

Abstract
Dihydrogen sulfide recently emerged as a biological signaling molecule with important physiological roles and significant pharmacological potential. Chemically plausible explanations for its mechanisms of action have remained elusive, however. Here, we report that H2S reacts with S-nitrosothiols to form thionitrous acid (HSNO), the smallest S-nitrosothiol. These results demonstrate that, at the cellular level, HSNO can be metabolized to afford NO+, NO, and NO– species, all of which have distinct physiological consequences of their own. We further show that HSNO can freely diffuse through membranes, facilitating transnitrosation of proteins such as hemoglobin. The data presented in this study explain some of the physiological effects ascribed to H2S, but, more broadly, introduce a new signaling molecule, HSNO, and suggest that it may play a key role in cellular redox regulation.
Introduction
Nitrogen monoxide and dihydrogen sulfide are two gaseous transmitters that regulate numerous physiological functions. Although the chemistry,1a−1d biochemistry,1d,1e and physiology1f,1g of NO• have been widely studied since its identification as the endothelium-derived relaxing factor,1h H2S has only recently been recognized as an important signaling molecule with physiological effects similar to those of nitrogen monoxide.2 A (bio)chemical understanding of its mechanism of action is minimal, however.3 Although some authors have proposed interplay between NO• and H2S signaling pathways,4 it is widely accepted that the physiological consequences of H2S, unlike NO, do not directly involve cyclic guanosine monophosphate (cGMP).5
One of the modes of NO• signaling is activation of soluble guanylate cyclase to produce cGMP.1f Another, equally important role of NO• action is S-nitrosation.6a Two decades after characterization of the first S-nitrosothiols in biological systems,1eS-nitrosation of ∼1000 proteins has been reported, and this posttranslational modification is increasingly considered to be as important as phosphorylation.6 The mechanism of S-nitrosation and signaling through formal transfer of the “NO+” moiety is still a matter of debate, however.6c It has been recently proposed4e that H2S may play a role in modulating the S-nitrosothiol profile in the cells, and we hypothesized that this mechanism may involve the reaction of hydrogen sulfide with low-molecular weight and/or protein S-nitrosothiols (RSNOs) to form the smallest S-nitrosothiol, thionitrous acid (HSNO).7
Although extensively examined by computational methods,8,9 HSNO has never been proved to exist in aqueous solution or characterized under physiologically relevant conditions. It has only been isolated and spectroscopically identified in an argon matrix at 12 K.10 In the reaction of elementary sulfur and bis(triphenylphosphine)iminium nitrite, (PNP+)(NO2–), upon addition of triphenylphosphine under vacuum in pure organic water free solvents, (PNP+)(SNO–) has been characterized.11
Here, we prove the existence of HSNO under physiologically relevant conditions and demonstrate in vitro and at the cellular level that HSNO can serve as a source of NO+, NO•, and NO– entities, all of which evoke their own distinctive physiological responses. Furthermore, we show that HSNO can freely diffuse through membranes facilitating transnitrosation of proteins.
Experimental Section
Materials
All chemicals were of the highest purity available and purchased from Sigma-Aldrich. Buffers were prepared with nanopure water and further purified and stored over Chelex-100 to remove traces of transition metals. All experiments were performed using anhydrous Na2S (Sigma Aldrich),12 which was stored in a glovebox under argon (<1 ppm O2 and <1 ppm H2O). Stock solutions (100 mM) of sodium sulfide were prepared in the glovebox using argon-saturated nanopure water and stored in glass vials with PTFE septa at 4 °C for no longer than 1 week. Gas-tight Hamilton syringes were used to transfer these solutions throughout our studies.13
Analysis of NO• and H2S
The fate of hydrogen sulfide and nitrogen monoxide during the course of the reaction was monitored with a 2 mm shielded H2S sensor and the ISO-NO probe (World Precision Instruments), connected to a Free Radical Analyzer (World Precision Instruments).13 Electrode responses were monitored using DataTrax software for signal processing. Experiments were performed in a four-channel chamber (WPI) with both electrodes running either simultaneously or individually. A 2 mL portion of 50 mM pH 7.4 potasium phosphate (KPi) buffer was added to the reaction chamber before immersion of the electrodes. Depending on the type of measurement, different concentrations of sodium sulfide (50–500 μM) were injected followed by addition of S-nitrosoglutathione (GSNO) or S-nitrosoalbumin.
UV/Vis Spectrometric Studies
All spectrophotometric studies were performed on a Hewlett-Packard 8452A diode array spectrophotometer connected to a Dell computer operating with Olis SpectralWorks software.
GC–MS Detection of N2O
A 5 mM GS15NO solution in 50 mM pH 7.4 potassium phosphate buffer was degassed with argon and kept in dark glass vials sealed with PTFE septa. Sodium sulfide was added to yield a final concentration of 5 or 17.5 mM. GC–MS analyses were performed on a Bruker GC 450 TQ MS 300 instrument. The gas chromatograph was equipped with a capillary column Varian, VF-5 m. A 50 μL volume of headspace gas was injected (splitless mode), and the following oven temperature program was used with helium as carrier gas: 5 min at 50 °C, then increased to 155 °C at a rate of 10 °C/min, and then increased to 260 °C at a rate of 30 °C/min. The positive electron impact ionization mode was used. Detector multiplier voltage was set to 1400 V, and the detection was performed by selected ion monitoring of m/z 46 (15N2O) and m/z 31 (15NO) using a dwell time of 50 ms and scan width for SIM of 0.7 au. Areas under the peaks were determined using software provided by the manufacturer.
GC–MS Detection of Hydroxylamine
A 100 μL sample of a hydroxylamine standard solution in 50 mM phosphate buffer at pH 7.4 was treated with 200 μL of cyclopentanone, 2 μL of concentrated sulphuric acid, and 1 mL of HPLC–MS grade methanol. After 1 h of incubation, the samples were diluted 10 times in methanol and injected into the GC–MS. The gas chromatograph was equipped with a Varian, VF-5 m capillary column. A 5 μL volume of solution was injected (splitless mode) and the sample eluted using a 15 °C/min ramp from 50 to 250 °C. The positive electron impact ionization mode was used. The detector multiplier voltage was set to 1400 V, and the detection was performed by selected ion monitoring of m/z 84 (cyclopentanone) and m/z 99 (cyclopentanone oxime) using a dwell time of 50 ms and scan width for SIM of 0.3 au. Linear dependence of the area under the peak versus concentration of hydroxylamine was observed in the 2–100 μM range.
15N NMR Spectroscopic Characterization of HSNO
Spectra of 15N-labeled S-nitrosoglutathione (GS15NO, 25 mM), either with or without addition of sodium sulfide (25 mM), in 300 mM potassium phosphate buffer (pH 7.4) were recorded using a Bruker 400 MHz spectrometer (reference 15N-nitromethane, at 50.67 MHz, 35° pulse width, 5 s relaxation delay).14
FTIR Spectroscopy for in Situ Characterization of HSNO
GSNO or GS15NO (120 mM) in 300 mM potassium phosphate buffer, pH 7.4, was monitored in solution (ReactIR 45 m, Mettler Toledo) utilizing a DiComp AgX fiber probe with diamond ATR element and MCT detector for 25 min to confirm stability prior to addition of Na2S (100 mM). Thereafter, spectra were recorded for 10 min, with each spectrum representing an average of 256 scans collected over 1 min in the range 900–1900 cm—1. A difference spectrum was obtained by subtracting the initial GSNO spectrum from all subsequent spectra.
Pulse Radiolysis
Generation of HS• and NO• by pulse radiolysis was performed as previously described.15 Briefly, these free radicals were generated by irradiation of 1.2 mM Na2S and 0.12 mM KNO2 in Ar-saturated water pH = 11 with electron pulses of 2 MeV energy and 20–50 ns duration generated by a Febetron 705 accelerator (Titan, San Leandro, CA).16 A 300 μL capacity quartz cell was used, and a flow system ensured that each sample was exposed only to a single electron pulse.
Cell Culture and HNO Bioimaging Studies
Human umbilical vein endothelial cells (HUVECs, passage 2–3) were obtained from PromoCell GmbH (Heidelberg, Germany) and cultured in 35 mm μ-Dishes (ibidi, Martinsried, Germany) in endothelial cell growth medium 2 (PromoCell GmbH) at 37 °C and 5% CO2. For HNO detection, HUVECs were incubated with 10 μM of a nitroxyl-responsive dye (CuBOT1)17 for 20 min. Cells were washed of excess fluorescent dye three times, placed into new medium, and further treated as specified in the figure legends. Fluorescence microscopy was carried out using an inverted microscope (Axiovert 40 CLF, Carl Zeiss), equipped with green fluorescent filters and ICm1 AxioCam. Images were postprocessed using ImageJ software, NIH, for semiquantitative determination of fluorescence intensity.
