

Abstract
Emotion is critical to most aspects of human behavior, and individual differences in systems recruited to process emotional stimuli, expressed as variation in emotionality, are characteristic of several neuropsychiatric disorders. We examine the genetic origins of individual differences in emotion processing by focusing on functional variants at five genes: catechol-O-methyl transferase (COMT), serotonin transporter (SLC6A4), neuropeptide Y (NPY), a glucocorticoid receptor-regulating co-chaperone of stress proteins (FKBP5) and pituitary adenylate cyclase-activating polypeptide (PACAP). These represent a range of effects of genes on emotion as well as the variety of mechanisms and factors, such as stress, that modify these effects. The new genomic era of genome-wide association studies (GWAS) and deep sequencing may yield a wealth of new loci modulating emotion. The effects of these genes can be validated by neuroimaging, neuroendocrine and other studies accessing intermediate phenotypes, deepening our understanding of mechanisms of emotion and variation in emotionality.
Candidate gene studies of emotion
Emotion, or the assignment of affective valence to objects, states and situations (see Glossary), is critical to most aspects of human behavior: all our actions and decisions occur in emotional contexts, and cognitive functions are colored by emotional states. Neurobiologically, emotion is linked to the response of neural circuits by which individuals assign intensity and valence to objects and situations in the environment, and to internal states. Emotionality represents variation in emotion: it varies between individuals and also varies within an individual, depending on context and life history. Variation in systems recruited to process stimuli that trigger emotion and regulate emotional outputs leads to differences in emotionality. Crucially, emotionality is a factor in a wide spectrum of psychiatric diseases including mood [1, 2] and anxiety disorders [3]. Significant progress has been made in identifying the brain structures that underlie affective processing, including the prefrontal cortex (PFC), anterior cingulate cortex, hippocampus and amygdala [4, 5], in visualizing and modifying the activity of the circuitry of emotion [6] and in identifying genetic variants that modulate inter-individual differences in emotionality. In this review, we focus on the genetic origins of individual differences in emotion processing.
Heritable individual differences in affect, temperament and personality shape other complex behaviors, as well as responses to an ever-changing environment. These differences can also be important predictors of vulnerability to neuropsychiatric disorders that are themselves genetically influenced. Functional variants at five genes (catechol-O-methyl transferase (COMT), serotonin transporter (SLC6A4), neuropeptide Y (NPY), a glucocorticoid receptor-regulating co-chaperone of stress proteins (FKBP5) and pituitary adenylate cyclase-activating polypeptide (PACAP)) will be used to illustrate a range of effects of ‘emotion genes’ and factors that alter or confound these effects. These genes have been selected on the basis of convergent neurobiological evidence in modulation of emotional processes. Notably, each of these represents a discovery made prior to a new era in which genome-wide association studies (GWAS) and deep sequencing are expected to yield a wealth of new loci modulating emotion. As will be described, the effects of these functional alleles has to some extent been explored, and validated, both via human brain imaging and in animal models, in which the genes have been genetically manipulated, although space considerations prevent full review of the functional connections to the neurobiology of emotion for any of these genes.
Emotionality is moderately heritable (40-60%) [7] but is also strongly influenced by exposure to stress in a pattern consistent with gene × environment interaction. Identification of the mechanisms that give rise to inter-individual differences in emotional stability and vulnerability to stress and anxiety will deepen our understanding of human behavior and predisposition to disease.
The effect sizes of genetic variants implicated in emotional responses measured as psychological traits have been small. For example, in a meta-analytical study, the serotonin transporter promoter polymorphism was found to have only a 0.106 standard deviation effect on neuroticism per copy of the low expression allele [8]. However, several of the same variants, including this serotonin transporter allele, have larger effects on metabolic responses of the brain to emotional stimuli accessed in real time by brain imaging. These studies using intermediate phenotypes have not only validated the effects of these genes in emotion, but deepened our understanding of mechanisms by which neural circuits alter emotional responses and cognition. As described in Boxes 1 and 2, variation in BOLD fMRI measures of emotional response is both heritable and associated with emotion in the rhesus macaque. In humans, these fMRI measures of emotion have not been shown to be heritable, but they are a temporally stable and reliable index of brain function [9, 10], and correlate with emotionality traits including diseases whose heritability has been demonstrated. Thus, they are defined as endophenotypes [11].
Box 1. Heritability of emotionality and emotion-induced fMRI activations.
Emotionality, measured as neuroticism, introversion or harm avoidance, is moderately to highly heritable in both sexes: 40-60%, with little effect of shared environment [7,76]. Behavioral inhibition is manifested early in life and predicts anxiety disorders and depression later in life [77-79]. In humans, trait emotionality has been tied to brain metabolic measures obtained by neuroimaging. However, while these measures show long-term stability and are thus trait-like [10], they have not been shown to be heritable [80]. Non-human primates represent an alternative model for the study of the relationship of brain metabolic activity to emotionality and direct evaluation of heritability. In non-human primates, emotionality and correlated brain activations are also stable and persistent across environments [81]. As shown in Figure I, in the rhesus macaque, the central nucleus of the amygdala and anterior hippocampus are components of a neural circuit predictive of anxious temperament [82]. In these monkeys, brain metabolic activity was measured by fludeoxyglucose–PET (FDG–PET) and anxious behavior was measured by observation under a threatening condition. Using a multigenerational pedigree, the heritability of anxious temperament was 0.36 [82-83]. Heritability of threat-provoked hippocampal metabolic activity, but surprisingly not for the amygdala, was even higher (0.65 and 0.76 in right and left hippocampus, respectively). Concerning the lack of heritability of amygdala FDG, a key question would be whether it reflects measurement properties of FDG in hippocampus vs. amygdala.
