

Background: Her-2-induced mammary tumor onset is significantly delayed in GnT-V knock-out mice.
Results: The gene expression of the Pcdhβ cluster is up-regulated in her-2-induced tumors with GnT-V deletion.
Conclusion: Up-regulation of the Pcdhβ cluster is one of the mechanisms for the reduced her-2-mediated tumorigenesis resulting from GnT-V deletion.
Significance: Our findings shed new light on the molecular mechanisms of the effects of GnT-V on mammary tumorigenesis.
Keywords: Cancer Biology, Gene Transcription, Glycosyltransferases, Oncogene, Receptor Regulation, GnT-V, Her-2, Protocadherin
Abstract
Changes in the levels of N-acetylglucosaminyltransferase V (GnT-V) can alter the function of several types of cell surface receptors and adhesion molecules by causing altered N-linked glycan branching. Using a her-2 mammary tumor mouse model, her-2 receptor signaling was down-regulated by GnT-V knock-out, resulting in a significant delay in the onset of her-2-induced mammary tumors. To identify the genes that contributed to this GnT-V regulation of early events in tumorigenesis, microarray analysis was performed using her-2 induced mammary tumors from wild-type and GnT-V-null mice. We found that 142 genes were aberrantly expressed (>2.0-fold) with 64 genes up-regulated and 78 genes down-regulated after deletion of GnT-V. Among differentially expressed genes, the expression of a subgroup of the cadherin superfamily, the protocadherin β (Pcdhβ) cluster, was up-regulated in GnT-V-null tumors. Altered expression of the Pcdhβ cluster in GnT-V-null tumors was not due to changes in promoter methylation; instead, impaired her-2-mediated signaling pathways were implicated at least in part resulting from reduced microRNA-21 expression. Overexpression of Pcdhβ genes inhibited tumor cell growth, decreased the proportion of tumor-initiating cells, and decreased tumor formation in vivo, demonstrating that expression of the Pcdhβ gene cluster can serve as an inhibitor of the transformed phenotype. Our results suggest the up-regulation of the Pcdhβ gene cluster as a mechanism for reduced her-2-mediated tumorigenesis resulting from GnT-V deletion.
Introduction
Studies have shown that changes in N-glycan structures on specific receptors were associated with abnormal receptor-mediated phenotypes by affecting cell adhesion, migration, cell survival, and tumorigenesis (1). A glycan whose expression is often up-regulated during malignant transformation contains N-linked β(1,6)-acetylglucosamine synthesized by N-acetylglucosaminyltransferase V (GnT-V2 or Mgat5; EC 2.4.1.155) (2, 3). Both in vitro and in vivo studies have implicated GnT-V in regulating tumorigenesis and invasiveness (4–9). Moreover, patients with colorectal or breast carcinomas that show expression of GnT-V glycan product have lowered 5-year survival rates (7, 10). Multiple cell surface receptors have been identified as substrates of GnT-V, including integrins (4, 6), cadherins (11, 12), and growth factor receptors (13, 14).
Her-2 (neu/ErbB2), a 185-kDa transmembrane glycoprotein and a member of the epidermal growth factor (EGF) receptor family, is overexpressed in ∼15–30% of human breast cancer and has been correlated with poor prognosis of cancer patients and therapeutic resistance (15, 16). The oncogenic potential of her-2 in mammary tumorigenesis has been confirmed in transgenic mouse models with overexpression or mutation of her-2 under the transcriptional control of the mouse mammary tumor virus promoter, and oncogenesis induced by her-2 in these transgenic mice has been shown to be quite similar to human breast cancer (17, 18). We recently showed that her-2 receptor signaling is modulated by GnT-V expression levels. Her-2-induced mammary tumor onset is significantly delayed in GnT-V knock-out mice coincident with the reversion of her-2-induced deregulation of acinar morphogenesis and a significantly reduced population of tumor-initiating cells (cancer stem cells) in isolated tumor cells with GnT-V deletion, resulting in reduced ability to form secondary tumors in NOD/SCID mice (9). These results indicate that GnT-V promotes mammary tumor development by regulating some early events during tumorigenesis.
In addition to altering growth factor receptor signaling, the function of cadherins is modulated by GnT-V expression levels. For example, knockdown of GnT-V reduces the expression of β(1,6) branching on the extracellular EC2–3 domains of N-cadherin, increasing N-cadherin-mediated cell-cell adhesion (11), whereas the deletion of GnT-V increases E-cadherin localization in cell adhesion junctions and cell-cell adhesion in both polyoma middle-T and her-2-induced mammary tumors (9, 13). The cadherins constitute a superfamily of single pass transmembrane glycoproteins mediating calcium-dependent cell-cell adhesion that plays an essential role in regulating major cellular behaviors, including cell growth, motility, and differentiation (19, 20). Several subgroups of cadherins have been defined based on shared properties and sequence similarity, including classical cadherins and protocadherins (21). Protocadherins (Pcdhs) are divided into clustered and non-clustered groups based on their genomic structures. Three closely linked protocadherin gene clusters, Pcdhα, Pcdhβ, and Pcdhγ, have been identified in both mouse and human (22, 23). Unlike classical cadherins that have five extracellular domains, a transmembrane domain, and a conserved cytoplasmic domain, Pcdhs have six extracellular domains encoded by one large exon and distinct intracellular domains (24). Protocadherins are predominantly expressed in the nervous system and appear to play an important role in regulating neuron development (21). However, epigenetic aberrations of protocadherin gene clusters caused by hypermethylation have been identified recently in some human tumors, including breast cancer (25–28), indicating that Pcdh genes may serve as tumor “suppressors” that can inhibit tumor development.
To identify the genes that could be contributive to early breast tumor progression regulated by GnT-V (9), microarray analysis was performed using mammary tumor tissues from GnT-V wild-type and knock-out her-2 mice, and qRT-PCR was used to confirm transcript differences. Here, we present evidence that members of the Pcdhβ gene cluster are implicated, functioning to suppress the malignant phenotype, in her-2-mediated mouse mammary tumorigenicity. We found that deletion of GnT-V caused enhanced gene expression of the Pcdhβ cluster in GnT-V knock-out tumors that contributes to the reduced her-2-induced tumorigenesis. Increased gene expression of the Pcdhβ cluster in GnT-V knock-out tumors was mediated by attenuated her-2-mediated signaling pathways caused by deletion of GnT-V. One of the downstream regulators of her-2 signaling, microRNA-21 (miR-21), was identified and implicated at least in part in the increased gene expression of Pcdhβ cluster.
EXPERIMENTAL PROCEDURES
Cell Lines and Materials
Human breast carcinoma cell lines MDA-MB231 and SK-BR3 were from the American Type Culture Collection (Manassas, VA). Mouse her-2 tumor cells with different GnT-V backgrounds were isolated from her-2 tumor tissues as described in our previous report (9). For silencing experiments, miRCURY locked nucleic acid-modified anti-miR-21 or control miRCURY knockdown oligonucleotides were purchased from Exiqon. Silencer® select neu/ErbB2 siRNA and scrambled control siRNA oligonucleotides were from Applied Biosystem (Ambion). pSuper vectors containing GnT-V siRNA and scrambled control siRNA were constructed and described in our previous report (14). 5-Aza-2′-deoxycytidine, PD98059, and wortmannin were purchased from Sigma; LipofectamineTM 2000 reagent was from Invitrogen. Anti-Pcdhβ4 and -Pcdhβ7 were products of Abcam. Antibodies against ErbB2/neu, ERK, phospho-ERK, PKB, and phospho-PKB and HRP-labeled anti-rabbit IgG and anti-mouse IgG were from Santa Cruz Biotechnology. Anti-c-myc tag (clone 9E10) was from Millipore.
Mouse Breeding and Tumor Tissue Isolation
All procedures used for this study were approved by the Institutional Animal Care and Use Committee of the University. The her-2/GnT-V(+/+) and her-2/GnT-V(−/−) mice were produced by breeding her-2/neu transgenic mice with GnT-V knock-out mice and genotyped by PCR as our previously described (9). Mice with tumors were euthanized at 10 weeks after the first detection of a palpable tumor. Mammary tumor tissues were collected from three her-2 mice with wild-type GnT-V(+/+) (mouse identification numbers 2319, 2328, and 2323) and GnT-V-null (−/−) (mouse identification numbers 2318, 2205, and 2414) backgrounds, respectively; immediately frozen in liquid nitrogen; and stored at −80 °C.
Microarray
Total RNA was isolated from tumor tissues using TRIzol reagent and cleaned using RNeasy columns (Qiagen). Using a random hexamer incorporating a T7 promoter, double-stranded cDNA was synthesized from total RNA. cRNA was generated from the double-stranded cDNA template through an in vitro transcription reaction and purified using the Ambion WT Expression kit and sample cleanup module. cDNA was then regenerated through a random primed reverse transcription using a dNTP mixture containing dUTP. Fragmented and biotinylated cDNA was used for hybridization with an Affymetrix Mouse GeneChip® Gene 1.0 ST Array according to the manufacturer's protocol.
