

Abstract
Despite its decreasing frequency in developed countries, gastric cancer remains a significant health burden. The aim of the present study was to construct cDNA libraries and analyze differentially expressed genes related to this disease. Gene expression profiles were generated with suppressive subtraction hybridization (SSH). We constructed eight SSH libraries, four representing up-regulated genes and four representing down-regulated genes in tumor tissues. Our approach revealed that several genes are abnormally expressed in gastric cancer. We also identified global deregulation of several pathways involved in the maintenance of normal gastric homeostasis. The results of this study support the view that, as a result of complex pathogenesis, diversity of genomic aberrations and multiplicity of carcinogenic causes, gastric cancer cannot be reduced to a single molecule. Our results may contribute new insight into molecular aspects of the disease and may prove advantageous for future development of therapeutic targets and diagnostic molecular markers.
Introduction
Gastric cancer is the second most common cause of cancer-related death worldwide (1). Although extensive studies have been performed to identify genes involved in the disease development and progression, the prognosis for patients with gastric cancer remains poor and little improvement of long-term survival has been achieved (2). Due to its non-specific symptoms, gastric adenocarcinoma is often diagnosed at an advanced stage (2). The estimated five-year survival rate for patients with advanced gastric cancer is only 24-27% (3). The main reason for this low survival rate is that the disease spreads via metastases to distant organs or invades through the stomach wall into adjacent structures, such as the soft tissues and fat surrounding the stomach as well as other organs including the spleen, pancreas, large intestine, small intestine, liver and large blood vessels. In Japan, where the incidence of gastric cancer is high, nationwide screening programs have been implemented in order to detect the disease in its early stages (3-5). The five-year survival rates improved and exceed 90% (3). However, these screening programs are not suitable for European and Western countries, where incidence is low, due to the cost and invasiveness of the screening methods. The most efficacious approach would be to discover reliable non-invasive biomarkers, which may be used in early detection of the disease. Pathological conditions such as cancer alter the expression profiles in tumor cells; thus identifying differentially expressed genes is a common starting point in understanding the molecular mechanism underlying the disease development (6,7). Comprehensive gene expression analyses identified relevant genes whose expression profiles appeared to have a role in gastric carcinogenesis (8). However, as new information was gained, it became evident that gastric cancer is a complex and heterogeneous disease. Despite the identification of a number of altered oncogenes, tumor suppressors, DNA repair genes, growth factors, cell adhesion molecules and other genes, it is evident that various genes involved in disease initiation and progression remain unidentified. Therefore, determining the molecular signatures of gastric cancer types is of key importance in understanding gastric cancer carcinogenesis (9). The present study aimed to construct cDNA libraries using suppression subtractive hybridization (SSH) and analyze differentially expressed genes related to gastric cancer.
Materials and methods
Tissue samples. Tumor and corresponding non-tumor tissues at least 7 cm away from the edge of the adenocarcinoma were obtained from 3 female patients who were admitted to the Clinical Department for Abdominal Surgery at the University Medical Center of Ljubljana. Samples were also obtained from 1 patient with chronic gastritis during routine gastroscopy performed at the Clinical Department for Gastroenterology at the University Medical Centre of Ljubljana, Slovenia. Specimens were stored in RNALater™ (Ambion, Austin, TX, USA) at -20˚C according to the manufacturer's recommendations. The study was approved by the Slovenian Medical Ethics Committee and confidentiality of personal medical data as well as other data relating to individual identification has been confirmed.
RNA isolation. RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA, USA) after grinding the tissues with glass homogenizers on ice. Approximately 5 µg of glycogen (Invitrogen) was added to the samples prior to the precipitation step. Its quality was assessed with RNA 6000 Nano LabChip Kit (Agilent Technologies, Santa Clara, CA, USA) on a 2100 Bioanalyzer (Agilent). Samples with RNA integrity numbers greater than 8 were subjected to subsequent procedures.
SSH. cDNA subtraction was carried out using a Clontech cDNA subtraction kit (BD Bioscience Clontech, San Jose, CA, USA) following the manufacturer's instructions with certain modifications. Tumor cDNA was used as the driver and counterpart non-tumor cDNA as the tester to construct the library of down-regulated genes. Non-tumor cDNA was used as the driver and tumor cDNA was used as the tester to construct the library of up-regulated genes.