These findings strongly suggest that brain metabolic responses measured by neuroimaging represent an endophenotype—an intermediate phenotype that is both heritable and disease-associated [11]—for emotionality and related disorders.
Figure I. Correlation between anxious temperament and glucose metabolism in (a) the amygdala and (b) the hippocampus in rhesus macaques.
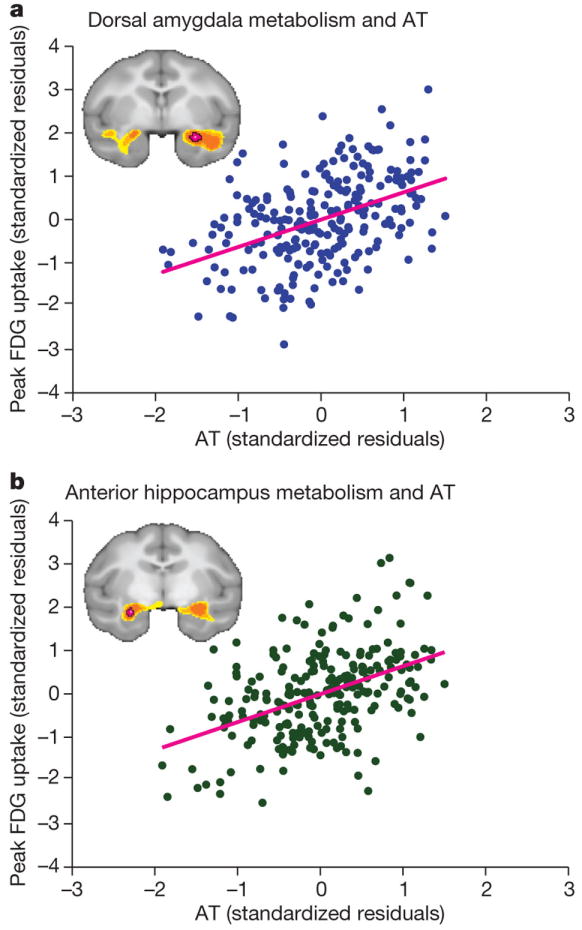
Reproduced, with permission, from [82].
Box 2. The power of endophenotypes for studying the effects of emotionality genes.
A functional promoter polymorphism of NPY, a gene that encodes an anxiolytic neuropeptide, appears to predict emotionality and stress response [58]. This study evaluated the effect of common functional NPY haplotypes on molecular, neuroimaging and behavioral phenotypes. Illustrated in Figure I, in one of the imaging genetic studies in this report, amygdala and hippocampal activations were measured in response to threat-related facial expressions. NPY diplotypes were grouped into low (LL), intermediate (LH) and high (HH) expression groups, based on the in vitro and in vivo RNA levels associated with different haplotypes, and the identification of a functional locus within the haplotype, and located in the NPY promoter region. Task-related reactivity was predicted in an allele-dosage fashion in both right dorsal amygdala and hippocampus, accounting for 6-9% of the variance in fMRI response to emotional challenge. Emotion-induced activations were highest in individuals with low NPY expression diplotypes (LL) and lowest in those with high-expression (HH) diplotypes. In contrast with large effects of NPY on brain functional responses and measures of NPY mRNA and protein levels, the effect on trait anxiety, measured as Harm Avoidance with the Tridimensional Personality Questionnaire, was directionally congruent in an independent dataset but accounted for only 3.3-3.4% of the variance. Intermediate phenotypes, if they are heritable endophenotypes, may, therefore, more closely reflect the effects of genes.
Figure I. Effect of diplotype-predicted NPY mRNA expression on fMRI measured amygdala and hippocampal activation in response to threat related facial expressions.
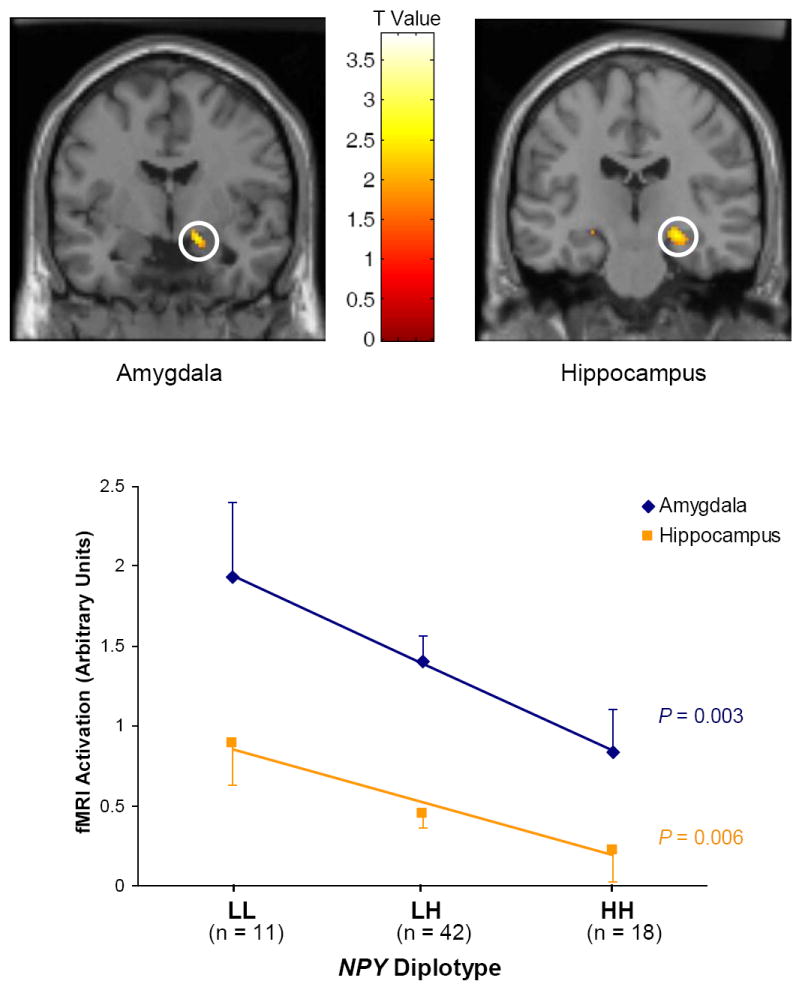
Top: Genotype-dependent NPY expression predicts the fMRI activation of amygdala and hippocampus in response to threat-related facial expressions. Bottom: Right amygdala (black diamonds) and hippocampus (grey squares) activation predicted by NPY genotype. The lower expression NPY haplotype predicted stronger threat-related activations. Reproduced, with permission, from [58].