Expression Analysis
Gene expression alterations were determined using the PARTEK Genomics Suite. The CEL files were imported from the Affymetrix Expression Console and background-corrected and quantile-normalized, and probe summarization was performed using Robust Multichip Analysis (RMA). A gene summarization was performed on the data that estimates the intensity of individual genes by averaging the intensities of all the probe sets comprising the gene followed by an n-way analysis of variance using a mixed model and methods of moment to equate analysis of variance mean sum of squares to their expected values. The data were then analyzed using a two-sample t test for significance at p = 0.05 and a -fold change cutoff of 2.0. To assess the possible functional connections between the differentially expressed genes, a pathway analysis, which assesses statistically overrepresented functional terms within a list, was conducted using Ingenuity Pathways Analysis (IPA) for all comparisons. The probability that a specific set of genes has a significant number of members in a canonical pathway is assigned a p value, which is calculated by Fisher's exact test (right tailed). The p value indicates the probability of observing the fraction of the focus genes in the canonical pathway compared with the fraction expected by chance in the reference set with the assumption that each gene is equally likely to be selected by chance.
qRT-PCR Analysis
TRIzol was used to isolate total RNA from tumor tissues and cell lines. Reverse transcription reactions were performed using cDNA synthesis kit (Bio-Rad). Primers used in the qRT-PCR analysis are listed in supplemental Table S1. Real time reactions were performed using the iQTM SYBR Green Supermix (Bio-Rad) as reported previously (29). All PCRs were performed in triplicate samples and repeated at least two times.
Genomic DNA Purification, Bisulfite Modification, and Methylation-specific PCR
Purification and bisulfite treatment of genomic DNA samples were performed using the DNeasy Tissue kit and the EpiTect Bisulfite kit (Qiagen), respectively, according to the manufacturer's instructions. Methylation-specific PCR was carried out using the following cycling conditions: 95 °C for 5 min; 40 cycles at 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 45 s; and a final cycle at 72 °C for 5 min. The primer sequences used for methylation-specific PCR are listed in supplemental Table S2. The PCR products were isolated on a 1.5% agarose gel and visualized by ethidium bromide staining.
Real Time PCR Detection of miR-21
Mature miR-21 was detected using the miR-Q method as described previously (30, 31). In brief, 500 ng of total RNA was used for reverse transcription of mature miR-21 using TaqMan reverse transcription reagents (Applied Biosystems) with a specific reverse primer (RT6-miR-21, tgtcaggcaaccgtattcaccgtgagtggttcaaca). The miR-cDNA generated was then quantitated using SYBR Green PCR Master Mix (Bio-Rad) with another specific primer (short-miR-21-rev, cgtcagatgtccgagtagagggggaacggcgtagcttatcagactga) and one pair of universal primers (MP-fw, tgtcaggcaaccgtattcacc and MP-rev, cgtcagatgtccgagtagag). The amplification was performed by a first step at 95 °C for 10 min followed by 40 cycles of 15 s at 95 °C, 10 s at 59 °C, and 20 s at 72 °C. Amplification of Gapdh was used as an internal control.
Construction of pcDNA3.1/myc-His/Pcdhβ Expression Plasmids and Cell Transfection
Mouse Pcdhβ4 cDNA clone (clone identification number 6401988) and Pcdhβ19 cDNA clone (clone identification number 9055902) were purchased from Source BioScience (Cambridge, UK). These cDNA clones were used as PCR templates to amplify the ORFs of Pcdhβ4 and Pcdhβ19 with the forward primer containing a HindIII site and the reverse primer containing an XbaI site. The sequences of forward and reverse primers were 5′-cccaagcttacaatggagacagcgcta-3′ and 5′-ctgtatctagaactattcaacatgt-3′, respectively, for Pcdhβ4 and 5′-tggaagcttactatggagaatcaagag-3′ and 5′-acatctagaattacagtccctaaat-3′, respectively, for Pcdhβ19. The final PCR product was ligated into a pcDNA3.1/myc-His expression vector (Invitrogen) digested with HindIII and XbaI according to the manufacturer's instructions. The resulting vectors were confirmed by sequencing and digested with HindIII and XbaI to release the DNA fragment, which was inserted into the pcDNA3.1 vector upstream from the myc-His tags.
Cell transfections were performed in 6-well plates with Lipofectamine 2000 according to the manufacturer's instructions using 4 μg of recombinant plasmids. 24 h after transfection, cells were selected for 3 weeks using G418 (800 μg/ml), and nonclonal populations of transfected cells were used for all experiments.
Colony Formation and Anchorage-independent Growth Assay
For the colony formation assay, cells were transfected with 4 μg of expression plasmids in 6-well plates and transferred into 100-mm culture dishes the next day. Selection was performed 2 days after transfection with 800 μg/ml G418 for 2 weeks. Colonies were stained with crystal violet and counted in 5–10 random fields under a phase-contrast microscope.
An assay of cell growth in soft agar was performed using 24-well culture plates (27). The wells were coated with two layers of agar in different concentrations. The lower layer was 0.7% agar in 0.9% sodium chloride, whereas the upper layer was 0.35% soft agar in complete culture medium. 3 × 104 cells were added into the upper layers of the wells. Plates were incubated at 37 °C in 5% CO2 for 2–3 weeks. Then the numbers of the colonies that developed in soft agar were counted in 5–10 random fields under a microscope.
Implantation of Tumor Cells in NOD/SCID Mice
All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Georgia. Subconfluent tumor cells were harvested and resuspended in serum-free Hanks' balanced salt solution in a 70-μl volume containing 2 × 106 cells. After NOD/SCID mice (The Jackson Laboratory) were anesthetized with isoflurane, a 70-μl single cell suspension mixed with 30 μl of Matrigel (BD Biosciences) was injected into flanks of mice of 6–8 weeks age using a 27-gauge needle (9). Tumor formation was monitored by palpation, and tumor size was measured with calipers once a week.
Western Blotting
Subconfluent cells were harvested and lysed. Total cell lysates containing 30 μg of protein were used for Western blotting as described in our earlier report (6).
ALDEFLUOR Assay
To detect tumor cells with high aldehyde dehydrogenase activity, the ALDEFLUOR assay was performed using an ALDEFLUOR kit from STEMCELL Technologies as described previously (9, 32). In brief, dissociated single cells (1 × 106 cells/ml) were incubated in ALDEFLUOR assay buffer containing aldehyde dehydrogenase substrate (1.5 μm) at 37 °C for 30 min. In each experiment, a fraction of cells was stained under identical conditions with a specific aldehyde dehydrogenase inhibitor, diethylaminobenzaldehyde (15 μm), as a negative control. After staining with propidium iodide, aldehyde dehydrogenase-positive (tumor-initiating cells) and -negative cells (non-tumor-initiating cells) were analyzed using flow cytometry.
Immunochemical and Fluorescent Staining
Immunochemical staining were performed using a VECTASTAIN® Elite ABC kit (Vector laboratories) following the manufacturer's instructions. For fluorescent staining, cells were cultured on chamber slides, fixed with 4% paraformaldehyde in PBS for 10 min, and permeabilized with 0.05% Triton X-100. After blocking with 10% goat serum, cells were stained with primary antibodies followed by incubation with secondary fluorescence-conjugated anti-mouse or rabbit IgG (1:250). After washing with PBS, the chamber slides were mounted, and the cells were subjected to fluorescence microscopy.
RESULTS
Microarray Analysis of Her-2-induced Mammary Tumors
We recently demonstrated that deletion of GnT-V reduces the size of the compartment of tumor-initiating cells in a her-2 mouse model, consequently leading to an inhibition of her-2-induced mammary tumor onset (9). To systematically study the mechanisms by which deletion of GnT-V inhibited her-2-induced tumor onset, microarray analyses were performed on the her-2-induced mammary tumors isolated from mice with wild-type (WT) and GnT-V-null (KO) genotypes. Three tumor tissues (T1–3) were collected, each from three her-2 mice with wild-type GnT-V(+/+) and GnT-V-null (−/−) backgrounds, respectively, and the GnT-V expression was confirmed by RT-PCR using total RNA (supplemental Fig. 1A). Using the normalized data from the Mouse GeneChip Gene 1.0 ST Array, gene lists were generated using the PARTEK Genomics Suite for differentially expressed genes in these two groups of tumors. There were 142 genes (0.4% of the total 35,556 genes) differentially expressed in GnT-V KO tumors compared with wild-type tumors using a 2-fold change as the ratio threshold. Among the 142 genes identified, 64 genes were up-regulated, and 78 genes were down-regulated (from 2.0- to 6.5-fold) (supplemental Table S3). Of the 142 differentially expressed genes, 57 genes (40.1%) were found to be significantly different (p < 0.05) (Table 1). An agglomerative clustering diagram (heat map) was generated from the microarray data of her-2 tumors based on the differentially expressed genes (Fig. 1A and supplemental Fig. 1B) in GnT-V KO tumors. Functional characterization of these differentially expressed genes by Ingenuity Pathways Analysis (IPA) classified them into different categories, including RNA damage and repair, cellular compromise, inflammatory response, and cell-to-cell signaling and interaction (supplemental Fig. 1B, left panel). The top 10 IPA-identified, differentially expressed genes (both up-regulated and down-regulated) are summarized in supplemental Table S4 and were validated by qRT-PCR as shown in supplemental Fig. 2.
TABLE 1.