S-Nitrosothiol Immunocytochemistry
HUVECs were exposed to medium supplemented with 1 mM L-NAME, 1 mM propargylglycine, or both for 2 h and then fixed with 4% formaldehyde. Cells were permeabilized, blocked, and incubated with primary monoclonal, mouse anti-S-nitrosocysteine antibodies (Abcam, Cambridge, UK) following the instructions of the manufacturer. FITC-labeled, goat antimouse Fc-IgG antibodies (Sigma Aldrich, St. Louis, MO) were used as secondary antibodies.
Transnitrosation of Albumin
GSNO (1 mM) was incubated with (1 mM) sodium sulfide in 300 mM KPi, pH 7.4, for 3 min. Spectrophotometric measurements confirmed that all GSNO had reacted within 1 min. A 200 μM portion of bovine serum albumin (BSA) was added to the solution and incubated for 15 min, dialyzed against water for 24 h, and prepared for MS analysis in acetonitrile/methanol (1:1, v/v) containing 0.1% formic acid.
Protein-to-Protein Trans-nitrosation Mediated by HSNO
To test the possibility that HSNO can serve as a small, easily diffusible trans-nitrosating agent, a special experimental setup was devised. Poly-S-nitrosoalbumin (BSA-SNO) was prepared by mixing 300 μM BSA with 1 mM GSNO in the dark and incubation for 15 min, followed by 4 h dialysis against Chelex-resin 100-containing 50 mM KPi, pH 7.4, with constant exchange, and divided into two dialysis bags (cut off 5 kDa). A 1.5 mL portion of this material, serving as a control, was placed into a 15 mL Falcon tube containing 30 μM BSA and incubated there for 5 min. To prove HSNO formation and that diffusion through the membrane takes place, 500 μM Na2S was added into the other 1.5 mL of 300 μM BSA-SNO, and the solution was dialyzed against 30 μM BSA for 5 min. The entire experimental procedure was performed in the dark. Subsequently, the dialysis bag was removed and the external solution was analyzed for RSNO content using NO• and H2S electrodes,18 Saville’s method,19 and the biotin-switch assay.20
Transnitrosation of Hemoglobin in Red Blood Cells
Red blood cells (RBCs) were obtained fresh from venous blood of healthy volunteers by centrifugation at 5000g for 5 min and washed three times with sterile PBS (Sigma Aldrich). RBCs were then resuspended in buffer to a cell count comparable to that of blood and used immediately. S-Nitrosoalbumin was prepared by mixing 300 μM BSA with 1 mM CysNO in the dark and incubated for 15 min, followed by 4 h dialysis against Chelex resin-100-containing 50 mM KPi, pH 7.4, with constant exchange. A 30 μM portion of this polynitroso-albumin was injected into 1 mL of RBC suspension, with or without further addition of 50 μM Na2S or glutathione. After 5 min of incubation at room temperature, RBCs were centrifuged at 10 000g for 1 min, and the supernatant was replaced with fresh PBS. This procedure was repeated five times to ensure complete removal of residual RSNOs. RBC hemolysis was performed by addition of 3 volumes of nanopure water containing 100 μM neocuproine, and obtained hemoglobin was further purified on Sephadex G-10 columns protected from light. All samples were adjusted to the same protein concentration and directly analyzed by Saville’s assay19 and/or ESI-TOF-MS.21
Additionally, 20 μM washed RBC was diluted 100× with PBS and exposed to 100 μM poly BSA-SNO (prepared by acidification of BSA/nitrite mixture and then further purified on micro Bio Spin column) without or with addition of 100 μM H2S, 100 μM GSH, or 100 μM Cys. Two minutes after the addition of BSA-SNO, the samples were centrifuged for 3 min at 3000g and then washed with PBS three times prior to the hemolysis with nanopure water supplemented with 100 μM neocuproine. Samples (20 μL) were separated on LC and subsequently analyzed by ESI-TOF-MS. LC separation was done using following protocol: column was equilibrated with a 50:50 mixture of buffer A (80:20, water:acetonitrile; 0.1% trifluoroacetic acid) and buffer B (40:60, water:acetonitrile; 0.1% trifluoroacetic acid) at a flow rate of 1 mL/min. Twenty microliters of each sample was loaded and eluted with a 2 min hold at 50% B, followed by a linear gradient to 66% B over 40 min.
Mass Spectrometric Characterization
All samples were analyzed using maXis (Bruker Daltonics), an ultrahigh-resolution ESI-TOF mass spectrometer operating in the positive ion mode. Data analysis, mass deconvolution, and spectra simulation were performed using data analysis software, provided by the manufacturer.
Computational Methods
Geometries of all structures were fully optimized at the B3LYP22a−22d level of theory using the aug-cc-pVTZ22e−22g basis set. Stationary points were confirmed to be minima or transition states by calculating the normal vibrations within the harmonic approximation. All computed relative energies are corrected for zero-point vibrational energies (ZPE). The Gaussian 09 program package was used for all of the above calculations.23
Thermochemical arguments are based on the group additivity approach described by Benson (see the Supporting Information) and standard electrode potentials.
Results and Discussion
Generation of HSNO/SNO– by Pulse Radiolysis
To test whether HSNO could be generated in an aqueous solution, we performed pulse radiolysis experiments in which HS• and NO• (Scheme 1, eq 1) were cogenerated from HS–/nitrite-containing solutions (pH 11).
Scheme 1. HSNO Generation (Eqs 1–3) and Reactivity (Eqs 4–6).

A broad peak around 370 nm is formed transiently after irradiation, reaching a maximum 5 μs after irradiation (see Figure 1A). However, this spectrum comprises not only HSNO/SNO– but also precursors of HSNO/SNO– and side products formed during pulse radiolysis of nitrite and sulfide solutions.
Figure 1.
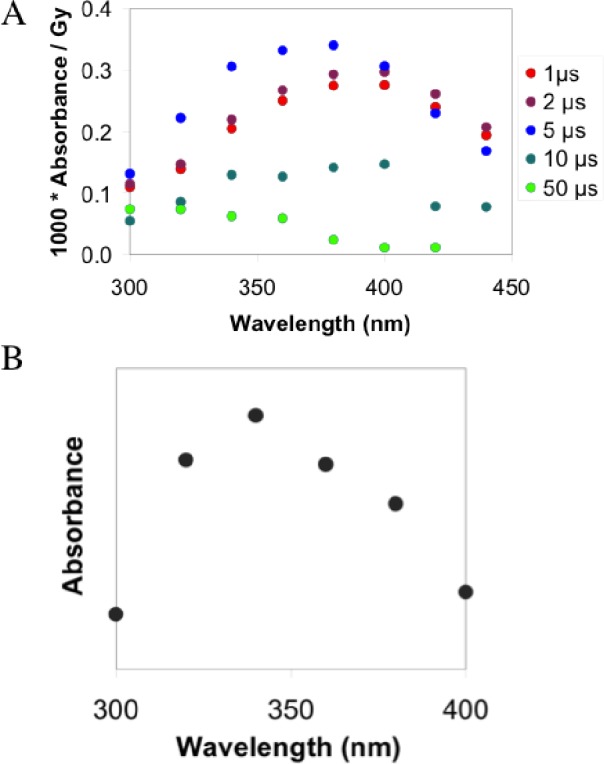
Generation of HSNO/SNO– by pulse radiolysis in argon-saturated water pH 11. (A) Time-resolved absorbance spectral investigation of HSNO/SNO– generated by pulse radiolysis. The spectra reveal formation of a peak at 370 nm consisting of HSSH•–/HSS•2– and HSNO/SNO–. (B) The actual spectrum of generated HSNO/SNO–, obtained by subtracting the first spectrum from that acquired after 5 μs.
HS– is oxidized to HS• via reactions given in eqs 7–9. Initially, HO• add to HS–, reaction 7,15 with a rate constant k1 = 5.4 × 109 M–1 s–1 and, given the concentrations used, with a half-life of 110 ns. This oxidation step is followed by addition of hydrogen sulfide anion to yield disulfanuidyl, or dihydrogen disulfide radical anion (HSSH•–).
| 7 |
| 8 |
| 9 |
The reaction given in eq 8 has a half-life of 300 ns (k2 ≈ 2 × 109 M–1 s–1).24a The HSSH•– product has an absorption maximum near 380 nm15 (Figure 1A, red dots) and equilibrates very rapidly with HS•/S•– (eq 9), with a half-life of ∼100 ns24a (k3 ≈ 5 × 105 s–1, k–3 = 5.4 × 109 M–1 s–1, K3 ≈ 10–4). With 1.2 mM HS– present, we expect 90% of the sulfur radicals to be present in the form of disulfanuidyl, which decays by mixed order.15 Therefore, we have to assume that at least two different decay mechanisms compete with the desired reaction with NO•. Dominant at high radiation doses and, consequently, large radical concentrations is radical recombination leading to disulfide, which may trigger polysulfide production with an ill-defined reaction order.