COMT
Catechol-O-methytransferase (COMT) metabolizes dopamine, noradrenaline and other catecholamines. This enzyme plays an important role in the prefrontal cortex where the dopamine transporter is not abundant [12]. Val158Met is a common functional polymorphism that alters enzyme stability [13] and is predicted to lower dopamine levels in the prefrontal cortex. Under normal conditions the COMT knockout mouse may not have altered catecholamine levels but under others, for example when challenged with dihydroxyphenylalanine, region and sex-specific changes have been observed [14]. Individuals with the Val158 allele perform less well on tests of working memory and executive cognition assessed by several methods [15-18]. On the other hand, the Met158 allele, although associated with better cognitive performance, leads to higher anxiety [19] and emotionality, as summarized in the ‘warrior’ (Val158) versus ‘worrier’ (Met158) model [20]. The two alleles potentially represent an example of balanced selection: counterbalancing effects of the Val and Met alleles in stress resiliency and cognition, respectively, may account for their conservation and high abundance across populations. This serves as a reminder that many of the common alleles with roles in diseases are not disease alleles per se.
Met158 leads to enhanced emotionality as visualized by emotional responses assessed by Positron Emission Tomography (PET) and functional Magnetic Resonance Imaging (fMRI). Met158 leads to reduced pain thresholds and increased emotional response to pain [21-22], apparently via diminished regional opioid responses [21]. Amygdala fMRI response to emotional challenge is stronger in Met158 carriers [23], and the effect also appears to be additive with the low expression allele of the serotonin transporter promoter polymorphism [24]. These fMRI measures of metabolic activity of regions, such as the amygdala and hippocampus, appear to have validity for understanding emotion [25], leading to their intensive use, and creating the opportunity for imaging genetic studies. Interestingly, COMT Met/Met individuals present increased reactivity and stronger connectivity of brain circuitry implicated in generating and regulating emotional responses [23]. This circuitry includes the amygdala, orbito-frontal and ventro-lateral prefrontal cortex and the hippocampus. It is clear that specific brain regions play an important role in emotion. However, they do not work in isolation, but are part of networks that are responsible for perception, emotion and cognitive processes [26].
Maladaptation to stress is critical in the development of many psychiatric disorders, including mood and anxiety disorders [27]. For COMT, the context-dependent effect of the Met158 allele on emotionality was demonstrated in a study conducted with Rwandan refugees [28]. Post-traumatic stress disorder (PTSD) was more common in carriers of the Met158 alleles, except that the highest levels of stress exposure overwhelmed the resiliency effect of the Val158 allele, as might have been expected. The effects of genes on emotion is strongly influenced by prior exposure to environmental stress [29], but this study of Kolassa and colleagues [28] provides evidence that the interactions between genes and environmental stress are furthermore not necessarily linear or monophasic. Of interest is recent evidence for epigenetic regulation of the COMT Val158 allele by lifetime stress exposure [30]. Greater stress correlates with reduced methylation at the CpG dinucleotide created by the Val158 allele, possibly altering the expression of COMT and traits associated with it.
5-HTTLPR
The serotonin transporter gene is responsible for the re-uptake of serotonin from the synaptic cleft and is a major target in the pharmacologic treatment of depression and anxiety. A common polymorphism (5-HTTLPR) is located upstream from the gene, SLC6A4 [31]. A 14-repeat allele (S) has reduced transcriptional efficiency compared to the 16 repeat allele (L). Moreover, the L allele frequently contains a relatively common, A→G substitution that makes it functionally equivalent to the low-expression S allele via binding of a defined transcription factor [32]. Subsequent to initial association studies to anxiety, Hariri and colleagues [33] observed that the low transcription allele (S) increased activation of the amygdala after passively viewed emotional stimuli. As mentioned above, the metabolic response of the amygdala to emotional challenge is contingent on modulation by other regions [23]. Consistent with this, connectivity studies revealed that the low expression allele altered the functional coupling between the amygdala and the ventro-medial prefrontal cortex (vmPFC) [34] and the perigenual cingulate [35], potentially impairing fear extinction by the medial prefrontal cortex. S allele carriers showed increased coupling between the amygdala and vmPFC and relative uncoupling between the amygdala and the perigenual region. At the neural level, multiple lines of evidence link the S allele to stronger emotional arousal (see [36], for a review). These include structural alterations in the uncinate fasciculus, which connects the amygdala and PFC [37], in the pulvinar [38] and in the amygdala itself [39].