Genes showing significant -fold changes by microarray analysis
| Gene | Symbol | RefSeq accession no. | p value (KO/WT) | -Fold change (KO/WT) |
|---|---|---|---|---|
| Enolase 2, γ neuronal | Eno2 | NM_013509 | 4.32e−05 | −2.24011 |
| Hedgehog-interacting protein-like 2 | Hhipl2 | BC034362 | 0.000242 | −6.52572 |
| Mannoside acetylglucosaminyltransferase 5 | Mgat5 | NM_145128 | 0.00044 | −2.11392 |
| CD14 antigen | Cd14 | NM_009841 | 0.000607 | 2.13547 |
| N-Acetylneuraminate pyruvate lyase | Npl | NM_028749 | 0.000753 | −3.64734 |
| —a | — | 0.000949 | −3.4074 | |
| Glucosaminyl (N-acetyl) transferase 1, core 2 | Gcnt1 | NM_173442 | 0.00149 | −3.37106 |
| — | — | 0.002031 | −2.03624 | |
| Regulator of G-protein signaling 2 | Rgs2 | NM_009061 | 0.003417 | −3.81328 |
| Adenylate kinase 3-like 1 | Ak3l1 | NM_009647 | 0.003423 | −2.26087 |
| RIKEN cDNA 1700040L02 gene | 1700040L02Rik | BC087900 | 0.004049 | 2.81943 |
| Interferon-activated gene 202B | Ifi202b | NM_008327 | 0.005126 | −2.69043 |
| — | — | 0.009093 | 2.03234 | |
| Solute carrier family 38, member 3 | Slc38a3 | NM_023805 | 0.00921 | 2.13304 |
| — | — | 0.009639 | 2.23546 | |
| Heat shock protein 1A | Hspa1a | NM_010479 | 0.010781 | −2.13604 |
| Potassium inwardly rectifying channel, subfamily J, member 13 | Kcnj13 | NM_001110227 | 0.012275 | −2.13735 |
| — | — | 0.01239 | −2.1894 | |
| Dipeptidylpeptidase 10 | Dpp10 | NM_199021 | 0.01564 | −4.71333 |
| RAS guanyl-releasing protein 1 | Rasgrp1 | NM_011246 | 0.015876 | −2.32995 |
| ENSMUSG00000072618 | ENSMUST00000100713 | 0.016753 | 3.72366 | |
| Caspase 12 | Casp12 | NM_009808 | 0.017624 | 2.50816 |
| d-Aspartate oxidase | Ddo | NM_027442 | 0.017712 | 2.01305 |
| Actin-binding LIM protein family, member 3 | Ablim3 | NM_198649 | 0.020009 | −2.25541 |
| Zinc finger protein 521 | Zfp521 | NM_145492 | 0.020746 | 3.12831 |
| Protocadherin β19 | Pcdhb19 | NM_053144 | 0.020934 | 2.02627 |
| NAD(P)H dehydrogenase, quinone 1 | Nqo1 | NM_008706 | 0.020957 | 2.4185 |
| — | — | 0.021262 | 2.29063 | |
| Protocadherin β18 | Pcdhb18 | NM_053143 | 0.021566 | 2.21905 |
| — | — | 0.022472 | 3.79211 | |
| Mohawk homeobox | Mkx | NM_177595 | 0.022573 | −2.66449 |
| V-set domain-containing T cell activation inhibitor 1 | Vtcn1 | NM_178594 | 0.022682 | 2.55331 |
| Scrapie-responsive gene 1 | Scrg1 | NM_009136 | 0.023531 | 3.86718 |
| — | — | 0.02368 | −2.07973 | |
| Major facilitator superfamily domain-containing 4 | Mfsd4 | NM_001114662 | 0.026284 | −3.09621 |
| Secernin 1 | Scrn1 | NM_027268 | 0.026819 | 2.32697 |
| Insulin-like growth factor-binding protein 4 | Igfbp4 | NM_010517 | 0.027816 | −2.30083 |
| SPARC-related modular calcium binding 1 | Smoc1 | NM_001146217 | 0.027821 | 2.88559 |
| Cadherin 19, type 2 | Cdh19 | NM_001081386 | 0.028015 | −3.07028 |
| — | — | 0.028619 | −2.0569 | |
| RIKEN cDNA 1700055N04 gene | 1700055N04Rik | AK081788 | 0.03032 | −2.96093 |
| — | — | 0.030372 | 2.20457 | |
| — | — | 0.034671 | −2.01378 | |
| Synaptic vesicle glycoprotein 2c | Sv2c | NM_029210 | 0.034726 | −2.22967 |
| — | — | 0.036855 | 2.21008 | |
| — | — | 0.037019 | 2.49757 | |
| — | — | 0.037103 | 2.077 | |
| Protocadherin β4 | Pcdhb4 | NM_053129 | 0.038903 | 2.04059 |
| Cytohesin 1-interacting protein | Cytip | NM_139200 | 0.039081 | 2.33386 |
| TOX high mobility group box family member 3 | Tox3 | NM_172913 | 0.040529 | −2.26008 |
| Thrombospondin, type I, domain-containing 4 | Thsd4 | NM_001040426 | 0.041443 | 2.06645 |
| Mucin 15 | Muc15 | NM_172979 | 0.043913 | 2.11666 |
| Claudin 8 | Cldn8 | NM_018778 | 0.045599 | 2.66327 |
| Protocadherin β2 | Pcdhb2 | NM_053127 | 0.048118 | 2.11683 |
| — | — | 0.048788 | 2.41597 | |
| Protocadherin β7 | Pcdhb7 | NM_053132 | 0.049552 | 2.15923 |
| Low density lipoprotein receptor class A domain-containing 3 | Ldlrad3 | NM_178886 | 0.050311 | −2.8352 |
a —, unidentified genes. SPARC, sereted protein, acidic, cystein-rich (osteonectin); LIM, lipophilic protein; TOX, thymocyte selection-associated high mobility group box.
FIGURE 1.

Microarray analysis of gene expression in her-2-induced tumors. A, total RNA was isolated from three wild-type and GnT-V knock-out tumor tissues and used for microarray analysis. A heat map of 57 transcripts differentially expressed (p < 0.05) between wild-type and GnT-V knock-out tumors was generated from the microarray data. Red indicates a high expression level, whereas green indicates a low expression level relative to wild-type. T1–3 represents three tumor tissues collected from three mice with GnT-V wild-type and knock-out backgrounds, respectively. B, total RNA was isolated from two wild-type and two GnT-V knock-out tumors and used for validation of transcript levels of the Pcdhβ cluster by qRT-PCR. For each transcript, the values are normalized to control (Gapdh or Rpl4) and expressed as means with error bars indicating ±1 S.D. C, comparison of -fold change (KO/WT) of transcripts from microarray and qRT-PCR analyses. D, mammary tumor sections from 10-week-old mice were immunostained with leukocytic phytohemagglutinin (L-PHA), anti-Pcdhβ4, and anti-Pcdhβ7, respectively.
Gene Expression of Pcdhβ Cluster Is Up-regulated in GnT-V KO Tumors
The functions of many cell surface receptors have been shown to be altered by the GnT-V expression level, including integrins (4, 6), cadherins (11, 12), and EGF family receptors (EGF receptor/ErbB1/her-1 and ErbB2/her-2) and TGFβ receptors (9, 13, 14). As listed in supplemental Table S5, compared with wild-type tumors, her-2-induced mammary tumors showed little change in the transcript abundances of other glycosyltransferases and lectins except for Gcnt1, the enzyme that synthesizes core 2-O-glycans. Gcnt1 was significantly down-regulated (>3-fold) in GnT-V-null tumors based on the microarray data. However, GnT-V expression was reduced by only 2.1-fold in GnT-V KO tumors, although undetectable levels of both GnT-V mRNA and N-linked β(1,6) branching were observed by RT-PCR and leukocytic phytohemagglutinin staining, respectively (supplemental Fig. 1, A and D) (9), indicating either that some probes in the GnT-V probe set used in the microarray analysis target the areas of GnT-V mRNA that is transcribed from the non-deleted portions of the GnT-V gene (5) or that nonspecific hybridization generated by a few of the probes in the GnT-V probe set may have occurred. Among the 142 differentially expressed genes in GnT-V KO tumors, neither integrins nor growth factor receptors were observed, indicating that deletion of GnT-V had no detectable effects on the transcript levels of these receptors (supplemental Table S6). Many differentially expressed genes, however, were found in the cadherin family, including classic cadherin 19 (Cdh19) and the Pcdhβ cluster (Pcdhβ2, Pcdhβ4, Pcdhβ7, Pcdhβ18, and Pcdhβ19) as shown in Fig. 1A (indicated by arrows) and Table 2. Interestingly, all gene members of the Pcdhβ group were up-regulated to a different degree in GnT-V-null tumors (Table 2), a result validated by qRT-PCR analyses (Fig. 1, B and C). These results suggested that the protocadherin β gene cluster may function to impede tumor progression and could be involved to delay the onset of her-2-induced mammary tumors in the mice with no GnT-V expression (9).
TABLE 2.