Briefly, total RNA (1 µg) was reverse transcribed with the SMART PCR cDNA synthesis kit (BD Bioscience Clontech) and the resulting cDNAs were purified using Qiagen Qiaquick columns (Qiagen, Valencia, CA, USA). Following the 3-h RsaI restriction, the cDNAs were purified with phenol extraction and ethanol precipitation. A total of 2 aliquots of the tester cDNAs were ligated overnight to adaptors 1 and 2R, respectively. Ligation was stopped by the addition of 1 µl of EDTA/glycogen and incubation of the reactions for 5 min at 72˚C. The efficacy of ligation was analyzed by polymerase chain reaction (PCR) using primers that span the adaptor/cDNA junctions of testers. The first hybridization was performed at 68˚C for 8 h. In this procedure, the excess of driver cDNA was added to each portion of tester cDNA. During the second hybridization, the two samples mentioned above were mixed together and fresh denatured driver cDNA was added to further enrich for differentially expressed fragments. Reactions were incubated overnight at 68˚C. Differentially expressed sequences were then selectively amplified with PCR. Only fragments containing different adaptors were amplified exponentially. The efficiency of subtraction was analyzed by comparing the abundance of cDNAs prior to and after subtraction by PCR using GAPDH and actin as controls. The subtracted libraries were cloned into pCRII vector using the TOPO TA dual promoter cloning kit (Invitrogen) and transformed into TOP10F' competent cells. Plasmids were isolated using NucleoSpin® Multi-96 plus plasmid (Macherey-Nagel, Germany) or QiaPrep Spin Miniprep Kit (Qiagen) and sequenced with M13 reverse primers at Macrogen, Korea or VBC-Biotech, Germany.
Analysis of subtracted libraries. The obtained cDNA sequences were manually edited in the Vector NTI Advance™ 9.0 (Invitrogen) software to remove vector and adapter sequences and to correct ambiguous base calls by comparison with the electropherograms to enhance the quality and reliability of the data. cDNA sequences were prepared in FASTA format in text files and batch-subjected to the web application Megablast available at the http://blast.ncbi.nlm.nih.gov/Blast.cgi. In the event that there was no hit in this application, the sequences were manually blasted with another web software, BLAT (10). For known gene homologues we obtained gene ontology (GO) information with the help of FatiGO and PubMed Gene to describe expressed sequence tags (ESTs) in functional terms (11). We also used Onto Tools and GeneInfoWiz to further evaluate functional profiles of obtained databases of cDNAs (12,13).
Quantitative real-time PCR (qRT-PCR) analysis. qRT-PCR was used to confirm the differential expression of selected clones identified by SSH. From the total population of clones we selected the following genes for analysis: B2M, CCNB1, CIP29, EEF1A1, FOS, HMGB1, GKN1, MALAT1, PGA5, RDX, SOX4, SPINK1, TBL1XR1, TFF1 and TPT1. RNAs used for qRT-PCR were treated with DNase I (Sigma Aldrich, Germany) prior to cDNA synthesis to remove DNA contamination. total RNA (1 µg) from each sample was used to create first strand cDNA using Superscript II (Invitrogen) according to the manufacturer's instructions. The reverse transcription was conducted with random hexamers (Promega, Madison, WI, USA). qRT-PCR was carried out in triplicate according to the manufacturer's instructions using Taq Man gene expression assays (Applied Biosystems, Carlsbad, CA, USA) and was performed on ABI PRISM 7900HT sequence detection system (Applied Biosystems). Gene expression was normalized to 18S rRNA content with Taq Man rRNA control reagent kit (Applied Biosystems). The level of each cDNA was determined using the 2-∆∆Ct method. The expression level of each gene was expressed as log2-∆∆Ct (14).
Results
Construction of SSH libraries. SSH was used to isolate differentially expressed genes from tumor and non-tumor tissues of gastric cancer patients and from precancerous lesions of the patient with chronic gastritis. We constructed eight libraries, four representing up-regulated genes and four representing down-regulated genes in tumor tissues. Subtraction efficiency analysis revealed the effectively reduced abundance of non-differentially expressed genes. In non-subtracted libraries, the control genes GAPDH or actin were generally visible after 18 cycles, while in subtracted libraries they were visible after 28 or 33 cycles. The depletion of control genes in subtracted libraries showed that the genes contained mostly differentially expressed fragments. The resulting libraries were cloned and propagated in TOP10F' competent cells.