Nitrogen monoxide is formed from NO2– according to eqs 10 and 11:
| 10 |
| 11 |
The rate constant of reaction 10 is 5 × 109 M–1 s–1, or, given the concentration of nitrite, that reaction has a half-life of approximately 1.2 μs. Under first-order conditions, 90% conversion is reached after roughly 3 half-lives. Therefore, we observe initially (1 μs after pulse) the spectrum of the disulfide radical anion with λmax ≈ 380 nm (Figure 1A). Only after the production of NO2•2– or NO• does the desired radical recombination product, HSNO/SNO–, appear (Figure 1A). We assign the difference between the spectra before quantitative formation of NO• (1 μs after pulse) and after its complete formation (5 μs after pulse) to thionitrite, as nitrogen monoxide itself is colorless. As shown in Figure 1B, this spectrum has an absorbance maximum at 340 nm, a feature characteristic of RSNOs that corresponds well to calculated values for SNO–.24b Very rapid disappearance of the reaction product is expected because HS– is in great excess under these experimental conditions and reacts with HSNO/SNO– to generate H2S2 and its deprotonated forms together with HNO (eq 5, Scheme 1). Furthermore, under the applied alkaline conditions, HSNO/SNO– could hydrolyze according to reaction 12.
| 12 |
These processes all contribute to the short half-life of HSNO/SNO– in the pulse radiolysis experiments. We therefore sought conditions for obtaining more stable HSNO/SNO– preparations.
HSNO/SNO– from the Reaction of Acidified Nitrite and H2S
As shown in Scheme 1, eq 2, S-nitrosothiols could be generated from the acidified mixture of corresponding thiol and nitrite.19 An attempt to synthesize HSNO by mixing sodium sulfide with acidified solution of sodium nitrite resulted in a transient color change, which was rapidly followed by decoloration and sulfur formation. Fast neutralization of the observed brownish reaction mixture gave a UV–vis spectrum with broad absorbance maximum at 370 nm, similar to that observed in pulse-radiolysis experiments (Figure S1A) consisting of dihydrogen disulfide radical anion, unreacted nitrite, and HSNO/SNO–.
When analyzed by ESI-TOF-MS in the positive ion mode, the acidic reaction mixture showed a dominant peak at m/z 64 (Figure S1B). Neutralization of the colored solution and subsequent MS analysis revealed the existence of the same peak at m/z 63.9898 (100% abundance), which we ascribe to [HSNO + H]+ (Figure 2).
Figure 2.

Mass spectrum of HSNO, that is, [HSNO + H+]+, prepared by acidification of nitrite in the presence of sulfide and then neutralized with the 300 mM potassium phosphate pH 7.4 buffer.
This might suggest that, at pH = 7.4, some HSNO present in solution. Recent experimental and computational work indicates that the pKa value of an iron-coordinated HSNO intermediate is 10.5.25 On the basis of that, free HSNO should have pKa >10.5 and HSNO would be the major form under physiological conditions. However, comparing HSNO with HNO2, one would expect a pKa < 3.2 and thus the anion as the predominant species at physiological pH. This issue remains to be resolved by future work.
HSNO/SNO–, a Product of Transnitrosation between S-Nitrosoglutathione and H2S
Although indicative of HSNO formation, the foregoing generation of the molecule under very acidic conditions is unlikely to be of physiological relevance except perhaps for the stomach or acidic cell compartments. We therefore investigated the ability of H2S to participate in transnitrosation reactions (Scheme 1, eq 3). Addition of equimolar amounts of Na2S to buffered GSNO (S-nitrosoglutathione) solutions led to rapid (<1 min) formation of a yellow color (see Figure S2). A very similar UV–vis spectrum reported in the literature26 was ascribed to SSNO– as a product of this reaction.27 However, the presence of other polysulfides in the reaction mixture can also be anticipated.
We analyzed the reaction mixture using ultrahigh-resolution ESI-TOF mass spectrometry. When mixed with Na2S, the intensities of the main GSNO peaks decreased (see Figure S3) and new peaks appeared. The most intense is a peak at m/z 345.0358, which we assign to [GS• + K]+ (or [GSSG + 2K]2+) (see Figure S3). More importantly, low-mass range spectral analysis revealed the formation of a new peak at m/z 63.9902 (∼5% of maximal intensity, similar to the spectrum observed in Figure 2).
Simulation of the isotopic distribution for [HSNO + H]+ and comparison of the calculated molecular weight (63.9852) with the obtained value unambiguously established its identity as HSNO (Figure 3A). The same experiment with GS15NO additionally confirmed generation of HS15NO (see Figure S4) in the reaction of GS15NO and H2S (Scheme 1, eq 3). No peaks of the putative [HSSNO + H]+ cation were detected, suggesting that this adduct is not present in solution.
Figure 3.

Characterization of HSNO/SNO– generated by transnitrosation of S-nitrosoglutathione (GSNO) and H2S. (A) ESI-TOF-MS spectrum of HSNO generated in transnitrosation reaction between GSNO and H2S. Experimental (black) and theoretical (red) isotope distribution of the detected m/z 64 peak of [HSNO + H]+. (B) Real-time FTIR confirms formation of a new nitrosothiol product. Differential IR spectrum of the reaction of 120 mM GSNO and 100 mM Na2S in 300 mM potassium phosphate buffer pH 7.4 (black, after 1 min; red, after 10 min). Inset: Spectral difference between 14N and 15N labeled HSNO/SNO–. (C) 15N NMR spectrum of HSNO/SNO– at pH 7.4. Black: Mixture of 15N-enriched GSNO with nitrite. Red: After addition of equimolar concentration (25 mM) of sulfide. Blue: After 1 h, only the nitrite signal remains. The reaction was performed in 300 mM potassium phosphate buffer, pH 7.4.
The transnitrosation reaction was also characterized by FTIR spectroscopy. Difference FTIR spectra (Figure 3B) clearly show the disappearance of the νNO vibration from GSNO at ∼1515 cm–1 with the appearance of a new signal of similar intensity at ∼1568 cm–1. This frequency is slightly lower than that calculated for HSNO in the gas phase and measured in an argon matrix (1596 cm–1 for the trans- and 1569 cm–1 for the cis-isomer),10 due to solvation effects.
When GS15NO was used to prepare HS15NO, the corresponding N=O vibration shifted to lower values (∼1549 cm–1 as compared to the calculated value of 1540 cm–1 from the simple harmonic oscillator model) (inset to Figure 3B).
Finally, HSNO prepared in the reaction of 15N-enriched GSNO and H2S was characterized by using 15N NMR spectroscopy. Addition of an equimolar amount of Na2S led to the immediate disappearance of the GSNO peak (409 ppm) and appearance of a previously unidentified 15N resonance at 322 ppm (Figure 3C), which we assign to HSNO. This new chemical entity was stable for less than 1 h at pH 7.4 and 21 °C, with subsequent measurements showing only the nitrite signal at 247 ppm. A linear relationship exists between chemical shifts of S-nitrosothiols and the pKa values of the corresponding thiols; a lower pKa of a starting thiol implies smaller ppm values of 15N chemical shifts of the resulting S-nitrosothiol.14 Therefore, the lower pKa of H2S in comparison to R-substituted thiols implies a smaller chemical shift of HSNO as compared to those of the substituted analogs. Furthermore, less electron density on S in the case of HSNO when compared to RSNO speaks in favor of a higher n → π* transition energy and a smaller 15N chemical shift.14
The yellow coloration of the solution, corresponding to the absorbance maximum at 412 nm (Figure S2), remains even after the 15N NMR peak and characteristic N=O vibration disappear from the solution spectra. This result, in agreement with our MS findings, excludes the presence of SSNO–, which as a nitrogen atom-containing species would have displayed a corresponding 15N peak and N=O vibration band. Thus, the band at 412 nm results predominantly from the mixture of polysulfides.
Collectively, these experiments unambiguously confirm the presence of HSNO/SNO– in aqueous solution at pH 7.4 and demonstrate that it can be formed in a transnitrosation reaction between H2S and RSNOs (Scheme 1, eq 3).
Chemical Biology of HSNO: Generation of NO•
RSNOs may spontaneously decompose to give NO•, nitrosate a thiol, or form nitroxyl (HNO), as originally proposed.28 Following the same reasoning (Scheme 1, eqs 4–6), we focused on characterizing the metabolic fate of HSNO.