Microarray analyses of cadherin genes
| Gene symbol | RefSeq accession no. | p value (KO/ WT) | -Fold change (KO/WT) |
|---|---|---|---|
| Cdh1 | NM_009864 | 0.0482811 | 1.19963 |
| Cdh10 | NM_009865 | 0.947114 | 1.00215 |
| Cdh11 | NM_009866 | 0.0454794 | −1.22638 |
| Cdh11 | NM_009866 | 0.190889 | 1.13634 |
| Cdh12 | NM_001008420 | 0.81751 | −1.01606 |
| Cdh13 | NM_019707 | 0.0121108 | −1.34055 |
| Cdh15 | NM_007662 | 0.513186 | 1.02147 |
| Cdh16 | NM_007663 | 0.221272 | 1.08585 |
| Cdh17 | NM_019753 | 0.00202595 | 1.09613 |
| Cdh18 | NM_001081299 | 0.447682 | 1.03206 |
| Cdh18 | NM_001081299 | 0.536517 | −1.05179 |
| Cdh19 | NM_001081386 | 0.0280145 | −3.07028 |
| Cdh2 | NM_007664 | 0.131852 | −1.35843 |
| Cdh20 | NM_011800 | 0.560637 | 1.05647 |
| Cdh22 | NM_174988 | 0.497218 | −1.02541 |
| Cdh23 | NM_023370 | 0.974851 | 1.00535 |
| Cdh24 | NM_199470 | 0.244223 | 1.04482 |
| Cdh24 | NM_199470 | 0.332417 | 1.05419 |
| Cdh26 | NM_198656 | 0.00252523 | 1.12984 |
| Cdh3 | NM_001037809 | 0.180038 | 1.39927 |
| Cdh4 | NM_009867 | 0.0425419 | −1.0686 |
| Cdh5 | NM_009868 | 0.0339117 | −1.50675 |
| Cdh6 | NM_007666 | 0.0109419 | −1.05413 |
| Cdh7 | NM_172853 | 0.503633 | 1.04568 |
| Cdh8 | NM_001039154 | 0.0832978 | −1.0873 |
| Cdh9 | NM_009869 | 0.616825 | 1.03588 |
| Pcdhb1 | NM_053126 | 0.0759057 | 1.16026 |
| Pcdhb10 | NM_053135 | 0.133674 | 1.68619 |
| Pcdhb11 | NM_053136 | 0.116768 | 2.33405 |
| Pcdhb11 | BC116321 | 0.918698 | 1.01082 |
| Pcdhb12 | NM_053137 | 0.208368 | 1.4337 |
| Pcdhb13 | NM_053138 | 0.404271 | 1.54499 |
| Pcdhb14 | NM_053139 | 0.266798 | 1.37775 |
| Pcdhb15 | NM_053140 | 0.155838 | 1.58338 |
| Pcdhb16 | NM_053141 | 0.0844595 | 1.8585 |
| Pcdhb17 | NM_053142 | 0.17262 | 1.54492 |
| Pcdhb18 | NM_053143 | 0.0215659 | 2.21905 |
| Pcdhb19 | NM_053144 | 0.0209341 | 2.02627 |
| Pcdhb2 | NM_053127 | 0.0481177 | 2.11683 |
| Pcdhb20 | NM_053145 | 0.0421417 | 1.75779 |
| Pcdhb21 | NM_053146 | 0.0452228 | 1.76552 |
| Pcdhb22 | NM_053147 | 0.275681 | 1.43601 |
| Pcdhb3 | NM_053128 | 0.143204 | 1.96662 |
| Pcdhb4 | NM_053129 | 0.0389026 | 2.04059 |
| Pcdhb5 | NM_053130 | 0.0149483 | 1.81789 |
| Pcdhb6 | NM_053131 | 0.0947665 | 2.38506 |
| Pcdhb7 | NM_053132 | 0.0495524 | 2.15923 |
| Pcdhb8 | NM_053133 | 0.185062 | 1.3181 |
| Pcdhb9 | NM_053134 | 0.0761935 | 1.66598 |
To test whether the expression of Pcdhβ was also increased at the protein level, tumor sections were used for immunochemical staining using antibodies against Pcdhβ4 and Pcdhβ7 (Fig. 1D). Consistent with increased gene expression, GnT-V KO tumors in which leukocytic phytohemagglutinin binding was totally suppressed due to deletion of GnT-V showed increased Pcdhβ4 and Pcdhβ7 staining compared with wild-type tumors, indicating increased protein expression of Pcdhβ4 and Pcdhβ7 in GnT-V KO tumors.
To test whether increased expression of Pcdhβ genes in GnT-V KO tumors was indeed caused by GnT-V expression differences, her-2 tumor cell lines, including GnT-V WT cells, KO cells, and rescued KO cells (KO cells transfected with GnT-V), were established from tumor tissues (9), and transcript levels of Pcdhβ2, Pcdhβ4, Pcdhβ18, and Pcdhβ19 were measured by qRT-PCR because these four genes showed significant -fold changes (-fold change ≥2.0, p < 0.05) in GnT-V KO tumors based on the microarray data. As show in Fig. 2A, increased expression of the Pcdhβ genes was observed in the cells derived from GnT-V-null tumors compared with WT tumor cells, consistent with the results obtained from microarray analyses of tumor tissues. After treatment with swainsonine, an inhibitor of N-linked β(1,6) branching, WT cells showed increased Pcdhβ gene expression (Fig. 2B); reintroduction of GnT-V cDNA into KO tumor cells significantly attenuated the expression of Pcdhβ genes in these cells (Fig. 2C), supporting the hypothesis that differential expression of Pcdhβ genes in GnT-V KO tumors was due to altered expression levels of GnT-V. A recent study has shown that Pcdh gene clusters, including the Pcdhβ family, are expressed at lower levels in human breast cancer tissues than in normal tissues, and these lower expression levels were linked to human breast tumorigenesis (26). To confirm and extend the observation concerning the differential expression of the Pcdhβ gene cluster in mouse mammary tumors after deletion of GnT-V, Pcdhβ gene expression was measured in the human breast cancer cell line MDA-MB231 after knockdown of GnT-V by siRNA. As shown in Fig. 2D, three members of the human PCDHβ cluster (PCDHβ3, PCDHβ6, and PCDHβ13), which were shown to be significantly down-regulated in human breast cancer tissues (26), were dramatically enhanced in MDA-MB231 cells with GnT-V knockdown, further confirming the ability of GnT-V expression levels to regulate the expression of the Pcdhβ cluster during breast tumorigenesis.
FIGURE 2.
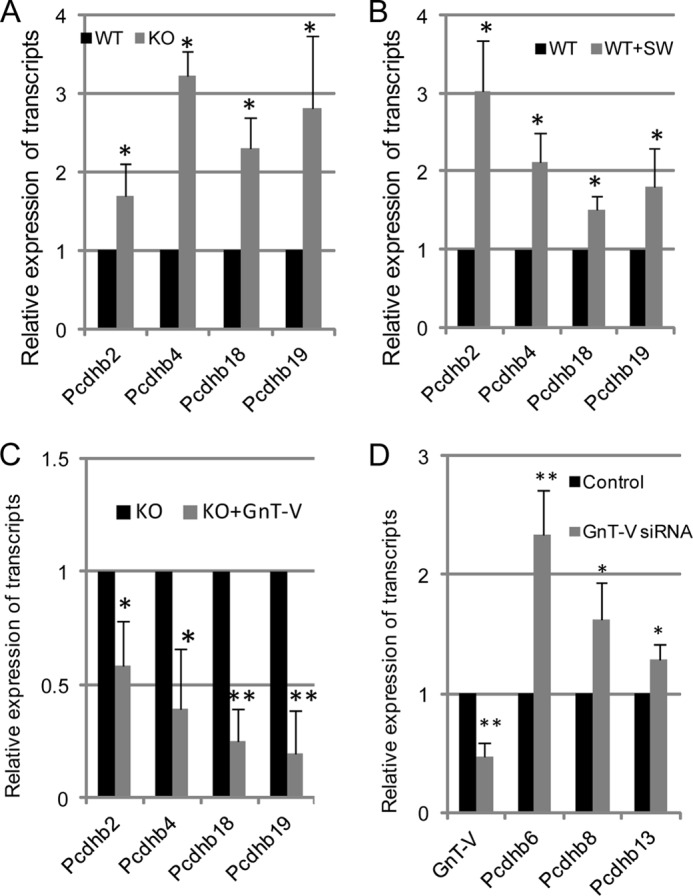
Increased transcript expression of the Pcdhβ gene cluster is related to absence of GnT-V activity. A, total RNA was isolated from wild-type and GnT-V knock-out tumor cells and used for detection of transcript levels of Pcdhβ genes. B, wild-type her-2 tumor cells were treated with swainsonine (SW; 1 μg/ml) for 24 h, and total RNA was isolated and used for detection of transcript levels of Pcdhβ by qRT-PCR. C, total RNA was isolated from GnT-V KO cells with GnT-V expression and used for detection of transcript levels of Pcdhβ. D, total RNA was isolated from control (scrambled siRNA)- and GnT-V siRNA-transfected MDA-MB231 cells and used for detection of transcript levels of GnT-V and Pcdhβ genes, respectively. For each transcript, the values are normalized to control (Gapdh or Rpl4) and expressed as means with error bars indicating ±1 S.D. of three independent experiments. *, Student's t test, p < 0.05; **, p < 0.01.
