (81%, A) |
13
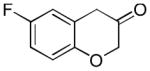 2n (54%, B) |
14
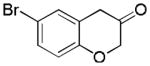 2o (50%, B) |
15
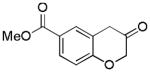 2p (43%, A) |
16
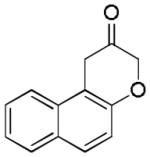 2q (68%, A) |
17
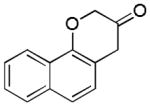 2r (81%, A) |
18
 2s (83%, A) |
19
 2t (66%, A) |
20[b]
 2u (60%, l:b = 8:1, B) |
21[b]
 2v (61%, l:b = 3.3 :1, B) |
The reactions were run in vial without exclusion of air and moisture, and the substrate concentration is 0.05 M. Isolated yields were reported.
The reaction was run at 0 °C.
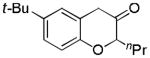
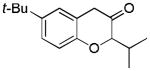
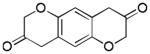
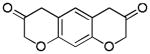
Aliphatic substitutions at the propargyl position including the sterically demanding isopropyl group were readily tolerated (entries 18 and 19). Notably, the diazo ketone approach could not afford this type of 2-alkyl substituted product cleanly due to a major side reaction leading to norcaradiene side products.[14] In the case of a phenyl group at the propargylic position, the yield was low (<40%), likely due to the sensitivity of the substrate in the presence of the Lewis acidic gold complex. With the bisether substrate derived from p-hydroquinone, double oxidative cyclization occurred smoothly, affording the linear tricycle 2u selectively in a respectable yield (entry 20). A similar reaction was achieved with the substrate derived from resorcinol although the regioselectivity was moderate (entry 21).
In summary, we have developed a one-step synthesis of chroman-3-ones from readily available propargyl aryl ethers. Comparing with literature protocols, this gold oxidation approach is step-economic and efficient, and should facilitate research involving this versatile heterocycle. A new gold catalyst, Me4tBuXPhosAuNTf2, is prepared and fully characterized. Its hindered nature is revealed by X-ray diffraction studies. This study as well Barriault’s work suggests that cationic gold complexes based on Me4tBuXPhos can be uniquely effective and should be included in the ligand repertoire of practitioners interested in gold catalysis. In addition, two easily accessible pyridine N-oxides derived from Hantzsch esters were shown as highly effective oxidants; they should help facilitate the development of gold-catalyzed alkyne oxidation reactions, which promise excellent step economy and synthetic efficiency.
Experimental Section
General procedure
Pyridine N-oxide 4d (0.65 mmol, 1.3 equiv) or 4e (0.6 mmol, 1.2 equiv) and Me4tBuXPhosAuNTf2 (0.025 mmol, 5 mol %) were added sequentially to a solution of the propargyl aryl ethers 1 (0.50 mmol) in DCE (10 mL. 0.05 M) at room temperature. The resulting reaction mixture was stirred at r.t., and the progress of the reaction was monitored by TLC. The reaction typically took 1–3 h. Upon completion, the mixture was concentrated and the residue was purified by silica gel flash chromatography (eluent: hexanes/ethyl acetate) to afford the desired products 2.
Supplementary Material
Acknowledgments
The authors thank NIGMS (R01 GM084254) and UCSB for generous financial support and Dr. Guang Wu for helping with X-ray crystallography.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Corey EJ, Wu LI. J Am Chem Soc. 1993;115:9327. [Google Scholar]
- 2.Maloney DJ, Deng JZ, Starck SR, Gao Z, Hecht SM. J Am Chem Soc. 2005;127:4140. doi: 10.1021/ja042727j. [DOI] [PubMed] [Google Scholar]
- 3.Tsantrizos YS, Bailey MD, Bilodeau F, Carson RJ, Coulombe R, Fader L, Halmos T, Kawai S, Landry S, Laplante S, Morin S, Parisien M, Poupart M-A, Simoneau B. PCT Int Appl. 2009 Copyright (C) 2011 American Chemical Society (ACS). All Rights Reserved. [Google Scholar]
- 4.Kawamura K, Ohta T, Otani G. Chem Pharm Bull. 1990;38:2092. [Google Scholar]
- 5.Cook AS, Miller DC. PCT Int Appl. 2006 Copyright (C) 2011 American Chemical Society (ACS). All Rights Reserved. [Google Scholar]
- 6.Rodriguez-Lopez JN, Sanchez dCFL, Cabezas-Herrera J, Tarraga TA, Saez AMM. PCT Int Appl. 2009 Copyright (C) 2011 American Chemical Society (ACS). All Rights Reserved. [Google Scholar]
- 7.Danan A, Kirkiacharian BS. Bull Soc Chim Fr. 1991:189. [Google Scholar]
- 8.Velkov J, Mincheva Z, Bary J, Boireau G, Fujier C. Synth Commun. 1997;27:375. [Google Scholar]
- 9.Pavé G, Chalard P, Viaud-Massuard MC, Troin Y, Guillaumet G. Synthesis. 2004;2004:121. [Google Scholar]
- 10.Wise LD, DeWald HA, Hawkins ES, Reynolds DM, Heffner TG, Meltzer LT, Pugsley TA. J Med Chem. 1988;31:688. doi: 10.1021/jm00398a032. [DOI] [PubMed] [Google Scholar]