Evidence for NO• production from HSNO was obtained by following the previously characterized transnitrosation reaction between GSNO and H2S, as a source of HSNO, in combination with simultaneous amperometric monitoring with the use of H2S- and NO•-sensitive electrodes. To minimize metal ion-catalyzed decomposition of S-nitrosothiols, buffers were thoroughly treated with Chelex 100 and supplemented with 100 μM neocuproine. Addition of GSNO to a buffered Na2S solution caused removal of free H2S with concomitant NO• formation (7 ± 3% based on total nitrogen) (Figure 4A). Similar results were obtained for the reaction of S-nitrosoalbumin and H2S (see Figure S5), confirming that the reaction also proceeds with high-molecular-weight RSNOs.
Figure 4.
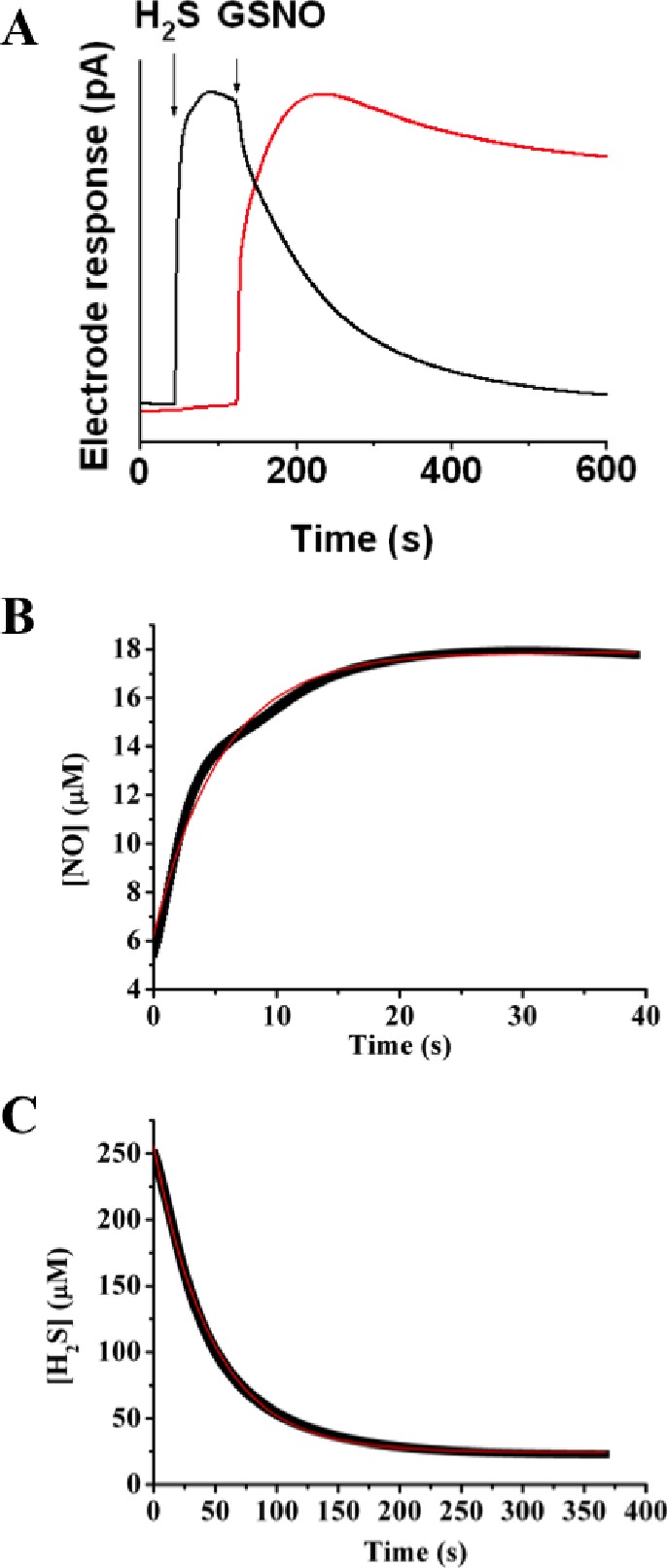
Kinetics of H2S consumption and NO generation in the reaction of GSNO with H2S. (A) Representative recordings by H2S (black) and NO• (red) electrodes, illustrating two processes described by eqs 3 and 4. A 400 μM Na2S solution was prepared in 50 mM potassium phosphate buffer, pH 7.4. When the electrode response reached its maximum, an equimolar amount of GSNO was added, triggering an immediate drop in current at the H2S electrode and a rise in the NO• signal. Kinetic traces of NO• release (B) and H2S consumption (C) from the reaction mixture containing 250 μM GSNO and 250 μM H2S at pH 7.4 at 25 °C. Red lines represent a first-order fit for (B) and a second-order fit for (C).
The release of NO• is a consequence of HSNO homolysis, as shown in eq 4 of Scheme 1. Taking into account the existence of the zwitterionic and ion-pair resonance structures (R–S+=N–O– and RS–/NO+, respectively), resulting in multireference character of the HSNO wave function, S–N bond length and dissociation energy for HSNO were computed to be 1.85 Å and 29.2 kcal/mol, respectively.8,9 As compared to other S-nitrosothiols, HSNO should be slightly less stable and more prone to homolysis.8,9 The kinetics of HSNO homolytic bond dissociation (eq 4, Scheme 1) can be determined on the basis of amperometric detection of NO release (Figure 4B).
For this first-order reaction, the corresponding rate constant was determined to be 0.12 + 0.01 s–1. Conversely, the kinetics of the HSNO/SNO– formation reaction (eq 3 in Scheme 1), assuming that the side reactions29a and the spontaneous release of NO from GSNO29b are slower than the main transnitrosation reaction, were monitored following H2S consumption (Figure 4C) under second-order reaction conditions (GSNO:H2S ratio was 1:1, 250 μM each). The corresponding rate constant is 84 ± 7 M–1 s–1 at 25 °C. In comparison with other transnitrosation reactions (for S-nitroso-N-acetylpenicillamine (SNAP) + GSH, k = 9 M–1 s–1; for SNAP + Cys, k = 21 M–1 s–1),29c higher reactivity of H2S is expected.
Chemical Biology of HSNO – Generation of HNO
Recent studies showed that sulfide is produced in tissues at a high metabolic rate,30a the concentration in plasma being in the micromolar range30b and therefore in excess of what has been proposed to represent the physiological concentration range for RSNOs.6a,6b Considering such concentrations, cellular HSNO/SNO– formation would be expected to yield nitroxyl (HNO/NO–) (Scheme 1, eq 5), the reduced form of NO• with distinct signaling properties.31a−31c To prove its existence, reductive nitrosylation with methemoglobin was used.31d When a mixture of GSNO and H2S (1:2 molar ratios) was added to a buffered methemoglobin solution, immediate formation of nitrosyl-hemoglobin occurred (Figure 5A), whereas no spectral changes were observed with GSNO or H2S alone (see Figure S6). Nitroxyl also converts to nitrous oxide, N2O (k = 8 × 106 M–1 s–1), reaction 13.31e
| 13 |
Figure 5.

Generation of HNO from HSNO in biological milieu. (A) Reductive nitrosylation as a proof of HNO formation. 100 μM GSNO and 200 μM Na2S solution were added to 50 μM metHb in 50 mM potassium phosphate buffer pH 7.4, and the reaction was followed every 10 s for a total of 5 min. Immediate formation of nitrosylhemoglobin is indicative of HNO. (B) GC–MS detection of hydroxylamine. A 60 μM GSNO solution was mixed, at different ratios indicated in the figure, with H2S at pH 7.4, and after 10 min the reaction mixture was treated with cyclopentanone and methanol. At 1 h after the incubation, the corresponding mixtures were analyzed by GC–MS (m/z 99 for cyclopentanone oxime). Buffer and a GSNO solution served as controls. (C) H2S and GSNO react in cells to yield HNO. Human umbilical vein endothelial cells, loaded with 10 μM CuBOT1, were treated with either 100 μM Na2S, 100 μM GSNO, or both for 20 min. Some cells were pretreated with 100 μM GSNO for 20 min to increase intracellular nitrosothiol content and then exposed to 100 μM Na2S. A 100 μM DEA/NONOate solution served as a negative control. (D) Fluorescence intensity was quantified using ImageJ, NIH (n = >30 cells).
We therefore wanted to confirm the generation of nitroxyl along with that of NO• from HSNO using GC–MS (see Figure S7). Fifteen minutes after addition of an equimolar amount of H2S to GSNO, a moderate increase of the NO• amount was detected in the gas phase above the reaction mixture, in agreement with the NO• release detected by NO• electrode (Figure 2A). More importantly, an increased H2S concentration caused a large increase in the peak area of N2O (yield of 17 ± 5% calculated on the basis of total N content in starting GSNO solution) (see Figure S7).