Differentially Expressed Pcdhβ Cluster in GnT-V KO Tumors Is Not Primarily Mediated by Altered DNA Methylation
Epigenetic aberrations are frequent events in human breast cancer development (26, 33), and promoter methylation has been implicated in the regulation of Pcdh clusters, including the Pcdhβ family (26). Also, an association between frequent CpG island methylation and her-2 expression status was observed in human breast cancer (33, 34). To determine whether altered expression of the Pcdhβ cluster in GnT-V KO tumors was caused by promoter methylation differences, the promoter methylation status of Pcdhβ genes was analyzed. We first searched the mouse Pcdhβ cluster for CpG islands around the transcription start site (TSS) (−1 to +1 kb) using CpGPlot. As shown in supplemental Fig. 3, few CpG islands around the TSS area were found in most of the Pcdhβ genes (Pcdhβ2, Pcdhβ4, Pcdhβ7, Pcdhβ18, and Pcdhβ19) that showed alterations in GnT-V-null tumors, consistent with reports that the mouse genome contains lower amounts of CpG islands than the human genome (22, 23). Interestingly, treatment of her-2 tumor cells with the demethylation drug 5-aza-2′-deoxycytidine increased expression of some Pcdhβ genes, such as Pcdhβ4 and Pcdhβ18 (Fig. 3A), indicating that these genes can still be regulated by methylation status regardless of the lower frequency of CpG islands. Because no CpG islands around the TSS area of Pcdhβ4 and Pcdhβ18 were found, it is likely that demethylation activated certain transcription activators of these genes, indirectly leading to increased gene expression. Methylation-specific PCR targeting the area surrounding the TSS was performed for the Pcdhβ genes that have predicted CpG island(s) around their TSS (Fig. 3B). Interestingly, Cdh1, whose gene expression is silenced due to promoter hypermethylation in many human tumors, including breast tumors (35), showed very low promoter methylation in the her-2-induced mouse mammary tumors. The same patterns of promoter methylation of Pcdhβ genes were observed in both wild-type and GnT-V KO cells, however, consistent with the unchanged expression of methylation-related genes in KO tumors observed in microarray analysis data (data not shown). These results indicated that knock-out of GnT-V had little effect on methylation levels of Pcdhβ genes. The altered gene expression of the Pcdhβ cluster following deletion of GnT-V probably does not primarily result from changes in methylation status.
FIGURE 3.
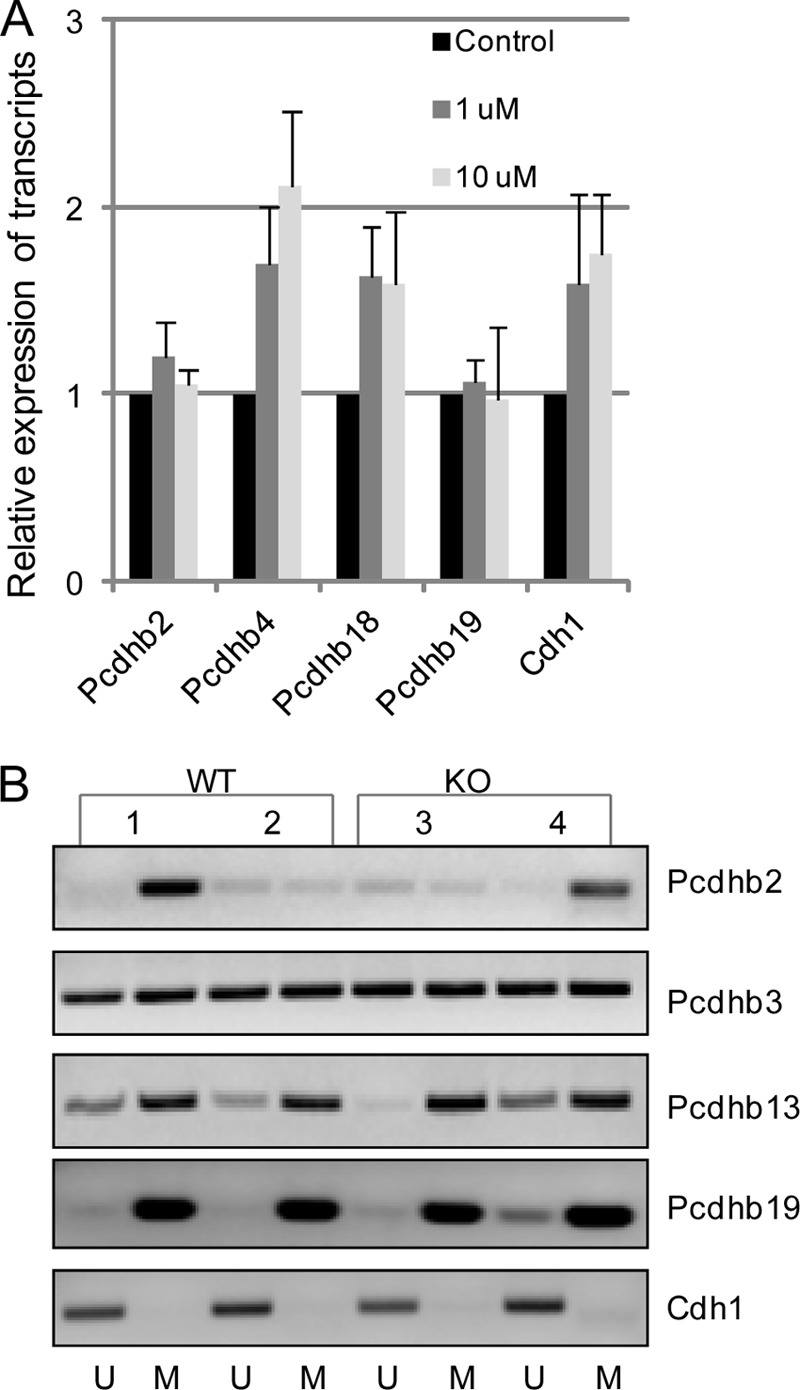
Increased transcript expression of the Pcdhβ gene cluster is not primarily mediated by altered promoter methylation. A, her-2 tumor cells were treated with the demethylating reagent 5-aza-2′-deoxycytidine at two different concentrations for 3 days, and total RNA was isolated and used for detection of transcript levels of Pcdhβ. For each transcript, the values are normalized to control (Gapdh or Rpl4) and expressed as means with error bars indicating ±1 S.D. of three independent experiments. B, after the genomic DNA samples were purified and bisulfate-treated, methylation-specific PCR for the Pcdhβ genes was performed in two wild-type (samples 1 and 2) and two GnT-V knock-out (samples 3 and 4) tumors. The presence of a PCR product band in lane M or U indicates methylated or unmethylated genes, respectively. Data are representative of two independent experiments.
Impaired Her-2-mediated Signaling Pathways Are Involved in Differentially Expressed Pcdhβ Cluster Transcripts in GnT-V KO Tumors
Deletion of GnT-V had no effect on her-2 expression levels but did cause inhibition of her-2-induced oncogenic signaling pathways (supplemental Fig. 4) (9). Therefore, the differentially expressed Pcdhβ cluster in GnT-V KO tumors may possibly result from attenuation of her-2-induced activation of either the PKB or ERK signaling pathways. To this end, inhibitors and activators of these two signaling pathways were used to study the effect of her-2 signaling on expression of the Pcdhβ cluster. As shown in Fig. 4A, treatment of WT tumor cells with PD98059 or wortmannin, inhibitors of MAPK and PI3K/PKB signaling pathways, respectively, led to increased gene expression of the Pcdhβ cluster in these cells. Stimulation of KO tumor cells with EGF and neuregulin, the ligand of her-1 (EGF receptor) and her-3, respectively, activated her-2 signaling pathways as evident by enhanced expression levels of phospho-ERK and phospho-PKB and attenuated gene expression of Pcdhβ (Fig. 4B). These results indicated that her-2-induced signaling pathways were implicated in GnT-V-mediated gene regulation of the Pcdhβ cluster. To further confirm that the MAPK and PI3K/PKB pathways involved in the regulation of gene expression of Pcdhβ were her-2 oncoprotein-related, knockdown of her-2 was performed using her-2/neu siRNA oligonucleotides. As shown in Fig. 4, C–E, a 60% reduction of her-2 in WT tumor cells treated with her-2 siRNA was accompanied by attenuated her-2-induced downstream signaling (phospho-ERK and phospho-PKB), significantly enhancing Pcdhβ gene expression. This result strongly supports the conclusion that gene expression of the Pcdhβ cluster is regulated by her-2-induced signaling pathways. Therefore, the deletion of GnT-V observed in the GnT-V-null tumors attenuated the her-2-mediated signaling pathways, resulting in increased gene expression of the Pcdhβ cluster.
FIGURE 4.