- 11.a) Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2007;41:40. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]; b) Newhouse T, Baran PS, Hoffmann RW. Chem Soc Rev. 2009;38:3010. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trost BM. Angew Chem, Int Ed. 1995;34:259. [Google Scholar]
- 13.Saba A. Synthesis. 1984;1984:268. [Google Scholar]
- 14.Pusino A, Saba A, Rosnati V. Tetrahedron. 1986;42:4319. [Google Scholar]
- 15.a) Ye L, He W, Zhang L. Angew Chem, Int Ed. 2011;50:3236. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye L, He W, Zhang L. J Am Chem Soc. 2010;132:8550. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ye L, Cui L, Zhang G, Zhang L. J Am Chem Soc. 2010;132:3258. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lu B, Li C, Zhang L. J Am Chem Soc. 2010;132:14070. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) He W, Li C, Zhang L. J Am Chem Soc. 2011:8482. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- 16.a) Vasu D, Hung H-H, Bhunia S, Gawade SA, Das A, Liu R-S. Angew Chem, Int Ed. 2011 doi: 10.1002/anie.201102581. [DOI] [PubMed] [Google Scholar]; b) Qian D, Zhang J. Chem Commun. 2011;47:11152. doi: 10.1039/c1cc14788a. [DOI] [PubMed] [Google Scholar]; c) Davies PW, Cremonesi A, Martin N. Chem Commun. 2011;47:379. doi: 10.1039/c0cc02736g. [DOI] [PubMed] [Google Scholar]; d) Mukherjee A, Dateer RB, Chaudhuri R, Bhunia S, Karad SN, Liu RS. J Am Chem Soc. 2011;133:15372. doi: 10.1021/ja208150d. [DOI] [PubMed] [Google Scholar]
- 17.a) Dörwald FZ. Metal carbenes in organic synthesis. Wiley-VCH, Weinheim; New York: 1999. [Google Scholar]; b) Doyle MP, McKervey MA, Ye T. Modern catalytic methods for organic synthesis with diazo compounds: from cyclopropanes to ylides. Wiley; New York: 1998. [Google Scholar]
- 18.a) Fructos MR, Belderrain TR, de Fremont P, Scott NM, Nolan SP, Díaz-Requejo MM, Perez PJ. Angew Chem Int Ed. 2005;44:5284. doi: 10.1002/anie.200501056. [DOI] [PubMed] [Google Scholar]; b) Prieto A, Fructos MR, Mar Diaz-Requejo M, Pérez PJ, Pérez-Galán P, Delpont N, Echavarren AM. Tetrahedron. 2009;65:1790. [Google Scholar]
- 19.Mézailles N, Ricard L, Gagosz F. Org Lett. 2005;7:4133. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]
- 20.a) Amijs CHM, López-Carrillo V, Raducan M, Pérez-Galán P, Ferrer C, Echavarren AM. J Org Chem. 2008;73:7721. doi: 10.1021/jo8014769. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Ye L, Zhang L. Chem Commun. 2011;47:7815. doi: 10.1039/c1cc12212f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Herrero-Gómez E, Nieto-Oberhuber C, López S, Benet-Buchholz J, Echavarren AM. Angew Chem, Int Ed. 2006;45:5455. doi: 10.1002/anie.200601688. [DOI] [PubMed] [Google Scholar]; b) Nieto-Oberhuber C, Lopez S, Echavarren AM. J Am Chem Soc. 2005;127:6178. doi: 10.1021/ja042257t. [DOI] [PubMed] [Google Scholar]
- 22.a) Burgos CH, Barder TE, Huang X, Buchwald SL. Angew Chem, Int Ed. 2006;45:4321. doi: 10.1002/anie.200601253. [DOI] [PubMed] [Google Scholar]; b) Vorogushin AV, Huang X, Buchwald SL. J Am Chem Soc. 2005;127:8146. doi: 10.1021/ja050471r. [DOI] [PubMed] [Google Scholar]; c) Barabé F, Levesque P, Korobkov I, Barriault L. Org Lett. 2011;13:5580. doi: 10.1021/ol202314q. [DOI] [PubMed] [Google Scholar]
- 23.It is unlikely that the gold arene interaction plays a role for the difference. For examples of gold arene complexes, see ref. 21a. For a detailed study on meta-arene interactions in dialkylbiarylphosphine complexes with coinage metals, see: Pérez-Galán P, Delpont N, Herrero-Gómez E, Maseras F, Echavarren AM. Chem Eur J. 2010;16:5324. doi: 10.1002/chem.200903507.
- 24.Calculated using SambVca using the standard parameters. For reference, see: Poater A, Cosenza B, Correa A, Giudice S, Ragone F, Scarano V, Cavallo L. Eur J Inorg Chem. 2009;2009:1759.
- 25.Clavier H, Nolan SP. Chem Commun. 2010;46:841. doi: 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- 26.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Nevado C, Echavarren AM. Chem Eur J. 2005;11:3155. doi: 10.1002/chem.200401069. [DOI] [PubMed] [Google Scholar]; b) Menon RS, Findlay AD, Bissember AC, Banwell MG. J Org Chem. 2009;74:8901. doi: 10.1021/jo902032p. [DOI] [PubMed] [Google Scholar]; c) Lykakis IN, Efe C, Gryparis C, Stratakis M. Eur J Org Chem. 2011;2011:2334. [Google Scholar]
- 28.a) Wang GW, Xia JJ, Miao CB, Wu XL. Bull Chem Soc Jpn. 2006;79:454. [Google Scholar]; b) Filipan-Litvi M, Litvi M, Vinkovi V. Tetrahedron. 2008;64:5649. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.