HNO also reacts rapidly with thiols to give hydroxylamine, reaction 14.31a
| 14 |
Assuming that excess H2S does react further with HNO generated in this manner, we wished to detect hydroxylamine. However, the use of standard colorimetric assays31f proved to be of no value for this purpose, because free thiols interfere with this assay, resulting in false negative results. For the present purpose, we therefore developed a rapid and reproducible GC–MS method for detecting hydroxylamine based on its ability to react with ketones, cyclopentanone in the present case, to give corresponding oximes. Different reaction conditions corresponding to different ratios of GSNO and H2S were analyzed. Selected ion monitoring for the m/z 99 (cyclopentanone oxime) peak was applied. The maximal yield, observed for 10-fold excess of H2S over GSNO, was 71 ± 4% (Figure 5B). Together with the detected N2O and NO, this value corresponds to 112 ± 12% of total nitrogen consumed in the reaction. Some hydroxylamine (2 ± 1%) was detected when a 1:1 ratio was used, presumably due to the side reactions.
Although endogenous generation of HNO has never been detected directly in vivo, the recent development of a nitroxyl-responsive dye, CuBOT1,17 enabled us to do so. When human umbilical vein endothelial cells, loaded with CuBOT1, were incubated with either GSNO or H2S (Figure S8), there was no change in fluorescence. Nitrogen monoxide releasing donor, diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEANONOate), also did not have any significant effect on CuBOT1 fluorescence, confirming the previous study of its selectivity.17 Addition of both GSNO and H2S provided clear proof of HNO generation (Figure 5C,D). The effect was even more pronounced when cells were pretreated with GSNO to increase intracellular RSNO content, washed, and then loaded with the probe before exposure to H2S (Figure 5C,D).
These experiments strongly support the hypothesis that cellular HSNO is involved in the formation of nitroxyl, but they cannot exclude the possibility that HNO is produced directly in the reaction of GSNO and H2S (eq 15).
| 15 |
In particular, previous studies suggest that GSNO decomposes in the presence of GSH with the formation of GSSG,32 which would imply the direct formation of HNO via eq 15. We found no evidence for such direct formation of HNO, for neither N2O nor NH2OH levels were significantly changed when GSNO was mixed with H2S in a 1:1 molar ratio (Figure 5B and Figure S7). Furthermore, no GSSH was detected in the ESI-TOF MS spectra (Figure S3). We further explored the possibility, however, by carrying out DFT calculations for the reaction of CH3SNO, as a model for S-nitrosothiols, with H2S/HS–.
Figure 6 (Table S1) shows the lowest-energy profile for the reaction of protonated (A) and deprotonated hydrogen sulfide (B). In both cases, the reaction can proceed by two pathways, H2S/HS– attack on either the N or the S atom of CH3SNO.
Figure 6.

B3LYP/aug-cc-pVTZ computed energy profile for the reaction between CH3SNO and H2S/HS–. Energies are in kcal mol–1; E+ZPE (first entry), G (second entry).
H2S attack on the N atom leads to the formation of CH3SH and HSNO molecules (Final 1) via transition state, TS1, with the activation barrier of 23.8 kcal mol–1 (Figure 6A). The reaction via attack on the S atom of CH3SH is characterized by a much higher activation barrier (TS2, Eact = 47.5 kcal mol–1) and leads to the formation of CH3S–SH and HNO (Final 2). Formation of CH3S–SH and HNO, Final 2, is also thermodynamically less favorable than formation of CH3SH and HSNO, Final 1 (13.9 and 7.3 kcal mol–1, respectively, Figure 6A).
With HS–, the reaction proceeds in the same manner: the activation barrier for the N attack (TS3) is lower than that for the S attack (TS4) (5.8 vs 39.8 kcal mol–1, respectively), with favorable formation of the N attack products (Final 3, Figure 6B).
In addition, we estimate that HNO generation by eq 15 is energetically unfavorable by 40 kJ/mol by using standard electrode potentials and known bond energies (see the Supporting Information).
Whereas RSNO can exist as two isomers (cis/trans, with an energy difference of 0.8 kcal mol–1), regioselective attack on the N atom leads only to cis-HSNO, in agreement with the observed IR spectra.
Whether the same kinetics and thermodynamics operate for larger intracellular proteins remains to be determined. In the light of recent suggestions that H2S acts though sulfhydrylation of protein thiol residues,3a,5b it is possible that direct reaction of RSNOs and H2S, with concomitant formation of HNO, takes place in vivo.
Sulfhydrylation of the proteins in vivo can be envisioned via another reaction mechanism. The reaction of hydroxylamine formation, given in eq 14, goes stepwise, and, for the case where H2S is reacting thiol, it would proceed as follows:
| 16 |
| 17 |
It is possible, however, that the product of the initial reaction, HS(O)NH2, reacts further with protein thiols, particularly when H2S is not present in large excess (eq 18), and form sulfhydrylated protein with elimination of hydroxylamine.
| 18 |
HSNO, a Diffusible Transnitrosating Agent
Given its small size and reactive nature, HSNO might serve as a shuttle for nitrosonium equivalents by supporting sequential transnitrosation reactions. Equation 3 in Scheme 1 is slightly, ca. 10 kJ mol–1, uphill energetically, which is readily calculated33a from the minor difference (ca. 12 kJ stronger) between bond strengths of GS–NO33b and HS–NO,8,9 and the electrode potentials, E°′(GS•,H+/GSH) = 0.94 V33c and E°′(S•–,2H+/H2S) = E°′(S•–,H+/HS–) = 0.92 V33d at pH 7. Consequently, eq 6 in Scheme 1 is energetically favorable by the same amount.
To examine the feasibility of this scenario, we first investigated transnitrosation reactions between GSNO and bovine serum albumin (BSA) as a model. Albumin is proposed to be one of the carriers/storages of bioactive NO• in human plasma.34 As a proof-of-concept experiment, H2S was incubated with GSNO at 37 °C for 5 min to allow complete GSNO decomposition prior to the addition of albumin. After addition of BSA and a further 15 min incubation, the reaction mixture was analyzed by ESI-TOF MS. The peak shifts imply that four nitroso equivalents per protein were added (see Figure S9). When the dialyzed HSNO-treated albumin sample was placed into buffer and irradiated with light, release of NO• was induced (see Figure S9), confirming S-nitrosation of BSA.
S-Nitrosoproteins are relatively stable and unless exposed to light or traces of heavy metals, they cannot exchange NO+ equivalents with other proteins through the membrane.6b Considering the potential significance of the shuttle function of HSNO, we designed an experiment specifically to address this notion (Figure 7A). Poly nitrosated albumin was placed inside a dialysis bag and inserted into a buffered solution of non-nitrosated albumin. In one of the samples, H2S was injected into the dialysis bag containing BSA-SNO, while the untreated one served as a control. If HSNO were to be formed as a small neutral species, it should have been able to freely diffuse through the membrane and reach the outside solution of BSA. Without such intermediate, the exchange of NO+ equivalents between the compartments would be unlikely. After 5 min of incubation, the dialysis bags were removed and the exterior albumin solution was dialyzed for 2 h against water to remove traces of small molecules. Subsequent addition of aliquots of these solutions into Cu2+-containing buffer (Figure 7B) revealed a clear NO• signal for H2S-treated samples as detected by an NO• electrode, whereas control incubates showed only minor signals, most likely the result of metal contamination. RSNO quantification by the Saville assay showed that 11 ± 1% of BSA became nitrosated in H2S-treated samples as compared to only 2 ± 1% in the controls (Figure 7C), and these results were qualitatively confirmed with the biotin-switch assay (Figure 7D).
Figure 7.

HSNO serves as a shuttle for NO+. (A) Experimental design for the protein-to-protein trans-nitrosation mediated by HSNO (see Experimental Section). (B) S-Nitrosothiol content in the sample obtained from protein-to-protein transnitrosation experiment. NO• electrode responses upon subsequent addition of aliquots of the control (black) or H2S-treated samples (red) into 500 μM ascorbate/Cu2+ containing solution. Upon addition of hemoglobin (Hb), all NO• was scavenged. (C) Total amount of RSNOs generated by H2S in protein-to-protein transnitrosation experiment (n = 4). (D) The results of the biotin switch assay for the same samples. (E,F) HSNO facilitated nitrosation of hemoglobin in human red blood cells. Deconvoluted mass spectra (E) of hemoglobin beta subunit isolated from RBC after the treatment with synthetic poly-S-nitrosoalbumin (BSA-SNO) in the presence or absence of H2S. Sample treated with poly-S-nitrosoalbumin in the presence of H2S exhibit another peak shifted by mass of 58 corresponding to [Hb – 2H + NO + K]+. S-Nitroso hemoglobin content (F) determined using Saville’s method. Hemoglobin concentration in all samples was 650 μM.