Increased transcript expression of the Pcdhβ gene cluster is caused by impaired her-2-mediated downstream signaling. A, wild-type tumor cells were grown for 2 days with or without MEK inhibitor PD98059 (PD; 10 mol/liter), PI3K inhibitor wortmannin (WM; 1 mol/liter), or both inhibitors. Cells were then collected, and total RNA was isolated and used for detection of transcript levels of Pcdhβ. B, GnT-V knock-out tumor cells were grown in serum-free medium for 2 days and stimulated with EGF (100 ng/ml), neuregulin (NRG) (50 ng/ml), or serum-containing medium for 2 days. Cells were collected for detection of phospho-PKB (p-PKB) and phospho-ERK (p-ERK) using immunoblot (left panel) and transcripts of the Pcdhβ cluster (right panel). After wild-type her-2 cells were treated with control (scrambled) and her-2/neu siRNA oligonucleotides (50 nm) for 48 h, respectively, cells were collected and subjected to detection of transcripts of her-2/neu using qRT-PCR (C); her-2/neu, phospho-PKB (p-PKB), and phospho-ERK (p-ERK) using immunoblot (D); and transcripts of the Pcdhβ gene cluster (E). For each transcript, the values are normalized to control (Gapdh or Rpl4) and expressed as means with error bars indicating ±1 S.D. of three independent experiments. *, Student's t test, p < 0.05; **, p < 0.01.
MiR-21 Is One of the Downstream Regulators of Her-2 Signaling Implicated in the Altered Gene Expression of Pcdhβ Cluster
Activation of her-2 signaling increases the expression of miR-21 (36, 37), which contributes to the increased invasion and metastatic potential of breast cancer cells (36). To further study whether miR-21 was involved in her-2-regulated gene expression of the Pcdhβ cluster, the expression of mature miR-21 and its effects on Pcdhβ gene expression were investigated. Consistent with inhibited her-2 signaling, approximately 75% down-regulation of miR-21 levels as detected by qRT-PCR was observed in GnT-V-null tumors compared with wild-type tumors (Fig. 5A). To verify that reduced miR-21 expression was GnT-V-related, we silenced GnT-V by siRNA expression in two human breast cancer lines, MDA-MB231 and SK-BR3, and measured miR-21 levels. We found that miR-21 expression was dramatically inhibited in both cell types after GnT-V knockdown as shown in Fig. 5B. These results demonstrated that miR-21 was regulated by expression levels of GnT-V in the breast cancer cells. To investigate the role of miR-21 in the altered gene expression of the Pcdhβ cluster, we transfected her-2 tumor cells (wild-type GnT-V) with a locked nucleic acid-modified anti-miR-21 siRNA oligonucleotide to inhibit miR-21 expression (Fig. 5C). Interestingly, these treatments caused to varying degrees increases in Pcdhβ cluster gene expression (Fig. 5D), indicating that the Pcdhβ cluster is the likely target of miR-21 because the expression of these genes was altered when miR-21 levels were reduced. Therefore, increased gene expression of the Pcdhβ cluster observed in GnT-V knock-out tumors most likely resulted at least to some extent from reduced expression of miR-21 caused by attenuated her-2 signaling. Based on computational target prediction by Targetscan, we investigated some of the predicted targets of miR-21 that either have been validated by experiments or showed significant changes by microarray analysis in our study (Table 3). From our microarray data, we identified several genes whose expression was altered, including Rasgrp1, Npas3, Reck, and Epha4, and changes in these genes were validated by qRT-PCR (data not shown). However, none of the Pcdhβ family members was among them, suggesting that the Pcdhβ gene cluster does not contain miR-21 binding sequences in their 3′-UTRs and that the altered gene expression of the Pcdhβ cluster in the GnT-V-null tumors is regulated indirectly by miR-21 expression. To further substantiate that altered expression of Rasgrp1, Npas3, Reck, and Epha4 resulted from reduced miR-21 expression, the expression of these predicted miR-21 targets were determined in tumor cells whose miR-21 expression was knocked down. As shown in Fig. 5E, the gene expression of Rasgrp1, Npas3, Reck, and Epha4 was all up-regulated after knockdown of miR-21, indicating that all genes, except for Rasgrp1, were indeed targets of miR-21 in the her-2 mammary tumors, which is likely responsible for the altered gene expression of the Pcdhβ cluster in the GnT-V-null tumors.
FIGURE 5.

MiR-21 is implicated in the regulation of her-2 signaling-mediated gene expression of Pcdhβ cluster. A, total RNA was pooled from four tumors of GnT-V wild-type and knock-out mice, and expression of mature miR-21 was detected by qRT-PCR. B, total RNA was isolated from MDA-MB231 and SK-BR3 tumor cells with and without GnT-V siRNA expression, and expression of GnT-V and mature miR-21 was detected by qRT-PCR. After wild-type her-2 tumor cells were treated with control and anti-miR-21 siRNA oligonucleotides (50 nm) for 48 h, respectively, cells were collected and subjected to detection for transcripts of miR-21 (C), the Pcdhβ cluster (D), and miR-21 target genes as indicated (E). For each transcript, the values are normalized to control (Gapdh or Rpl4) and expressed as means with error bars indicating ±1 S.D. of three independent experiments. *, Student's t test, p < 0.05; **, p < 0.01.
TABLE 3.
MiR-21 target genes based on Targetscan
| Gene symbol | p value | -Fold change (KO/WT) |
|---|---|---|
| Jag1 | 0.065 | −1.3 |
| Rasgrp1 | 0.02 | −2.3 |
| Sox2 | 0.032 | −1.37 |
| Npas3 | 0.002 | 1.87 |
| Btg2 | 0.032 | −1.29 |
| Reck | 0.087 | 1.45 |
| Sox7 | 0.0008 | −1.26 |
| Epha4 | 0.004 | 1.55 |
| Nfat5 | 0.025 | 1.31 |
| Pdcd4 | 0.25 | −1.14 |
| Tgfb1 | 0.14 | −1.46 |
| Nfib | 0.029 | 1.09 |
| Bcl2 | 0.71 | −1.02 |
| Pten | 0.69 | −1.01 |
| Spry2 | 0.9 | 1.01 |
| Tpm1 | 0.15 | 1.29 |
Tumor-suppressive Functions of Pcdhβ Cluster
Pcdh gene clusters have characteristics of suppressors of the transformed phenotype, and their epigenetic silencing has been implicated in some human cancer development, including breast cancer (26, 27). To determine whether the differential expression of the Pcdhβ cluster following deletion of GnT-V contributed to the delayed onset of her-2-mediated tumorigenesis, cell culture-based assays of colony formation and anchorage-independent cell growth commonly utilized for assessing tumor suppressor function were used to evaluate the ability of the Pcdhβ cluster to inhibit tumor progression (27). Based on the microarray data and commercial availability of clones (Source BioScience), Pcdhβ4 and Pcdhβ19 were chosen for these experiments. Overexpression of either Pcdhβ4 or Pcdhβ19 in both mouse her-2 tumor cells (wild-type) and human MDA-MB231 cells, which was confirmed by qRT-PCR and immunofluorescent staining (supplemental Fig. 5), significantly inhibited colony formation after 2 weeks compared with control cells (Fig. 6A). Similarly, anchorage-independent cell growth in soft agar was markedly reduced in both cell lines after transfection of Pcdhβ4 and Pcdhβ19 (Fig. 6B), indicating inhibition of cell proliferation in these cells due to the exogenous expression of Pcdhβ genes. To further confirm the ability of Pcdhβ genes to inhibit tumor progression in vivo, we next tested the ability of tumor cells overexpressing Pcdhβ to form tumors in NOD/SCID mice. Slower tumor growth was observed in xenografts resulting from injection of either her-2 tumor cells (Fig. 6, C and D) or MDA-MB231 cells (supplemental Fig. 6A), each of which was overexpressing either Pcdhβ4 or Pcdhβ19, compared with mock-transfected cells. These results indicated reduced tumorigenesis due to Pcdhβ expression. Hematoxylin/eosin staining of tumor sections showed a reduced number of mitotic cells and less tumor necrosis in tumors resulting from injection of Pcdhβ-expressing cells (supplemental Fig. 6, B and C). Consistent with inhibited tumorigenesis, reduced populations of tumor-initiating cells as detected by the ALDEFLUOR assay (9) were also observed in both Pcdhβ-expressing her-2 tumor cells (1.47 versus 4.04% in control) and their xenograft tumors (2.04 versus 11.6% in control) in NOD/SCID mice (Fig. 6E). Our results are supportive of reports that Pcdh clusters function to suppress tumor progression and suggest that the delayed her-2-induced mammary tumor onset observed after deletion of GnT-V (9) might be attributed at least in part to enhanced expression of Pcdhβ cluster transcripts.
FIGURE 6.