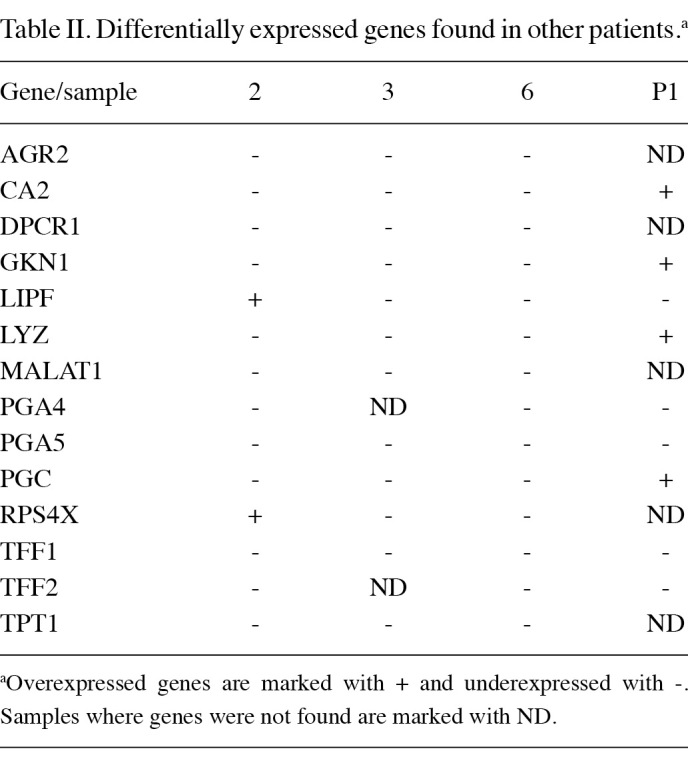
Sequencing and annotation of SSH cDNA libraries. Randomly chosen subsets of the clones were sequenced. We sequenced 621 clones from the up-regulated and 1797 clones from the down-regulated SSH libraries. Approximately 1500 good quality sequences were obtained. Of these, we were able to align approximately 1370 clones with MEGABLAST or BLAT. The sequences were deposited with the dbEST database (NCBI). We calculated the percentage of mitochondrial fragments, which were non-specifically amplified in the SSH process. The percentages ranged from 0 to 13.2%, which was considered to be acceptable. Unidentified and mitochondrial sequences, clones with short sequences and clones without inserts were excluded from further analyses. Unidentified sequences are probably redundant DNA or transcripts from regions that have yet to be annotated. Given that each cDNA clone obtained using SSH was digested with the restriction enzyme RsaI, certain ESTs correspond to partial regions of the same full-length cDNAs. Of the identified gene fragments, the sequences of certain gene fragments were repeated more than 5 times. These genes (Table I) are probably the most abundant genes in the obtained SSH cDNA libraries. It was shown previously that the frequency of a gene in the SSH library was correlated with the degree of the differential expression of the gene (15). According to fatigoGO biological process (at level 3), the most abundant over- and under-expressed ESTs were assigned to the following groups: cellular metabolic process (GO:0044237), regulation of biological process (GO:0050789), primary metabolic process (GO:0044238), cellular developmental process (GO:0048869), macromolecule metabolic process (GO:0043170), response to stress (GO:0006950), cell communication (GO:0007154), cellular component organization and biogenesis (GO:0016043), response to chemical stimulus (GO:0042221) and biosynthetic process (GO:0009058). Notably, the most abundant down-regulated genes were classified into the following groups: death (GO:0016265), defense response (GO:0006952), immune response (GO:0006955) response to external stimulus (GO:0009605), digestion (GO:0007586), establishment of localization (GO:0051234), cell proliferation (GO:0008283), response to biotic stimulus (GO:0009607), cell cycle (GO:0007049), cell adhesion (GO:0007155) cell proliferation (GO:0008283), catabolic process (GO:0009056), behaviour (GO:0007610) and protein localization (GO:0008104). We also identified genes that were differentially expressed in all of the examined samples (Table II).
Table 1. Most abundant transcripts in SSH libraries obtained from tissues of Slovenian patients with gastric cancer.

Table 2. Differentially expressed genes found in other patients.a.