Additional experiments were carried out using human red blood cells (RBCs). S-Nitrosation of hemoglobin (Hb) is important for the regulation of oxygen delivery,35 but the chemical basis for this process is still a matter of debate.6b,6c To test whether HSNO could be a carrier from plasma S-nitrosoproteins to Hb, washed RBCs were incubated for 5 min in PBS supplemented with 30 μM poly-S-nitrosoalbumin (BSA-SNO),34a in the absence or presence of 50 μM Na2S. Hemoglobin was obtained after extensive washing of cells with PBS followed by hemolysis in nanopure water containing 100 μM neocuproine and analyzed by ESI-TOF-MS and Saville assay. Deconvoluted mass spectra (Figure 7E) revealed that the hemoglobin β-subunits (obtained mass 15 866.3 ± 1, expected 15 867.2) of RBCs treated with nitrosoalbumin and Na2S exhibit an additional peak shifted by a mass of 58, indicative of the addition of 2NO moieties/Hb ([Hb – 2H + 2NO + K]+). For comparison, the total S-nitrosoprotein concentration was determined in the samples of Hb treated with GSNO, BSA-SNO, and BSA-SNO + H2S (Figure 7F).
There are several pathways by which GSNO or CysNO could traverse biological membranes and deliver the “NO+” moiety. Some of them require a specific transporter or a thiol-to-thiol cascade transfer of “NO+”.6b,6c,37 We wanted to test the observed superiority of HSNO over other low-molecular weight thiols to pass freely through membranes and S-nitrosate the targets faster. A 20 μL portion of washed packed RBC was diluted 100 times with PBS and exposed to 100 μM poly BSA-SNO in the presence or absence of 100 μM GSH, cysteine, or H2S for 2 min. After hemolysis, Hb was analyzed by LC-ESI-TOF-MS. Only in the case of H2S treatment was doubly S-nitrosated Hb β-subunit present in detectable quantities (Fig. S10). The alpha subunit remained unchanged. These data imply that, unlike GSNO and CysSNO, HSNO can freely diffuse to reach its intracellular target and increase the intracellular HbSNO content.
Physiological Significance
Given the universal abundance of H2S and RSNOs in biological systems, it is plausible that HSNO is produced in vivo. The direct in vivo detection of HSNO, however, seemed challenging considering its demonstrated reactivity and relative instability. Nonetheless, we designed experiment to investigate its role in intracellular transnitrosation.36
If produced in cells, HSNO would be capable of further trans-nitrosation, so it is reasonable to assume that DAF-2 dye could serve as a sensor for its formation as well. Mechanistically, the principle of this imaging technique involves a nitrosation step of DAF-2 to produce the fluorescence. We therefore tested the role of intracellularly produced H2S on transnitrosation of DAF-2 induced by GSNO.
Cells were incubated in buffer supplemented with the inhibitor of H2S producing enzyme (CSE), propargyl glycine (PG), washed and then exposed to GSNO. Whereas endothelial cell pretreatment with PG had no significant effects on basal DAF-2 fluorescence, it markedly blocked the GSNO-induced fluorescence increase (Figure 8A). CSE inhibition followed by sulfide treatment before exposure to GSNO restored GSNO-induced fluorescence, confirming that the effects were not due to unspecific quenching.
Figure 8.
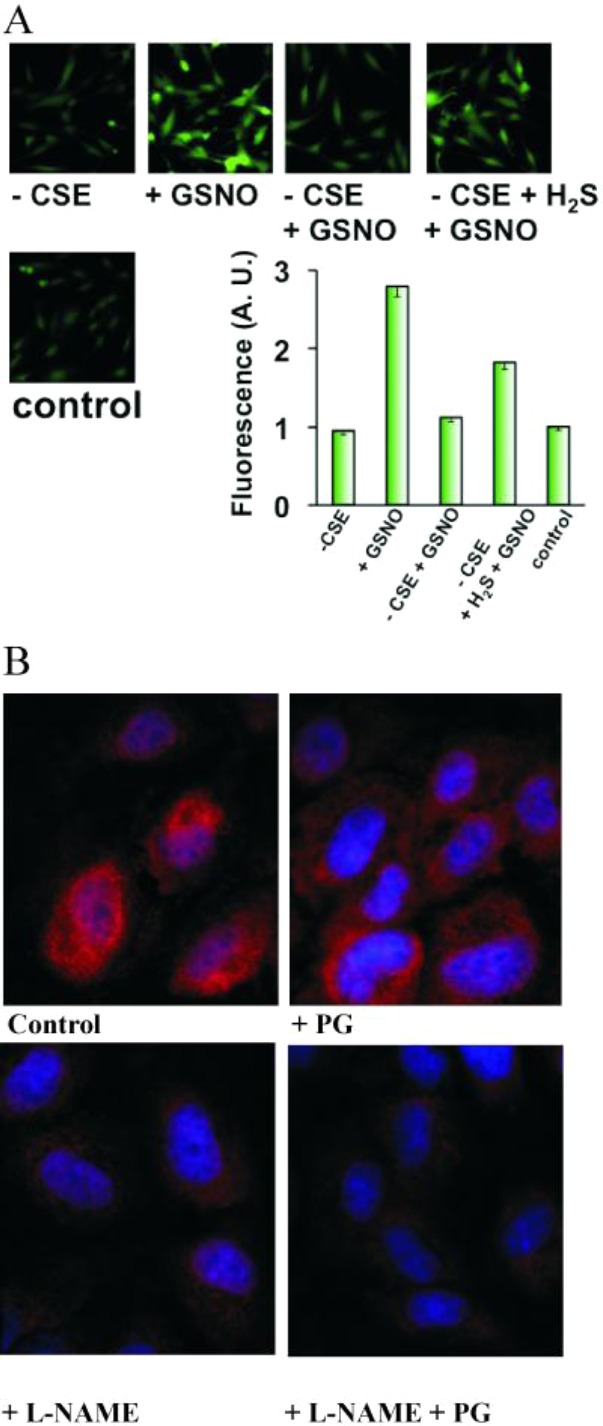
Endogenous H2S controls transnitrosation and S-nitrosothiol formation. (A) H2S-dependent GSNO-induced transnitrosation of DAF. HUVECs were incubated for 2 h in medium with or without CSE-inhibitor propargylglycine (1 mM). Cells were then exposed to 50 μM GSNO. Some cells were additionally incubated with 100 μM Na2S for 20 min prior to GSNO addition. NO-induced fluorescence was detected by fluorescence microscopy and quantified using ImageJ (n = 20–30 cells). (B) The effect of inhibitors of endogenous NO and H2S production on intracellular S-nitrosation. HUVECs were exposed to medium supplemented without or with 1 mM L-NAME, 1 mM PG, or both for 2 h. Cells were fixed and intracellular RSNOs visualized by immunocytochemistry using anti-S-nitrosocysteine antibodies.
We demonstrate that endogenous H2S is indeed involved in regulation of the intracellular S-nitrosation by an experiment in which HUVECs were exposed to the inhibitor of NO production (L-NG-nitroarginine methyl ester, L-NAME) or/and inhibitor of H2S production (propargylglycine, PG). Exposure to PG lowered the level of intracellular SNOs as detected using specific S-nitrosocysteine antibodies (Figure 8B). L-NAME had a much stronger effect, as expected, but did not completely block S-nitrosation because it is a competitive inhibitor of nitric oxide synthase. An inhibitory effect was, however, most obvious when cells were exposed to both L-NAME and PG (Figure 8B). These results suggest an important role of intracellular H2S in transnitrosation reactions.