As with COMT, there is accumulating evidence that 5-HTTLPR and stress interact to determine susceptibility to disorders later in life [29]. In the case of the serotonin transporter, however, the evidence is both more extensive and more conflicting, with contradictory meta-analyses [40-41]. Carriers of the low transcribing allele appear to be more likely to be depressed and suicidal following stressful life events as compared to individuals with two copies of the high expression allele [29]. The same effect was observed in other populations, including substance abusers, who overall have high exposure to stress and a high risk of suicidality [42]. As reviewed in [36], functional findings providing clues to the mechanisms of effects of 5-HTT reduction-of-function genotypes on emotion include increased acquisition of conditioned fear responses [43], increased auditory startle [44-45] and increased hypothalamic pituitary adrenal (HPA) axis activation to aversive stimuli [46-49].
The findings on human 5-HTTLPR are paralleled by studies in the rhesus macaque, mice and rats. To some extent, these studies are important as validating the sometimes conflicting human findings, but they have also enabled further exploration of the effects of low and high expression serotonin transporter genotypes. The rhesus macaque has an orthologous rh-HTTLPR polymorphism that leads to increased stress responses including cortisol release, and again only in the context of early life stress exposure, namely maternal separation [50]. In the rhesus macaque, the lower expression allele is associated with lower volumes of several of the same brain regions that are affected in humans, including the amygdala, PFC and pulvinar [51], and increased metabolic activity was observed in these regions in a stress paradigm involving relocation [52]. There is no rodent orthologue of 5-HTTLPR, but in both mice and rats silencing of 5-HTT heightens anxiety-like behaviors, impairs fear extinction and exaggerates HPA axis response [53, 54]. The finding that 5-HTT null mice have higher excitatory dendritic spine density on amygdala neurons and increased PFC dendritic arborization [55] again points to a developmental effect of this gene and its functional variants. The plasticity of this interaction is underlined by the ability of SSRIs administered early in development to mimic the 5-HTT knockout mouse, at least in terms of anxiety behaviors [56].
NPY
Neuropeptide Y (NPY) is an anxiolytic neuropeptide whose release is induced by stress. The release of NPY influences stress response and vulnerability to PTSD, for example in combat veterans [57]. At the molecular level, NPY RNA and neuropeptide expression is modulated by variation in the NPY promoter region, as shown in vitro and in vivo [58]. Individuals with low expression haplotypes exhibit stronger hemodynamic responses in the amygdala when presented with threat-related stimuli (facial expressions), lower endogenous opioid release during a pain stressor, and greater trait anxiety [58]. Also, amygdala activation to angry faces is greater in normal [58] and depressed patients [59] with low expression NPY genotypes. The dilution of effect of this functional locus from molecule to complex behavior is described in Box 2.
FKBP5
Acute stress activates hypothalamic release of corticotropin-releasing hormone (CRH) from the paraventricular nulecus to the pituitary, where it stimulates the secretion of the adrenocorticotropic hormone (ACTH). CRH directly, and through the action of ACTH, regulates adrenal cortisol release, steroidogenesis and catecholamine synthesis and release by the adrenal gland. Negative feedback to block release of CRH is necessary for normal function of the HPA axis, preventing prolonged or excessive activation [60]. Glucocorticoids mediate physiologic responses to stress but also reduce release of these neuropeptides by activation of glucocorticoid receptors (GR) in the paraventricular nucleus [60]. GR is a ligand-activated transcription factor that translocates from the cytosol to the nucleus after binding cortisol. GR function is regulated by a large molecular complex [61]. This molecular machinery includes hsp90/hsp70 chaperones and a number of co-chaperones, including FKBP5, a co-chaperone of hsp90 [62]. When FKBP5 is bound to the GR complex, the receptor has lower affinity for cortisol. Increased expression of FKBP5 therefore leads to cellular glucocorticoid resistance [63]. Although the precise functional locus has not been mapped, functional variation in the FKBP5 gene has been associated with response to antidepressants, recurrence of depressive episodes [64], suicide attempts in bipolar patients [65] and incomplete normalization of stress-elicited cortisol secretion [66]. Moreover, it has been shown that FKBP5 interacts with childhood trauma to predict PTSD [67] and suicidal behavior [68]. In controls, several FKBP5 loci are associated with high protein expression, increased glucocorticoid resistance, and thus reduced dexamethasone suppression [64]. However, in the presence of PTSD this functional association is altered. An interaction between high expression FKBP5 alleles and childhood trauma increases risk for PTSD and these alleles are associated with increased glucocorticoid sensitivity [67]. A similar relationship is observed in depressed patients [64]. Thus, genetic variation in FKBP5 may modulate effects of childhood trauma on cortisol release and abnormal protein expression may lead to altered GR responsiveness in target organs and long-lasting alterations in HPA axis reactivity.
PACAP
Among the genes that modulate stress response, the pituitary adenylate cyclase-activating polypeptide (PACAP) and its selective PAC1 receptor have recently been demonstrated to play a role in abnormal stress response underlying PTSD in females [69]. In this study, PACAP blood levels were correlated with the diagnosis and symptoms of PTSD. Ressler et al. [69] found that a single nucleotide polymorphism (SNP) within the PAC1 receptor gene (ADCYAP1R1) was associated with PTSD in females but not males, in two distinct samples of highly traumatized urban subjects. Expression of this gene is induced in the amygdala by fear [69]. The SNP, which alters mRNA expression, is located in an oestrogen response element, which could explain the striking gender difference. In female rats, oestradiol was shown to increase both Adcyap1 and Adcyap1r1 transcript levels in the bed nucleus of the stria terminalis, a component of the extended amygdala. Finally, methylation of ADCYAP1R1 was associated with PTSD.