To further characterize the SSH libraries, we generated lists of differentially expressed genes from which we removed repeated genes. The lists were subjected to analysis in Onto-Tools. Figs. 1-4 show that classification according to biological process and molecular function in all patients identified a large fraction of differentially expressed genes involved in cellular, developmental and immune systems, metabolic processes, biological regulation, binding, response to stimulus and localization. Other differentially expressed transcripts were distributed into several GO categories (Figs. 1-4).
Figure 1. Distribution of GO terms corresponding to the differentially expressed genes in patient 2 with gastric cancer. Functional profiles were generated with an Onto-Express tool from Onto-Tools software package. Numbers are the number of genes in the GO groups. (A) Up-regulated and (B) down-regulated groups of genes.

Figure 2. Distribution of GO terms corresponding to the differentially expressed genes in patient 3 with gastric cancer. Functional profiles were generated with an Onto-Express tool from the Onto-Tools software package. Numbers are the number of genes in the GO groups. (A) Up-regulated and (B) down-regulated groups of genes.

Figure 3. Distribution of GO terms corresponding to the differentially expressed genes in patient 6 with gastric cancer. Functional profiles were generated with an Onto-Express tool from the Onto-Tools software package. Numbers are the number of genes in the GO groups. (A) Up-regulated and (B) down-regulated groups of genes.

Figure 4. Distribution of GO terms of differentially expressed genes in patient with chronic gastritis (CG). Numbers are the number of genes in the GO groups. (A) Up-regulated and (B) down-regulated groups of genes.

The gene lists were then subjected to analyses in the Pathway program in Onto-Tools. This application searches the genes in the list and constructs a list of all associated pathways. The results are presented in Table III. Functional profiling revealed deregulation of a number of pathways, involved in genetic information processing, signal transduction, repair, cell motility, disease and carcinogenesis.
Table 3(i). Some of the noteworthy pathways involving differentially expressed genes found in Slovenian gastric cancer patients.

Table 3(ii). Continued.

Table 3(iii). Continued.

Table 3(iv). Continued.

qRT-PCR. To verify the results of SSH we randomly selected 15 genes and verified their expression in all samples. The quantitative RT-PCR was used to compare expression levels of selected genes in tumors with corresponding non-tumor samples from the same patient. We used 18S rRNA as a reference to establish an independent test for quantification. The expression levels of 11 candidate markers (B2M, FOS, GKN1, MALAT1, PGA5, RDX, SOX4, SPINK1, TBL1XR1, TFF1 and TPT1) were consistent in all patients with the expression profiles obtained with SSH. CCNB1, CIP29 and EEF1A1 were found to be up-regulated in tumor tissues of patient 6, and in patient 3 higher levels of EEF1A and HMGB1 were detected. However, with regard to SSH these genes were not detected in patients 6 and 3. Expression of these genes in patient 2 was consistent with the SSH profile. The reason for this discrepancy (patients 3 and 6) may be that we did not obtain all the clones after SSH or that the single-pass sequencing was not successful. However, our results indicated that a marked correlation exists between the quantitative RT-PCR and SSH.
Discussion
These are the first gene expression profiles of tumor and corresponding non-tumor gastric tissues in Slovenian patients. We have generated six libraries of over- and underexpressed genes of adenocarcinoma and two libraries of over- and underexpressed genes of chronic gastritis.
We have identified a number of overexpressed (ANXA2, BUB3, GUCY2C and NDUFA9) and underexpressed (CDKN1A, CTNNA1, CD44, LIPF, GKN1, PGA5 and TFF1) genes, which were previously described (16-24). Non-tumor tissues from all patients expressed high numbers of ESTs in the digestion, cellular metabolic and primary metabolic and regulation of metabolic process GO categories. Certain genes, including CA2, GKN1, LYZ, PGC and PGA5, which are involved in protein, lipid and carbohydrate metabolism, also occurred abundantly in the examined tissues. Their down-regulation may indicate loss of normal gastric function, suggesting that dedifferentiation is a common characteristic of stomach carcinogenesis (25). CA2, GKN1, LYZ and PGC were up-regulated in the patient with chronic gastritis as expected, since chronic gastritis involves inflammation of the stomach lining and in most cases does not permanently damage the lining. The normal stomach function is generally not substantially impaired. These findings are in concordance with those of previous studies (22,26). A markedly high number of ESTs from all libraries was associated with the immune system, binding, response to stimulus and cellular and developmental processes. These ESTs are different in up- and down-regulated libraries. Defence against microorganisms is a crucial role of the gut (22). In the present study, we found several down-regulated genes involved in immune response, including LYZ, DEFB4, IGHM, IGKC and IGJ. Again, the patient with chronic gastritis had only a a small number of down-regulated ESTs associated with immune defence and high levels of LYZ. Analyses also revealed deregulation of ribosomal and cytoskeletal genes, signal transducers, and genes involved in apoptosis, cell communication, cell migration, response to stress and growth, which is consistent with other studies (17,18,22,25,26). Impaired expression of these genes and/or their combinations may disturb the balance between proliferation and cell death, which is instrumental to gastric homeostasis (27). These events may lead to the establishment of immortal cells and consequently to tumor development.