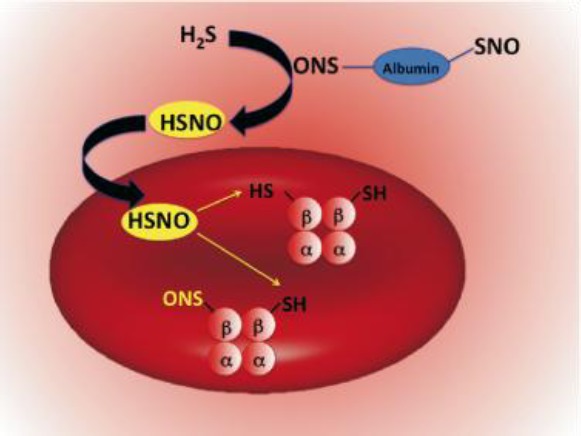
The unique physicochemical properties of HSNO make it an excellent candidate for redox sensing and regulation of metabolic activities. In particular, HSNO could lead to the generation of HNO, which has significant cardio-pharmacological potential, and herein we provide the first direct intracellular detection of its generation. HNO has been also suggested to be involved in H2S induced cardioprotection.4b Additionally, HSNO/SNO– might serve to facilitate further trans-nitrosation/denitrosation of other targets in the cell or even intercellularly, due to the facile diffusion ability of HSNO (as the neutral form of the HSNO/SNO– acid–base couple). Hemoglobin is one possible target, and the chemical mechanism of its S-nitrosation is still a matter of debate. S-Nitrosoproteins (RSNOs) cannot cross cell membranes by free diffusion,6b,37 and “NO+” transport has long been proposed to be facilitated by formation of smaller S-nitrosothiols, especially those of cysteine and/or glutathione, which may require specific transporters.37 Formation of HSNO/SNO– might link plasma S-nitrosothiols to intracellular S-nitrosohemoglobin. Circulating H2S could react with RSNOs forming HSNO/SNO– by transnitrosation, where HSNO would then freely diffuse across the RBC membrane to transfer its “NO+” moiety to the β-subunit of hemoglobin forming S-nitrosohemoglobin (Figure 9), known to have important physiological functions.35
Figure 9.
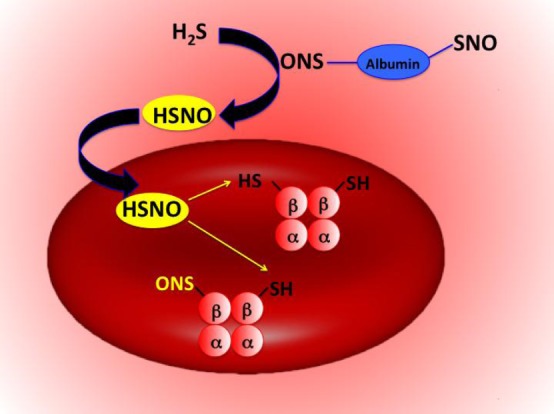
Proposed reaction scheme for HSNO-induced nitrosation of hemoglobin.
Three key proteins presently accepted as major targets for H2S and its subsequent regulation of blood pressure,2 neuronal activity,38a and cardioprotection,38b the KATP channel, NMDA receptor, and NRF-2, respectively, are shown to be regulated by S-nitrosation.39 Early5a and recent4d work suggests that activity of NO• synthase is a prerequisite for H2S to exhibit its effect. The present study thus offers a chemical perspective into the metabolic fate of H2S and opens a new chapter in biological chemistry of redox signaling.
Acknowledgments
We thank Dr. Leeane Nye (University of Erlangen-Nürnberg) for technical help with ESI-TOF MS measurements and Dr. Achim Zahl (University of Erlangen-Nürnberg) for help with 15N NMR measurements. This work was supported by the intramural grant from University Erlangen-Nürnberg (Emerging Field Initiative: Medicinal Redox Inorganic Chemistry). T.N. and W.H.K. acknowledge the support from ETH Zürich. Work in the lab of S.J.L. was supported by a grant from the National Science Foundation. M.R. acknowledges the NIH for a postdoctoral fellowship.
Supporting Information Available
Supporting text, supporting table, and supporting Figures S1–S10. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Koppenol W. H. Free Radical Biol. Med. 1998, 25, 385–391. [DOI] [PubMed] [Google Scholar]; b Ford P. C.; Lorkovic I. Chem. Rev. 2002, 102, 993–1018. [DOI] [PubMed] [Google Scholar]; c McCleverty J. A. Chem. Rev. 2002, 102, 403–418. [DOI] [PubMed] [Google Scholar]; d Tennyson A. G.; Lippard S. J. Chem. Biol. 2011, 18, 1211–1220. [DOI] [PubMed] [Google Scholar]; e Stamler J. S.; Singel D. J.; Loscalzo J. Science 1992, 258, 1898–1902. [DOI] [PubMed] [Google Scholar]; f Moncada S.; Palmer R. M. J.; Higgs E. A. Pharmacol. Rev. 1991, 43, 109–142. [PubMed] [Google Scholar]; g Calabrese V.; Cornelius C.; Rizzarelli E.; Owen J. B.; Dinkova-Kostova A. T.; Butterfield D. A. Antioxid. Redox Signaling 2009, 11, 2717–2739. [DOI] [PubMed] [Google Scholar]; h Palmer R. M. J.; Ferrige A. G.; Moncada S. Nature 1987, 327, 524–526. [DOI] [PubMed] [Google Scholar]
- Yang G.; Wu L.; Jiang B.; Yang W.; Qi J.; Cao K.; Meng Q.; Mustafa A. K.; Mu W.; Zhang S.; Snyder S. H.; Wang R. Science 2008, 322, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li L.; Rose P.; Moore P. K. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [DOI] [PubMed] [Google Scholar]; b Kabil O.; Banerjee R. J. Biol. Chem. 2010, 285, 21903–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li L.; Hsu A.; Moore P. K. Pharmacol. Ther. 2009, 123, 386–400. [DOI] [PubMed] [Google Scholar]
- a Whiteman M.; Li L.; Kostetski I.; Chu S. H.; Siau J. L.; Bhatia M.; Moore P. K. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [DOI] [PubMed] [Google Scholar]; b Yong Q. C.; Hu L. F.; Wang S.; Huang D.; Bian J. S. Cardiovasc. Res. 2010, 88, 482–491. [DOI] [PubMed] [Google Scholar]; c Ondrias K.; Stasko A.; Cacanyiova S.; Sulova Z.; Krizanova O.; Kristek F.; Malekova L.; Knezl V.; Breier A. Pfluegers Arch. 2008, 457, 271–279. [DOI] [PubMed] [Google Scholar]; d Sojitra B.; Bulani Y.; Putcha U. K.; Kanwal A.; Gupta P.; Kuncha M.; Banerjee S. K. Mol. Cell. Biochem. 2012, 60, 61–69. [DOI] [PubMed] [Google Scholar]; e Bertova A.; Cacanyiova S.; Kristek F.; Krizanova O.; Ondrias K.; Tomaskova Z. Gen. Physiol. Biophys. 2010, 29, 402–410. [DOI] [PubMed] [Google Scholar]
- a Zhao W.; Wang R. Am. J. Physiol.: Heart Circ. Physiol. 2002, 283, H474–H480. [DOI] [PubMed] [Google Scholar]; b Mustafa A. K.; Sikka G.; Gazi S. K.; Steppan J.; Jung S. M.; Bhunia A. K.; Barodka V. M.; Gazi F. K.; Barrow R. K.; Wang R.; Amzel L. M.; Berkowitz D. E.; Snyder S. H. Circ. Res. 2011, 109, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Foster M. W.; Hees D. T.; Stamler J. S. Trends Mol. Med. 2009, 15, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hogg N. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 585–600. [DOI] [PubMed] [Google Scholar]; c Keszler A.; Zhang Y.; Hogg N. Free Radical Biol. Med. 2010, 48, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lima B.; Forrester M. T.; Hess D. T.; Stamler J. S. Circ. Res. 2010, 106, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hess D. T.; Stamler J. S. J. Biol. Chem. 2012, 287, 4411–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Nakamura T.; Lipton S. A. Antioxid. Redox Signaling 2011, 14, 1479–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Tegeder I.; Scheving R.; Wittig I.; Geisslinger G. Pharmacol. Rev. 2011, 63, 366–389. [DOI] [PubMed] [Google Scholar]; h Stamler J. S.; Hess D. T. Nat. Cell Biol. 2010, 12, 1024–1026. [DOI] [PubMed] [Google Scholar]; i Benhar M.; Forrester M. T.; Hess D. T.; Stamler J. S. Science 2008, 320, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IUPAC nomenclature of the chemical compounds: HSNO, nitrososulfane or (hydridosulfanido)oxidonitrogen; NO·, oxidonitrogen(•); HNO2, hydroxidooxidonitrogen; H2S, dihydridosulfur; HNO, hydridooxidonitrogen; NO–, oxidonitrate(1−); NO+, oxidonitrogen(+); N2O, oxidodinitrogen.