Concluding remarks
Emotional differences arise because of the action of hundreds of genes, complex circuitries and environmental exposures. One can reasonably argue that it will not be possible to disentangle these factors or that, if it is possible, this will require far better measurement of behavior, intermediate processes and environmental exposures. However, the fact that emotionality is genetically driven—in other words, heritable—has enabled genetic approaches to be successfully applied against that large portion of the variance in emotionality which is driven by allelic differences. The five genes discussed here represent instances in which common functional variants alter pathways of emotion and stress response. Their naturally occurring genetic variants in humans, and parallel genetic models in other animals, represent a unique opportunity to disentangle the complexity of emotion and identify specific origins of individual differences in emotionality.
Multiple factors complicate the task of relating genes to emotion and suggest that future efforts to relate genes to emotion should be better focused, larger in scale, as well as deeper in genomic coverage. Gene × environment interactions play a critical role, suggesting that it is vital to study exposed populations, as exemplified here by the studies on Rwandan refugees [28], and that at other times environmental exposures may mask genetic effects. Most genes and environmental factors that alter brain function, alter the function of multiple neurons and circuits and may have effects elsewhere in the body, a phenomenon known as pleiotropy (see Box 3), suggesting that the genetics of emotionality is likely to be tied to several other problems.
Box 3. COMT and pleiotropy.
The COMT Val158Met polymorphism has pleiotropic effects on both emotion and cognition and it represents an example of a multilevel confirmation of the effect of a functional polymorphism. COMT was not genome-wide significant on any GWAS conducted so far, but has a functional locus that coherently influences several aspects of emotion and cognition. The understanding of this polymorphism’s effects was extended via pharmacological challenge studies and use of transgenic animal models. The effects of COMT Val158Met are pleiotropic, consistent with the multiple functions of the enzyme and its widespread distribution in the brain and body.
In numerous studies and different populations including normal controls [15-17], head-injured patients [84], schizophrenic patients and their healthy siblings [16], the Met158 allele is associated with better performance on neuropsychological tests of executive cognition. The role of COMT as a cognitive predictor has been comprehensively reviewed [85] and meta-analyzed with variable results, as in Barnett et al [86], where the genotype was associated with IQ but not with other cognitive measures. Subsequently, Met158 was again associated with higher IQ in a large longitudinal dataset [87]. However, all observations made on the effects of the Val158Met polymorphism are consistent with the role played by dopamine in regulating frontal cortical function. COMT inhibitors improve working memory and attention in animals [88] and in humans [89], but the improvement is lost in Met158/Met158 homozygotes, who tend to have superior baseline prefrontal function assayed by fMRI [90]. Also, under easy task conditions, where cognitive performance of individuals with different genotypes is equivalent, there is reduced cortical efficiency in Val158/Val158 homozygotes, consistent with lower dopamine levels.
Gene × gene interaction is a territory largely unexplored (see also Box 4). The complexity of the brain and its cellular molecular networks seems to suggest that variation in emotionality should arise due to the simultaneous action of variants at many genes. Under a heterogeneity model, different variants play a role in different individuals. Under a polygenic model, inheritance of many genetic variants is necessary and the effects may or may not be additive. Under the epistatic model, effects are non-additive—it is unique combinations of alleles at different loci that lead to the phenotype. Based on ratios of trait concordances between individuals at different degrees of relationship, emotionality, other personality traits and various psychiatric diseases may be predominantly additive in their inheritance (see, for example, [70] for a review of the MZ:DZ ratios for ten different addictive disorders). As mentioned above, the effect of COMT and 5-HTTLPR on fMRI emotion response appears to be additive [24]. However, it would be unsurprising if instances of epistatic interaction were identified: for example such instance has been reported between COMT Val158Met and various other genes [71], although the functional variants have not been identified at these other genes. Replication will be particularly important for gene × gene interactions, as will care in correcting for the multiple comparisons necessitated by the search for epistatic interactions.
Box 4. Priorities for future research.
Achieve deep (i.e., rich) phenotyping that includes the use of intermediate phenotypes or endophenotypes.
Establish consistency in definition of phenotypes across studies.
Carry out GWAS using intermediate phenotypes or endophenotypes.
Perform genome sequencing of individuals with extreme phenotypes, including intermediate phenotypes.
Identify, evaluate, select and prioritize rare and uncommon variants.
Establish the function of variants.
Perform gene-wide studies that incorporate multiple rare variants at the same gene.
Use population isolates with founder characteristics and families for the identification of rare and uncommon genetic variants with large effects on disease.
Study exposed populations and develop new measures of exposure, including epigenetic signatures.
GWAS is a hypothesis-free search strategy with great power to detect effects of relatively common alleles of moderate effect. However, and as may be a particular problem for genetic studies on emotionality, GWAS is unlikely to detect the effects of rare and uncommon alleles because, as sample size is increased, various types of within-sample heterogeneity, including consistency of phenotyping, ascertainment, environmental exposure and genetic background, are likely to detract from power. Different rare alleles at the same gene are likely to reside on different genetic backgrounds (haplotypes). A GWAS of neuroticism, a personality trait that increases the likelihood of internalizing disorders, which include depression and anxiety disorders, failed to identify any genome-wide significant loci, despite the fact that it was based on the genotyping of individuals with the most extreme phenotypes from a total sample approaching 88,000 [72]. Furthermore, this study failed to detect—at a genome-wide level—the influence on emotion of several genes that have been discovered and otherwise validated, as the genes discussed in this review. The genetic factors underlying neuroticism are largely shared with those that influence liability to internalizing disorders [73]. A powerful implication of these heritability findings is that it should be feasible to map the locations of genes that lead to neuroticism, to identify the relevant functional loci and define a pathway of causation from gene to behavior. The lack of genome-wide significant loci for this heritable trait could be due to the fact that heritability of neuroticism arises from multiple common alleles explaining a small percentage of the variance, an even larger number of rare alleles, the need for a larger sample, the strategy or implementation of this study, or as is most likely, a combination of factors. It is likely that increasing the sample size will result in the genome-wide capture of some genes influencing emotion, but the question is how many, and how much of the heritable trait variance. Instead of performing new genome scans of even larger samples, it would perhaps be more important to shift attention to the use of intermediate phenotypes, or to endophenotypes, for the identification of functional common variation as of rare and uncommon variation.