Tumor-suppressive effects of the Pcdhβ cluster. A, wild-type her-2 tumor cells and MDA-MB231 cells were transfected with either Pcdhβ4 or Pcdhβ19 cDNA and grown in selection medium for 2–3 weeks for colony formation. The number of colonies in six random fields was counted and expressed as the mean with error bars indicating ±1 S.D. * represents Student's t test, p < 0.001. B, transfected cells were grown in soft agar for 2–3 weeks, and the number of colonies in six random fields was counted and expressed as the mean with error bars indicating ±1 S.D. *, p < 0.05. C, Pcdhβ4-transfected wild-type her-2 tumor cells (2 × 106) were injected into mammary fat pads of SCID mice (n = 5), and secondary tumor growth was observed for up to 8 weeks. *, p < 0.05. D, secondary tumors in SCID mice formed by injection of Pcdhβ4-expressing cells were dissected at week 8 and photographed (left panel), and the weight of tumors was quantified and expressed as the mean with error bars indicating ±1 S.D. (n = 5; right panel). *, p < 0.05. E, ALDEFLUOR assays of Pcdhβ4-expressing her-2 tumor cells (left panel) and xenografts formed by injection of Pcdhβ4-expressing her-2 cells (right panel) were performed, and the percentage of ALDEFLUOR-positive cells (tumor initiating cells) was determined using similar gating criteria. Data are representative of two independent experiments. DEAB, diethylaminobenzaldehyde.
DISCUSSION
Aberrant N-glycosylation has been documented during tumorigenesis and tumor progression (38). We recently reported that a specific posttranslational modification by deletion of GnT-V disrupts mammary acinar formation and delays her-2-induced mouse mammary tumor onset by down-regulating the relative size of the compartment of tumor-initiating cells (9). In the present study, we show by microarray analysis that 142 differentially expressed genes were observed in GnT-V knock-out her-2-induced tumors compared with wild-type GnT-V her-2-induced tumors, including one group in the cadherin gene cluster, termed Pcdhβ, which was significantly up-regulated. Two members of this cluster were chosen and shown to suppress several characteristics of the transformed phenotype that could contribute at least in part to the reduced her-2-mediated mammary tumorigenesis observed in GnT-V-null mice.
Pcdh gene clusters consist of three closely linked family members designated Pcdhα, Pcdhβ, and Pcdhγ (22). The mouse Pcdhβ gene family contains 22 members (Pcdhβ1–22) that are organized in a tandem array on chromosome 18, and each member consists of a single exon that encodes the extracellular, transmembrane, and short cytoplasmic protein domains (22). Although Pcdh clusters play potential roles in neuron development (21), little is known about the cellular functions of these proteins. Unlike classic cadherins, Pcdhs appear not to function via the strong interaction of their extracellular domains as do the classic cadherins because their interactions appear to be much weaker (39, 40). Like the classic cadherins, Pcdhs are glycoproteins with N-linked glycan sequons located in their extracellular domains. Studies have shown that Pcdhs are proteolytically cleaved by γ-secretase complex, producing soluble intracellular fragments that may enter the nucleus and affect gene expression (41, 42). Of note, several studies have shown that the Pcdh superfamily may be implicated in tumor development and function as tumor suppressors. Dallosso et al. (27) reported that PCDH gene clusters, including PCDHα, PCDHβ, and PCDHγ, were epigenetically silenced in Wilms tumor, and ectopic expression of PCDHγ showed growth inhibition of tumor cells in vitro. Another study found that the expression of PCDH cluster transcripts was reduced due to promoter hypermethylation in human breast tumors compared with normal breast tissues and concluded that aberrant DNA hypermethylation of multiple PCDH CpG islands is a common event in human breast cancer (26). In our study, up-regulation of gene expression of the Pcdhβ cluster as detected by microarray analyses was observed in her-2-mediated breast tumors with GnT-V deletion compared with GnT-V wild-type tumors. These changes were further confirmed by qRT-PCR and proved to be GnT-V-related by both rescue experiments in which GnT-V cDNA was reintroduced into GnT-V knock-out tumor cells and inhibition experiments in which swainsonine was used to inhibit the biosynthesis of N-linked β(1,6) branching in GnT-V wild-type cells. Supporting these results, a human breast cancer cell line, MDA-MB231, also showed increased gene expression of some Pcdhβ family members after inhibition of GnT-V in these cells by GnT-V siRNA expression. These results indicate that expression levels of GnT-V regulate gene expression of the Pcdhβ cluster in mammary tumor tissues and cells, and the altered gene expression of Pcdhβ is implicated in the attenuation of her-2-induced tumorigenesis caused by deletion of GnT-V (9).
To further understand the role of altered gene expression of Pcdhβ cluster in the inhibition of her-2-induced tumor development that we observed previously, we subcloned two family members of the Pcdhβ cluster, Pcdhβ4 and Pcdhβ19, into an expression vector and stably transfected mouse and human breast cancer cells. Results obtained from these experiments strongly suggest that Pcdhβ genes display some features of genes that suppress or inhibit tumor progression. First, anchorage-independent cell growth in soft agar and colony formation ability after transfection, two commonly used methods to evaluate tumor suppressor function, were remarkably inhibited when Pcdhβ was expressed in both mouse and human breast cancer lines. Second, in vivo tumor growth in NOD/SCID mice that resulted from injection of tumor cells expressing Pcdhβ was significantly suppressed compared with that observed from injection of control mock-transfected cells. Third, a reduced proportion of tumor-initiating cells was observed in tumor cells with Pcdhβ expression, consistent with the inhibited tumor growth of these cells injected in NOD/SCID mice. Based on these observations, it is reasonable to conclude that Pcdhβ expression may function to inhibit some aspects of the transformed phenotype. The decreased tumorigenesis and the observed inhibition of her-2-induced breast tumor onset associated with GnT-V deletion (9), therefore, can be attributed at least in part to the increased gene expression of the Pcdhβ cluster.
Aberrant levels and patterns of DNA methylation have been found as ubiquitous events in many human cancers (43). Hypermethylation of promoter CpG islands, frequently observed in human breast cancer, is related to transcriptional silencing of some genes, including tumor suppressor genes (44). Although studies have shown that gene expression of PCDH clusters was epigenetically regulated through changes in promoter methylation patterns in some human cancers (26, 27, 33), the altered expression of the Pcdhβ cluster caused by GnT-V deletion in our study appeared not to result from aberrant DNA methylation of Pcdhβ genes. Despite the fact that fewer CpG islands are predicted in promoter areas of mouse Pcdh gene clusters compared with human PCDH clusters (22, 23), higher DNA methylation levels as detected by methylation-specific PCR were still observed in her-2 tumors or cells derived from these tumors after treatment with a methylation inhibitor, consistent with the previous observation that DNA methylation levels are increased in her-2-positive primary breast cancers (33). However, deletion of GnT-V had little effect on the DNA methylation status of Pcdhβ genes because the same patterns of methylation in GnT-V knock-out tumors were observed as those in wild-type tumors. These results indicated that DNA methylation of the Pcdhβ cluster was not regulated by GnT-V expression levels, and altered gene expression of the Pcdhβ cluster caused by null GnT-V was not likely due to the changes in DNA methylation. Supporting this conclusion, no significant changes in expression of DNA methylation-related enzymes, such as DNMT1, DNMT3A, and DNMT3B, were observed in the comparison of GnT-V knock-out and wild-type tumors by microarray analyses.
Although results from microarray analyses showed that deletion of GnT-V had little effect on expression of her-2 and other oncoproteins from the EGF family, such as her-3 and EGF receptor (her-1), consistent with our previous results (9), we found that two major downstream signaling pathways mediated by her-2 activation, the ERK and PI3K/PKB pathways, are significantly inhibited in GnT-V knock-out tumors (9). This result prompted us to investigate further whether aberrant her-2-mediated signaling pathways were implicated in altered gene expression of the Pcdhβ cluster observed in GnT-V knock-out tumors. We observed that stimulation of GnT-V knock-out cells with growth factors (EGF and neuregulin) significantly inhibited gene expression of the Pcdhβ cluster, whereas treatment of GnT-V wild-type tumor cells with PD98059 and wortmannin, inhibitors of ERK and PKB, respectively, remarkably enhanced gene expression of the Pcdhβ family, indicating the direct involvement of these two pathways in regulating Pcdhβ gene expression. The regulation of her-2-mediated signaling pathways on the gene expression of the Pcdhβ cluster was further confirmed by silencing her-2 oncoprotein by her-2 siRNA expression, demonstrating that altered gene expression of the Pcdhβ family closely followed changes in her-2 expression levels. These results strongly argue that her-2-mediated downstream signaling pathways negatively regulated the expression of the Pcdhβ cluster. Deletion of GnT-V impaired her-2 signaling pathways, therefore attenuating the inhibition of her-2 signaling on the gene expression of the Pcdhβ family. The involvement of other signaling pathways in regulating gene expression of the Pcdhβ family in GnT-V knock-out tumors cannot be ruled out, however, because of the ability of GnT-V glycan products to modify the function of other glycoprotein receptors.