- Timerghazin Q. K.; English A. M.; Peslherbe G. H. Chem. Phys. Lett. 2008, 454, 24–29. [Google Scholar]
- Timerghazin Q. K.; Peslherbe G. H.; English A. M. Phys. Chem. Chem. Phys. 2008, 10, 1532–1539. [DOI] [PubMed] [Google Scholar]
- a Nonella M.; Huber J. R.; Ha T.-K. J. Phys. Chem. 1987, 91, 5203–5209. [Google Scholar]; b Structural isomer of HSNO is known as thionylimide (HNSO).Kirchhoff W. H. J. Am. Chem. Soc. 1969, 91, 20437–20442. [Google Scholar]
- Seel F.; Kuhn R.; Simon G.; Wagner M.; Krebs B.; Dartmann M. Z. Naturforsch., B 1985, 40b, 1607–1617. [Google Scholar]
- Hughes M. N.; Centelles M. N.; Moore K. P. Free Radical Biol. Med. 2009, 47, 1346–1353. [DOI] [PubMed] [Google Scholar]
- Filipovic M. R.; Miljkovic J.; Algaeuer A.; Chario R.; Shubina T.; Herrmann M.; Ivanovic-Burmazovic I. Biochem. J. 2012, 441, 609–621. [DOI] [PubMed] [Google Scholar]
- Wang K.; Hou Y.; Zhang W.; Ksebeti M. B.; Xian M.; Cheng J.-P.; Wang P. G. Bioorg. Med. Chem. Lett. 1999, 9, 2897–2902. [DOI] [PubMed] [Google Scholar]
- Karmann W.; Meissner G.; Henglein A. Z. Naturforsch., B 1967, 22, 273–282. [Google Scholar]
- Nauser T.; Dockheer S.; Kissner R.; Koppenol W. H. Biochemistry 2006, 45, 6038–6043. [DOI] [PubMed] [Google Scholar]
- Rosenthal J.; Lippard S. J. Am. Chem. Soc. 2010, 132, 5536–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorren A. C.; Schrammel A.; Schmidt K.; Mayer B. Arch. Biochem. Biophys. 1996, 330, 219–228. [DOI] [PubMed] [Google Scholar]
- Stamler J. S.; Feelisch M.. Preparation and Detection of S-Nitrosothiols. In Methods in Nitric Oxide Research; Feelisch M., Stamler J. S., Eds.; John Wiley & Sons: Chichester, England, 1996; pp 521–539. [Google Scholar]
- Wang X.; Kettenhofen N. J.; Shiva S.; Hogg N.; Gladwin M. T. Free Radical Biol. Med. 2008, 44, 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamone G.; Sannolo N.; Malorni A.; Ferranti P. FEBS Lett. 1999, 462, 241–245. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. In The Challenge of d- and f-electrons: Theory and Computation; Salahub D. R., Zerner M. C., Eds.; American Chemical Society: Washington, DC, 1989; Chapter 12, p 165. [Google Scholar]; b Vosko S. H.; Wilk L.; Nusair M. Can. J. Phys. 1980, 58, 1200. [Google Scholar]; c Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]; d Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; e Kendall R. A.; Dunning T. H. Jr.; Harrison R. J. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar]; f Woon D. E.; Dunning T. H. Jr. J. Chem. Phys. 1993, 98, 1358–13571. [Google Scholar]; g Dunning T. H. Jr. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision B.1; Gaussian, Inc.: Wallingford, CT, 2009.
- a Mills G.; Schmidt K. H.; Matheson M. S.; Meisel D. J. Phys. Chem. 1987, 91, 1590–96. [Google Scholar]; b Chivers T.; Da Silva A. B. F.; Treu O. Jr.; Trsic M. J. Mol. Struct. 1987, 162, 351–357. [Google Scholar]
- Quiroga S. L.; Almaraz A. E.; Amorebieta V. T.; Perissinotti L. L.; Olabe J. A. Chem.-Eur. J. 2011, 17, 4145–4156. [DOI] [PubMed] [Google Scholar]
- Seel F.; Wagner M. Z. Anorg. Allg. Chem. 1988, 558, 189–192. [Google Scholar]
- Munro A. P.; Williams L. H. J. Chem. Soc., Perkin Trans. 2 2000, 1794–1797. [Google Scholar]
- Arnelle D. R.; Stamler J. S. Arch. Biochem. Biophys. 1995, 318, 279–285. [DOI] [PubMed] [Google Scholar]
- a Sonnenschein K.; de Groot H.; Kirsch M. J. Biol. Chem. 2004, 279, 45433–45440. [DOI] [PubMed] [Google Scholar]; b Meyer D. J.; Kramer H.; Özer N.; Coles B.; Ketterer B. FEBS Lett. 1994, 345, 177–180. [DOI] [PubMed] [Google Scholar]; c Singh R. J.; Hogg N.; Joseph J.; Kalyanaraman B. J. Biol. Chem. 1996, 271, 28596–18603. [DOI] [PubMed] [Google Scholar]
- a Vitvitsky V.; Kabil O.; Banerjee R. Antioxid. Redox Signaling 2012, 17, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shen X.; Pattillo C. B.; Pardue S.; Bir S. C.; Wang R.; Kevil C. G. Free Radical Biol. Med. 2011, 50, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Flores-Santana W.; Salmon D. J.; Donzelli S.; Switzer C. H.; Basudhar D.; Ridnour L.; Cheng R.; Glynn S. A.; Paolocci N.; Fukuto J. M.; Miranda K. M.; Wink D. A. Antioxid. Redox Signaling 2011, 14, 1659–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fukuto J. M.; Carrington S. J. Antioxid. Redox Signaling 2011, 14, 1649–1658. [DOI] [PubMed] [Google Scholar]; c Fukuto J. M.; Switzer C. H.; Miranda K. M.; Wink D. A. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 335. [DOI] [PubMed] [Google Scholar]; d Bazylinski D. A.; Hollocher T. C. J. Am. Chem. Soc. 1985, 107, 7982–7986. [Google Scholar]; e Miranda K. M.; Dutton A. S.; Ridnour L. A.; Foreman C. A.; Ford E.; Paolocci N.; Katori T.; Tocchetti C. G.; Mancardi D.; Thomas D. D.; Espey M. G.; Houk K. N.; Fukuto J. M.; Wink D. A. J. Am. Chem. Soc. 2005, 127, 722–731. [DOI] [PubMed] [Google Scholar]; f Wink D. A.; Feelisch M.. Formation and detection of nitroxyl and nitrous oxide. In Methods in Nitric Oxide Research; Feelisch M., Stamler J. S., Eds.; John Wiley & Sons: Chichester, England, 1996; pp 403–412. [Google Scholar]
- Singh S. P.; Wishnok J. S.; Keshive M.; Deen W. M.; Tannenbaum S. R. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Koppenol W. Inorg. Chem. 2012, 51, 5637–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bartberger M. D.; Mannion J. D.; Powell S. C.; Stamler J. S.; Houk K. N.; Toone E. J. J. Am. Chem. Soc. 2001, 123, 8868–8869. [DOI] [PubMed] [Google Scholar]; c Folkes L. K.; Trujillo M.; Bartesaghi S.; Radi R.; Wardman P. Arch. Biochem. Biophys. 2011, 506, 242–249. [DOI] [PubMed] [Google Scholar]; d Das T. N.; Huie R. E.; Neta P.; Padmaja S. J. Phys. Chem. A 1999, 103, 5221–5226. [Google Scholar]
- a Simon D. I.; Mullins M. E.; Jia L.; Gaston B.; Singel D. J.; Stamler J. S. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 4763–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Orie N. N.; Vallance P.; Jones D. P.; Moore K. P. Am. J. Physiol.: Heart Circ. Physiol. 2005, 289, H916–H923. [DOI] [PubMed] [Google Scholar]
- a Singel D. J.; Stamler J. S. Annu. Rev. Physiol. 2005, 67, 99. [DOI] [PubMed] [Google Scholar]; b Jia L.; Bonaventura C.; Bonaventura J.; Stamler J. S. Nature 1996, 380, 221–226. [DOI] [PubMed] [Google Scholar]; c Reynolds J. D.; Hess D. T.; Stamler J. S. Transfusion 2011, 51, 852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchet E.; Keiser W. E. J. Exp. Bot. 2006, 57, 3043–3055. [DOI] [PubMed] [Google Scholar]
- a Zhang Y.; Hogg N. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7891–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bronlowska K. A.; Zhang Y.; Hogg N. J. Biol. Chem. 2006, 45, 33835–33841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gadalla M. M.; Snyder S. H. J. Neurochem. 2010, 113, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Calvert J. W.; Jha S.; Gundewar S.; Elrod J. W.; Ramachandran A.; Pattillo C. B.; Kevil C. G.; Lefer D. J. Circ. Res. 2009, 105, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kawano T.; Zoga V.; Kimura M.; Liang M. Y.; Wu H. E.; Gemes G.; McCallum J. B.; Kwok W. M.; Hogan Q. H.; Sarantopoulos C. D. Mol. Pain 2009, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lipton S. A.; Choi Y. B.; Takahashi H.; Zhang D.; Li W.; Godzik A.; Bankston L. A. Trends Neurosci. 2002, 25, 474–480. [DOI] [PubMed] [Google Scholar]; c Um H. C.; Jang J. H.; Kim D. H.; Lee C.; Surh Y. J. Nitric Oxide 2011, 25, 161–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.