KEGG pathway analyses showed that several of the differentially expressed genes have well-recognized roles in genetic information processing, signal transduction, repair, cell adhesion, cell motility, disease and carcinogenesis. Notably, certain differentially expressed genes were correlated to non-carcinogenic diseases including Parkinson's, Alzheimer's and Huntington's diseases, diabetes, systemic lupus erythematosis and bacterial infections. The reason may be that these genes are involved in common cell pathways leading to immune evasion, proteotoxic and oxidative stress response, mitochondrial dysfunction and rejection of foreign tissues. The mentioned pathways probably reflect disturbed cell homeostasis. It is also hypothesized that some of these pathways activate other signaling pathways, including Erk and MAPK pathways. MAPK cascade is a highly conserved pathway involved in various cell functions, including cell proliferation, differentiation and migration. In many different species and organs, the Wnt pathway is a complex network of proteins that plays a role in basic developmental processes, including cell-fate specification, progenitor-cell proliferation and in the control of asymmetric cell division. It is also the main pathway in stomach epithelial maintenance (28). The ErbB family of receptor tyrosine kinases (RTKs) couples the binding of extracellular growth factor ligands to intracellular signaling pathways regulating diverse biological responses, including proliferation, differentiation, cell motility and survival. The convergence of these pathways may contribute to the development and maintenance of malignant cells. Notably, all of the patients exhibited deregulated MAPK, Wnt and ErbB pathways, although the genes differentially expressed were not identical. The cause for this difference may be the heterogeneity of gastric adenocarcinoma: predicting pathway activity by expression signatures may therefore permit the study of multiple cancer-related pathways interacting simultaneously in primary cancers (29).
A number of the identified genes were also previously associated with the development of various other types of cancer. Notably, in the patient with chronic gastritis we found only one potentially oncogenic gene, which probably reflects the fact that chronic gastritis is an inflammatory disease. In all gastric tumor tissues we observed down-regulation of TFF1, which is generally markedly expressed in normal gastric mucosa and in the regenerative tissues surrounding ulcerous lesions of the gastrointestinal tract. This observation is consistent with various previously reported results (17,22,25). The functions of this gene have yet to be defined, but it may protect the mucosa from insults, stabilize the mucus layer, and affect healing of the epithelium. In general, our analyses revealed deregulation of several crucial pathways required for normal gastric homeostasis. Certain differentially expressed genes (GUCY2C, FABP1 and KLF4) are normally expressed predominantly in the intestine, demonstrating the intestinal differentiation of gastric cancer cells (18,30).
Our analysis also identified genes that have not previously been reported to be differentially expressed in gastric cancer (information obtained using PubMed mining systems), including DPCR1, MALAT1 and TPT1. MALAT1 is a long non-coding RNA that was originally identified in early-stage non-small cell lung cancers, and possesses a propensity for metastasis (31). Subsequently, it was shown to be overexpressed in numerous types of human carcinoma, including those of the breast, pancreas, lung, colon, prostate and liver (32). In the present study we observed reduced levels of this gene in tumor tissues. The reason for this discrepancy has yet to be clarified. The role of DPCR1 is not known and the gene was found to be overexpressed in diffuse panbronchiolitis, an unusual form of bronchiolar disease exclusively affecting individuals of East Asian origin (33). TPT1 is frequently differentially expressed in tumor tissues and recent studies have shown that it may be play a role in a variety of intracellular and extracellular functions, including microtubule stabilization, cell cycle, apoptosis, growth regulation and cytokine release, although its exact biological function remains to be determined (34).
In conclusion, searches of the enriched GO terms associated with differentially expressed transcripts are beneficial in for understanding the biological significance of large sets of data. The limitations of these methods are incomplete annotation databases and unidentified genes. Nevertheless, incorporation of additional biological information can enhance the interpretability of data.