The search for rare variation will also require the use of massively parallel sequencing technologies, the selection of appropriate phenotypes and the insightful evaluation, prioritization and follow-up of the great number of rare variants that will be identified [74]. The use of isolated populations with founder characteristics, as well as individual families, is likely to be highly beneficial to reduce genetic heterogeneity, as with an HTR2B stop codon predictive of severe impulsivity, but only common in individuals of Finnish descent [75].
These observations suggest that emotion and diseases associated with emotional dysregulation will not be “solved” by a one-to-one correspondence of gene to behavior as currently defined. This encourages a multilevel approach to the genetic analysis of emotion, and a stepwise deconstruction of current typology based on predictive genetic and psychophysiologic measures, together with observed clinical state and history of exposure.
Glossary
- Allele
any of the alternative variants at a given locus
- Candidate gene study
a study focused on genes thought to have a causal role
- Diplotype
a genotype composed of haplotypes
- Emotion
the assignment of affective valence to objects, states and situations
- Emotionality
the tendency to assign strong emotional valence to objects, states or situations
- Endophenotype
a heritable, disease associated, intermediate phenotype
- Epistasis
non-additive interaction between alleles at different loci affecting a phenotype
- Genome-wide association study
allele-based linkage using markers genotyped across the entire genome
- Haplotype
a combination of alleles at loci located on the same chromosome
- Intermediate phenotype
mechanism-related manifestation of a complex phenotype
- Pleiotropy
different phenotypic effects of the same allele
- Polymorphism
inter-individual genetic variation occurring at a frequency of >1%
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Phillips ML, et al. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003;54:515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 2.Whalen PJ, et al. Functional neuroimaging studies of the amygdala in depression. Semin Clin Neuropsychiatry. 2002;7:234–242. doi: 10.1053/scnp.2002.35219. [DOI] [PubMed] [Google Scholar]
- 3.Etkin A, et al. Individual differences in trait anxiety predict the response of the basolateral amygdala to unconsciously processed fearful faces. Neuron. 2004;44:1043–1055. doi: 10.1016/j.neuron.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Etkin A, et al. Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn Sci. 2011;15:85–93. doi: 10.1016/j.tics.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elliott R, et al. Affective cognition and its disruption in mood disorders. Neuropsychopharmacology. 2011;36:153–182. doi: 10.1038/npp.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hariri A. The neurobiology of individual differences in complex behavioral traits. Annu Rev Neurosci. 2009;32:225–247. doi: 10.1146/annurev.neuro.051508.135335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchard TJ, Jr, Loehlin JC. Genes, evolution, and personality. Behav Genet. 2001;31:243–273. doi: 10.1023/a:1012294324713. [DOI] [PubMed] [Google Scholar]
- 8.Sen S, et al. Meta-analysis of the association between a serotonin transporter promoter polymorphism (5-HTTLPR) and anxiety-related personality traits. Am J Med Genet B Neuropsychiatr Genet. 2004;127:85–89. doi: 10.1002/ajmg.b.20158. [DOI] [PubMed] [Google Scholar]
- 9.Johnstone T, et al. Stability of amygdala BOLD response to fearful faces over multiple scan sessions. Neuroimage. 2005;25:1112–1123. doi: 10.1016/j.neuroimage.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 10.Manuck SB, et al. Temporal stability of individual differences in amygdala reactivity. Am J Psychiatry. 2007;164:1613–1614. doi: 10.1176/appi.ajp.2007.07040609. [DOI] [PubMed] [Google Scholar]
- 11.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 12.Yavich L, et al. Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J Neurosci. 2007;27:10196–10209. doi: 10.1523/JNEUROSCI.0665-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scanlon PD, et al. Catechol-O-methyltransferase: thermolabile enzyme in erythrocytes of subjects homozygous for allele for low activity. Science. 1979;203:63–65. doi: 10.1126/science.758679. [DOI] [PubMed] [Google Scholar]
- 14.Huotari M, et al. Brain catecholamine metabolism in catechol-O-methyltransferase (COMT)-deficient mice. Eur J Neurosci. 2002;15:246–256. doi: 10.1046/j.0953-816x.2001.01856.x. [DOI] [PubMed] [Google Scholar]
- 15.Barnett JH, et al. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol Psychiatry. 2007;12:502–509. doi: 10.1038/sj.mp.4001973. [DOI] [PubMed] [Google Scholar]
- 16.Egan MF, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci, U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malhotra AK, et al. A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. Am J Psychiatry. 2002;159:652–654. doi: 10.1176/appi.ajp.159.4.652. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg TE, et al. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Arch Gen Psychiatry. 2003;60:889–896. doi: 10.1001/archpsyc.60.9.889. [DOI] [PubMed] [Google Scholar]
- 19.Enoch MA, et al. Genetic origins of anxiety in women: a role for a functional catechol-O-methyltransferase polymorphism. Psychiatr Genet. 2003;13:33–41. doi: 10.1097/00041444-200303000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Ducci F, Goldman D. Genetic approaches to addiction: genes and alcohol. Addiction. 2008;103:1414–1428. doi: 10.1111/j.1360-0443.2008.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zubieta JK, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]
- 22.Diatchenko L, et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 23.Drabant EM, et al. Catechol-O-methyltransferase val158met genotype and neural mechanisms related to affective arousal and regulation. Arch Gen Psychiatry. 2006;63:1396–1406. doi: 10.1001/archpsyc.63.12.1396. [DOI] [PubMed] [Google Scholar]
- 24.Smolka MN, et al. Gene-gene effects on central processing of aversive stimuli. Mol Psychiatry. 2007;12:307–317. doi: 10.1038/sj.mp.4001946. [DOI] [PubMed] [Google Scholar]
- 25.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 26.Pessoa L. Emergent processes in cognitive-emotional interactions. Dialogues Clin Neurosci. 2010;12:433–448. doi: 10.31887/DCNS.2010.12.4/lpessoa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McEwen BS. Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- 28.Kolassa IT, et al. The risk of posttraumatic stress disorder after trauma depends on traumatic load and the catechol-o-methyltransferase Val(158)Met polymorphism. Biol Psychiatry. 2010;67:304–308. doi: 10.1016/j.biopsych.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Caspi A, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 30.Ursini G, et al. Stress-related methylation of the catechol-O-methyltransferase Val 158 allele predicts human prefrontal cognition and activity. J Neurosci. 2011;31:6692–6698. doi: 10.1523/JNEUROSCI.6631-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lesch KP, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 32.Hu X, et al. Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006;78:815–826. doi: 10.1086/503850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hariri AR, et al. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297:400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- 34.Heinz A, et al. Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat Neurosci. 2005;8:20–21. doi: 10.1038/nn1366. [DOI] [PubMed] [Google Scholar]
- 35.Pezawas L, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 36.Caspi A, et al. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim MJ, Whalen PJ. The structural integrity of an amygdala-prefrontal pathway predicts trait anxiety. J Neurosci. 2009;29:11614–11618. doi: 10.1523/JNEUROSCI.2335-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young KA, et al. 5HTTLPR polymorphism and enlargement of the pulvinar: unlocking the backdoor to the limbic system. Biol Psychiatry. 2007;61:813–818. doi: 10.1016/j.biopsych.2006.08.047. [DOI] [PubMed] [Google Scholar]
- 39.Pezawas L, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 40.Risch N, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karg K, et al. The Serotonin Transporter Promoter Variant (5-HTTLPR), Stress, and Depression Meta-analysis Revisited: Evidence of Genetic Moderation. Arch Gen Psychiatry. 2011;68:444–454. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy A, et al. Interaction between childhood trauma and serotonin transporter gene variation in suicide. Neuropsychopharmacology. 2007;32:2046–2052. doi: 10.1038/sj.npp.1301331. [DOI] [PubMed] [Google Scholar]
- 43.Lonsdorf TB, et al. Genetic gating of human fear learning and extinction: possible implications for gene-environment interaction in anxiety disorder. Psychol Sci. 2009;20:198–206. doi: 10.1111/j.1467-9280.2009.02280.x. [DOI] [PubMed] [Google Scholar]
- 44.Armbruster D, et al. Serotonin transporter gene variation and stressful life events impact processing of fear and anxiety. Int J Neuropsychopharmacol. 2009;12:393–401. doi: 10.1017/S1461145708009565. [DOI] [PubMed] [Google Scholar]
- 45.Brocke B. Serotonin transporter gene variation impacts innate fear processing: acoustic startle response and emotional startle. Mol Psychiatry. 2006;11:1106–1112. doi: 10.1038/sj.mp.4001908. [DOI] [PubMed] [Google Scholar]
- 46.Way B, Taylor S. The serotonin transporter promoter polymorphism (5-HTTLPR) is associated with cortisol response to psychosocial stress. Biol Psychiatry. 2010;67:487–492. doi: 10.1016/j.biopsych.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gotlib IH, et al. HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol Psychiatry. 2008;63:847–851. doi: 10.1016/j.biopsych.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexander N, et al. Gene-environment interactions predict cortisol responses after acute stress: implications for the etiology of depression. Psychoneuroendocrinology. 2009;34:1294–1303. doi: 10.1016/j.psyneuen.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 49.Mueller A, et al. The role of the serotonin transporter polymorphism for the endocrine stress response in newborns. Psychoneuroendocrinology. 2010;35:289–296. doi: 10.1016/j.psyneuen.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Spinelli S, et al. Association between the recombinant human serotonin transporter linked promoter region polymorphism and behavior in rhesus macaques during a separation paradigm. Dev Psychopathol. 2007;19:977–987. doi: 10.1017/S095457940700048X. [DOI] [PubMed] [Google Scholar]
- 51.Jedema HP, et al. Cognitive impact of genetic variation of the serotonin transporter in primates is associated with differences in brain morphology rather than serotonin neurotransmission. Mol Psychiatry. 2010;15:512–22. doi: 10.1038/mp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalin NH, et al. The serotonin transporter genotype is associated with intermediate brain phenotypes that depend on the context of eliciting stressor. Mol Psychiatry. 2008;13:1021–1027. doi: 10.1038/mp.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci. 2008;9:85–96. doi: 10.1038/nrn2284. [DOI] [PubMed] [Google Scholar]
- 54.Homberg J, et al. Characterization of the serotonin transporter knockout rat: a selective change in the functioning of the serotonergic system. Neuroscience. 2007;146:1662–1672. doi: 10.1016/j.neuroscience.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 55.Hariri AR, Holmes A. Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn Sci. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 56.Ansorge M, et al. Early-life blockade of 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- 57.Morgan CA, 3rd, et al. Plasma neuropeptide-Y concentrations in humans exposed to military survival training. Biol Psychiatry. 2000;47:902–909. doi: 10.1016/s0006-3223(99)00239-5. [DOI] [PubMed] [Google Scholar]