The limitations of the SSH method include high costs in time and labour; on the other hand, it is most adequate in identifying novel genes and low-abundant transcripts. Additionally, it may be a preferable method for transcript profiling of clinical tissues, due to their heterogeneity. Other methods, such as microarrays, function adequately only when RNA is derived from homogeneous cell populations (35). Therefore, extended studies with SSH may show a global expression profile associated with carcinogenesis.
In the present study, we identified a number of genes previously correlated to gastric carcinogenesis, as well as genes that have no defined relationship to malignant disease. The latter genes are potentially noteworthy in identifying new clinical and biological markers of gastric cancer. We identified the global deregulation of several pathways involved in the maintenance of normal gastric homeostasis. This study also confirmed the hypothesis that due to the complex pathogenesis, diversity of genomic aberrations and multiplicity of carcinogenic causes, gastric cancer cannot be reduced to a single molecule (9). Therefore, global investigations into the molecular aspects of gastric cancer with high throughput methods is of high priority to identify clusters of predictor genes that may be used in order to establish a molecular system for gastric cancer classification and detection.
Acknowledgments
This study was supported by grant P1-0104 from ARRS to author R.K. and a Young Investigator grant to P.H.
Contributor Information
Petra Hudler, Medical Centre for Molecular Biology, Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, SI-1000 Ljubljana, Slovenia.
Stanislav Repše, University Medical Centre of Ljubljana, Department of Abdominal Surgery, SI-1000 Ljubljana, Slovenia.
Robert Juvan, University Medical Centre of Ljubljana, Department of Abdominal Surgery, SI-1000 Ljubljana, Slovenia.
Radovan Komel, Medical Centre for Molecular Biology, Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, SI-1000 Ljubljana, Slovenia.
References
- Tahara E: Abnormal growth factor/cytokine network in gastric cancer. Cancer Microenviron 1: 85-91, 2008. [DOI] [PMC free article] [PubMed]
- Zheng L, Wang L, Ajani J and Xie K: Molecular basis of gastric cancer development and progression. Gastric Cancer 7: 61-77, 2004. [DOI] [PubMed]
- Lochhead P and El-Omar EM: Gastric cancer. Br Med Bull 85: 87-100, 2008. [DOI] [PubMed]
- Lee KJ, Inoue M, Otani T, Iwasaki M, Sasazuki S and Tsugane S: Gastric cancer screening and subsequent risk of gastric cancer: a large-scale population-based cohort study, with a 13-year follow-up in Japan. Int J Cancer 118: 2315-2321, 2006. [DOI] [PubMed]
- Yamamoto H and Kita H: Endoscopic therapy of early gastric cancer. Best Pract Res Clin Gastroenterol 19: 909-926, 2005. [DOI] [PubMed]
- Rhodes DR and Chinnaiyan AM: Bioinformatics strategies for translating genome-wide expression analyses into clinically useful cancer markers. Ann N Y Acad Sci 1020: 32-40, 2004. [DOI] [PubMed]
- Slonim DK: From patterns to pathways: gene expression data analysis comes of age. Nat Genet 32: 502-508, 2002. [DOI] [PubMed]
- Takenawa H, Kurosaki M, Enomoto N, et al: Differential gene-expression profiles associated with gastric adenoma. Br J Cancer 90: 216-223, 2004. [DOI] [PMC free article] [PubMed]
- Zhang YJ and Fang JY: Molecular staging of gastric cancer. J Gastroenterol Hepatol 23: 856-860, 2008. [DOI] [PubMed]
- Kent WJ: BLAT – the BLAST-like alignment tool. Genome Res 12: 656-664, 2002. [DOI] [PMC free article] [PubMed]
- Al-Shahrour F, Diaz-Uriarte R and Dopazo J: FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 20: 578-580, 2004. [DOI] [PubMed]
- Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA and Tainsky MA: Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res 31: 3775-3781, 2003. [DOI] [PMC free article] [PubMed]
- Zhou M and Cui Y: GeneInfoViz: constructing and visualizing gene relation networks. In Silico Biol 4: 323-333, 2004. [PubMed]
- Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408, 2001. [DOI] [PubMed]
- Diatchenko L, Romanov S, Malinina I, et al: Identification of novel mediators of NF-kappaB through genome-wide survey of monocyte adherence-induced genes. J Leukoc Biol 78: 1366‑1377, 2005. [DOI] [PubMed]
- Grabsch H, Takeno S, Parsons WJ, et al: Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer – association with tumour cell proliferation. J Pathol 200: 16-22, 2003. [DOI] [PubMed]
- Hasegawa S, Furukawa Y, Li M, et al: Genome-wide analysis of gene expression in intestinal-type gastric cancers using a complementary DNA microarray representing 23,040 genes. Cancer Res 62: 7012-7017, 2002. [PubMed]
- Hippo Y, Taniguchi H, Tsutsumi S, et al: Global gene expression analysis of gastric cancer by oligonucleotide microarrays. Cancer Res 62: 233-240, 2002. [PubMed]
- Johnson AH, Frierson HF, Zaika A, et al: Expression of tight-junction protein claudin-7 is an early event in gastric tumorigenesis. Am J Pathol 167: 577-584, 2005. [DOI] [PMC free article] [PubMed]
- Meireles SI, Cristo EB, Carvalho AF, et al: Molecular classifiers for gastric cancer and nonmalignant diseases of the gastric mucosa. Cancer Res 64: 1255-1265, 2004. [DOI] [PubMed]
- Montgomery E, Abraham SC, Fisher C, et al: CD44 loss in gastric stromal tumors as a prognostic marker. Am J Surg Pathol 28: 168-177, 2004. [DOI] [PubMed]
- Oien KA, Vass JK, Downie I, Fullarton G and Keith WN: Profiling, comparison and validation of gene expression in gastric carcinoma and normal stomach. Oncogene 22: 4287-4300, 2003. [DOI] [PubMed]
- Ossandon FJ, Villarroel C, Aguayo F, et al: In silico analysis of gastric carcinoma Serial Analysis of Gene Expression libraries reveals different profiles associated with ethnicity. Mol Cancer 7: 22, 2008. [DOI] [PMC free article] [PubMed]
- Yu CD, Xu SH, Mou HZ, Jiang ZM, Zhu CH and Liu XL: Gene expression profile differences in gastric cancer, pericancerous epithelium and normal gastric mucosa by gene chip. World J Gastroenterol 11: 2390-2397, 2005. [DOI] [PMC free article] [PubMed]
- Jinawath N, Furukawa Y, Hasegawa S, et al: Comparison of gene-expression profiles between diffuse- and intestinal-type gastric cancers using a genome-wide cDNA microarray. Oncogene 23: 6830-6844, 2004. [DOI] [PubMed]
- Yasui W, Oue N, Ito R, Kuraoka K and Nakayama H: Search for new biomarkers of gastric cancer through serial analysis of gene expression and its clinical implications. Cancer Sci 95: 385-392, 2004. [DOI] [PMC free article] [PubMed]
- Potten CS, Wilson JW and Booth C: Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells 15: 82-93, 1997. [DOI] [PubMed]
- Katoh Y and Katoh M: Hedgehog signaling pathway and gastrointestinal stem cell signaling network (Review). Int J Mol Med 18: 1019-1023, 2006. [PubMed]
- Ooi CH, Ivanova T, Wu J, et al: Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet 5: e1000676, 2009. [DOI] [PMC free article] [PubMed]
- Chen X, Leung SY, Yuen ST, et al: Variation in gene expression patterns in human gastric cancers. Mol Biol Cell 14: 3208-3215, 2003. [DOI] [PMC free article] [PubMed]
- Wilusz JE, Freier SM and Spector DL: 3' end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135: 919-932, 2008. [DOI] [PMC free article] [PubMed]
- Guffanti A, Iacono M, Pelucchi P, et al: A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics 10: 163, 2009. [DOI] [PMC free article] [PubMed]
- Matsuzaka Y, Tounai K, Denda A, et al: Identification of novel candidate genes in the diffuse panbronchiolitis critical region of the class I human MHC. Immunogenetics 54: 301-309, 2002. [DOI] [PubMed]
- Choi KW and Hsu YC: To cease or to proliferate: new insights into TCTP function from a Drosophila study. Cell Adh Migr 1: 129-130, 2007. [DOI] [PMC free article] [PubMed]
- Cao W, Epstein C, Liu H, et al: Comparing gene discovery from Affymetrix GeneChip microarrays and Clontech PCR-select cDNA subtraction: a case study. BMC Genomics 5: 26, 2004. [DOI] [PMC free article] [PubMed]