- 58.Zhou Z, et al. Genetic variation in human NPY expression affects stress response and emotion. Nature. 2008;425:997–1001. doi: 10.1038/nature06858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mickey BJ, et al. Emotion Processing, Major Depression, and Functional Genetic Variation of Neuropeptide Y. Arch Gen Psychiatry. 2011;68:158–66. doi: 10.1001/archgenpsychiatry.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovacs JK, et al. Glucocorticoid negative feedback selectively targets vasopressin transcription in parovocellular neurosecretory neurons. J Neurosci. 2000;20:3843–3852. doi: 10.1523/JNEUROSCI.20-10-03843.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pratt WB, et al. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol. 2006;172:111–138. doi: 10.1007/3-540-29717-0_5. [DOI] [PubMed] [Google Scholar]
- 62.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 63.Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34S:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 64.Binder EB, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- 65.Willour VL, et al. Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry. 2008;14:261–268. doi: 10.1038/sj.mp.4002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ising M, et al. Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur J Neurosci. 2008;28:389–398. doi: 10.1111/j.1460-9568.2008.06332.x. [DOI] [PubMed] [Google Scholar]
- 67.Binder EB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1–15. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roy A, et al. Interaction of FKBP5, a Stress-Related Gene, with Childhood Trauma Increases the Risk for Attempting Suicide. Neuropsychopharmacology. 2010;35:1674–1683. doi: 10.1038/npp.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ressler KJ, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldman D, et al. The genetics of addictions: uncovering the genes. Nat Rev Genet. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- 71.Nicodemus KK, et al. Evidence for statistical epistasis between catechol-O-methyltransferase (COMT) and polymorphisms in RGS4, G72 (DAOA), GRM3, and DISC1: influence on risk of schizophrenia. Hum Genet. 2007;120:889–906. doi: 10.1007/s00439-006-0257-3. [DOI] [PubMed] [Google Scholar]
- 72.Shifman S, et al. A whole genome association study of neuroticism using DNA pooling. Mol Psychiatry. 2008;13:302–312. doi: 10.1038/sj.mp.4002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hettema JM, et al. A population-based twin study of the relationship between neuroticism and internalizing disorders. Am J Psychiatry. 2006;163:857–864. doi: 10.1176/ajp.2006.163.5.857. [DOI] [PubMed] [Google Scholar]
- 74.Goldstein DB. Common genetic variation and human traits. N Eng J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 75.Bevilacqua L, et al. A population-specific HTR2B stop codon predisposes to severe impulsivity. Nature. 2010;468:1061–1066. doi: 10.1038/nature09629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eaves LJ, Eysenck HJ, Martin NG. Genes, Culture and Personality: An Empirical Approach. Academic Press; New York: 1989. [Google Scholar]
- 77.Biederman J, et al. Further evidence of association between behavioral inhibition and social anxiety in children. Am J Psychiatry. 2001;158:1673–1679. doi: 10.1176/appi.ajp.158.10.1673. [DOI] [PubMed] [Google Scholar]
- 78.Caspi A, Silva PA. Temperamental qualities at age three predict personality traits in young adulthood: Longitudinal evidence from a birth cohort. Child Dev. 1995;66:486–498. doi: 10.1111/j.1467-8624.1995.tb00885.x. [DOI] [PubMed] [Google Scholar]
- 79.Fox NA, et al. Behavioral inhibition: Linking biology and behavior within a developmental framework. Annu Rev Psychol. 2005;56:235–262. doi: 10.1146/annurev.psych.55.090902.141532. [DOI] [PubMed] [Google Scholar]
- 80.Glahn DC, et al. Neuroimaging endophenotypes: strategies for finding genes influencing brain structure and function. Hum Brain Mapp. 2007;28:488–501. doi: 10.1002/hbm.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fox AS, et al. Trait-like brain activity during adolescence predicts anxious temperament in primates. PLoS ONE. 2008;3:e2570. doi: 10.1371/journal.pone.0002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oler JA, et al. Amygdalar and hippocampal substrates of anxious temperament differ in their heritability. Nature. 2010;466:864–867. doi: 10.1038/nature09282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rogers J, et al. Genetic influences on behavioral inhibition and anxiety in juvenile rhesus macaques. Genes Brain Behav. 2008;7:463–469. doi: 10.1111/j.1601-183X.2007.00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lipsky RH, et al. Association of COMT Val158Met genotype with executive functioning following traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2005;17:465–471. doi: 10.1176/jnp.17.4.465. [DOI] [PubMed] [Google Scholar]
- 85.Tunbridge EM, et al. Catechol-O-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60:141–151. doi: 10.1016/j.biopsych.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 86.Barnett JH, et al. Meta-analysis of the cognitive effects of the Catechol-O-Methyltransferase gene Val158/108Met polymorphism. Biol Psychiatry. 2008;64:137–144. doi: 10.1016/j.biopsych.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 87.Barnett JH, et al. Cognitive effects of genetic variation in monoamine neurotransmitter systems: a population-based study of COMT, MAOA, and 5HTTLPR. Am J Med Genet B Neuropsychiatr Genet. 2011;156:158–167. doi: 10.1002/ajmg.b.31150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tunbridge EM, et al. Catechol-o-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci. 2004;24:5331–5335. doi: 10.1523/JNEUROSCI.1124-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Apud JA, et al. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology. 2007;32:1011–1020. doi: 10.1038/sj.npp.1301227. [DOI] [PubMed] [Google Scholar]
- 90.Mattay VS, et al. Catechol-O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci, U S A. 2003;100:6186–6191. doi: 10.1073/pnas.0931309100. [DOI] [PMC free article] [PubMed] [Google Scholar]