To further investigate the mechanisms governing how impaired her-2-mediated downstream signaling positively regulates gene expression of the Pcdhβ cluster in GnT-V-null tumors, we explored the possible downstream effectors that are regulated by her-2-mediated signaling pathways that could be involved in the regulation of Pcdhβ cluster expression in GnT-V-null tumors. MicroRNAs constitute an abundant class of non-coding RNAs of about 21–23 nucleotides that negatively regulate protein expression by targeting mRNA transcripts and mediate either translational repression or degradation of targeted mRNA (45, 46). There is increasing evidence that microRNAs are key molecules involved in cancer initiation and progression (47, 48). Functioning as an oncogene (49), miR-21 is one of the most studied microRNAs associated with cancer and is highly up-regulated in breast cancer (50, 51). Studies have shown that her-2 signaling activates several transcription factors, such as Ets-1 and Ap-1, that stimulate the expression of miR-21 (36, 37), and increased metastatic potential of her-2-expressing breast cancer cells is mediated by up-regulation of miR-21 (36). Altered expression of miR-21 may, therefore, be one of the downstream regulators of her-2 signaling implicated in her-2-mediated up-regulation of the Pcdhβ cluster. Consistent with impaired her-2 signaling pathways, the expression of miR-21 was significantly reduced in GnT-V knock-out tumors. Mostly importantly, silencing of miR-21 using a locked nucleic acid-modified anti-miR-21 resulted in the enhancement to different degrees of the expression of the Pcdhβ gene cluster, indicating the involvement of miR-21 in the altered gene expression of Pcdhβ caused by inhibited her-2 signaling at least in part. Interestingly, the effect of regulation of miR-21 on Pcdhβ expression appeared to be indirect because Pcdhβ genes are not among the predicted potential targets of miR-21 due to the lack of binding sequences for miR-21 in their 3′-UTRs. Although some of the miR-21 target genes that have been experimentally confirmed were not affected by reduced miR-21 expression in our study, including Pdcd4, Pten, Tpm1, and Spry2 (49), effects on the expression of other target genes, for example Npas3, a newly confirmed tumor suppressor gene (52); Reck, a membrane-associated inhibitor of metalloproteinases and recently confirmed miR-21 target gene (53); and Epha4, a member of the Eph receptor tyrosine kinases (54), were observed. These targets were further validated by miR-21 silencing experiments (Fig. 5E) and likely mediate miR-21 regulation of the expression of the Pcdhβ gene cluster. Indirect regulation of genes by miR-21 has been documented; for example, an array expression analysis of MCF-7 cells depleted of miR-21 by siRNA identified miR-21 target genes that were subsequently experimentally validated, including targets that did not contain miR-21 binding sequences in their 3′-UTRs (55). Also, miR-21 was found to indirectly inhibit the expression of DNMT1 by targeting Rasgrp1, a critical upstream regulator of the Ras-MAPK signaling cascade, which modulates DNMT1 levels in lupus CD4+ T cells (56). Another indirect regulation of Bcl-2 by miR-21 has also been reported in breast cancer (57).
The attenuation of her-2-mediated signaling pathways observed in GnT-V KO tumor cells is likely to be the result of aberrant N-glycosylation of her-2 and/or the ErbB family of receptors that can lead to altered ligand (EGF and neuregulin) binding, regulation of the endocytosis of signaling complexes, and/or inhibition of dimer and multimer formation among members of this family (13, 58, 59). Experiments are in progress to determine the mechanisms by which β(1,6) N-linked glycosylation regulates her-2 function.
In conclusion, we provide evidence that a gene cluster in the cadherin superfamily, Pcdhβ, functions to inhibit her-2-mediated mouse mammary tumorigenicity. Our results show that GnT-V expression levels regulate the expression of members of the Pcdhβ cluster by affecting her-2-mediated downstream signaling pathways. This increased Pcdhβ expression is mediated at least in part by miR-21, one of the downstream effectors regulated by her-2 signaling (Fig. 7). Because up to 30% of human breast tumors are her-2-positive (15, 16), our findings shed new light on the molecular mechanisms of the suppressive effects of GnT-V deletion on mammary tumorigenesis and progression and illuminate GnT-V as a potential target for an inhibitor with therapeutic utility for her-2-positive human breast cancer.
FIGURE 7.

Schematic indicating how the expression of the Pcdhβ gene cluster is regulated by GnT-V. Pathways implicated in the regulation of the Pcdhβ gene cluster are depicted based on the observations presented in this study. GnT-V expression levels regulate N-glycosylation of her-2 and her-2-induced signaling pathways. Knock-out of GnT-V results in inhibited expression of N-linked β(1,6) branching on her-2 and impaired her-2-induced signaling pathways, which leads to up-regulation of the Pcdhβ gene cluster. The increased Pcdhβ expression is mediated indirectly at least in part by miR-21, one of the downstream effectors regulated by her-2 signaling, and contributes to the inhibition of her-2-induced tumor onset.
Supplementary Material
Acknowledgments
We thank the Cancer Research Center-Integrated Genomics Core at the Georgia Health Sciences University, Nikki Harvel, and Dr. Lesleyann Hawthorn for kind help with microarray analysis and Drs. Jin-kyu Lee, Karen Abbott, Hong Qiu, and Bing Zhang for informative discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants U01CA128454 (to M. P.) and P41RR018502 (to M. P. and K. M.).

This article contains supplemental Figs. 1–6 and Tables S1–S6.
- GnT-V
- N-acetylglucosaminyltransferase V
- NOD/SCID
- non-obese diabetes/severe combined immunodeficiency
- Pcdh
- protocadherin
- qRT-PCR
- quantitative RT-PCR
- miR
- microRNA
- phospho
- phosphorylated
- TSS
- transcription start site.
REFERENCES
- 1. Boscher C., Dennis J. W., Nabi I. R. (2011) Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 23, 383–392 [DOI] [PubMed] [Google Scholar]
- 2. Hakomori S. (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U.S.A. 99, 10231–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao Y. Y., Takahashi M., Gu J. G., Miyoshi E., Matsumoto A., Kitazume S., Taniguchi N. (2008) Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demetriou M., Nabi I. R., Coppolino M., Dedhar S., Dennis J. W. (1995) Reduced contact-inhibition and substratum adhesion in epithelial cells expressing GlcNAc-transferase V. J. Cell Biol. 130, 383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Granovsky M., Fata J., Pawling J., Muller W. J., Khokha R., Dennis J. W. (2000) Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat. Med. 6, 306–312 [DOI] [PubMed] [Google Scholar]
- 6. Guo H. B., Lee I., Kamar M., Akiyama S. K., Pierce M. (2002) Aberrant N-glycosylation of β1 integrin causes reduced α5β1 integrin clustering and stimulates cell migration. Cancer Res. 62, 6837–6845 [PubMed] [Google Scholar]
- 7. Handerson T., Camp R., Harigopal M., Rimm D., Pawelek J. (2005) β1,6-Branched oligosaccharides are increased in lymph node metastases and predict poor outcome in breast carcinoma. Clin. Cancer Res. 11, 2969–2973 [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto H., Swoger J., Greene S., Saito T., Hurh J., Sweeley C., Leestma J., Mkrdichian E., Cerullo L., Nishikawa A., Ihara Y., Taniguchi N., Moskal J. R. (2000) β1,6-N-Acetylglucosamine-bearing N-glycans in human gliomas: implications for a role in regulating invasivity. Cancer Res. 60, 134–142 [PubMed] [Google Scholar]
- 9. Guo H. B., Johnson H., Randolph M., Nagy T., Blalock R., Pierce M. (2010) Specific posttranslational modification regulates early events in mammary carcinoma formation. Proc. Natl. Acad. Sci. U.S.A. 107, 21116–21121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seelentag W. K., Li W. P., Schmitz S. F., Metzger U., Aeberhard P., Heitz P. U., Roth J. (1998) Prognostic value of β1,6-branched oligosaccharides in human colorectal carcinoma. Cancer Res. 58, 5559–5564 [PubMed] [Google Scholar]
- 11. Guo H. B., Johnson H., Randolph M., Pierce M. (2009) Regulation of homotypic cell-cell adhesion by branched N-glycosylation of N-cadherin extracellular EC2 and EC3 domains. J. Biol. Chem. 284, 34986–34997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinho S. S., Reis C. A., Paredes J., Magalhães A. M., Ferreira A. C., Figueiredo J., Xiaogang W., Carneiro F., Gärtner F., Seruca R. (2009) The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum. Mol. Genet. 18, 2599–2608 [DOI] [PubMed] [Google Scholar]
- 13. Partridge E. A., Le Roy C., Di Guglielmo G. M., Pawling J., Cheung P., Granovsky M., Nabi I. R., Wrana J. L., Dennis J. W. (2004) Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 306, 120–124 [DOI] [PubMed] [Google Scholar]
- 14. Guo H. B., Randolph M., Pierce M. (2007) Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J. Biol. Chem. 282, 22150–22162 [DOI] [PubMed] [Google Scholar]
- 15. Carlsson J., Nordgren H., Sjöström J., Wester K., Villman K., Bengtsson N. O., Ostenstad B., Lundqvist H., Blomqvist C. (2004) HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br. J. Cancer 90, 2344–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Slamon D. J., Clark G. M., Wong S. G., Levin W. J., Ullrich A., McGuire W. L. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 [DOI] [PubMed] [Google Scholar]
- 17. Muller W. J., Sinn E., Pattengale P. K., Wallace R., Leder P. (1988) Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54, 105–115 [DOI] [PubMed] [Google Scholar]
- 18. Guy C. T., Webster M. A., Schaller M., Parsons T. J., Cardiff R. D., Muller W. J. (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. U.S.A. 89, 10578–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeichi M. (1995) Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7, 619–627 [DOI] [PubMed] [Google Scholar]
- 20. Yagi T., Takeichi M. (2000) Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 14, 1169–1180 [PubMed] [Google Scholar]
- 21. Morishita H., Yagi T. (2007) Protocadherin family: diversity, structure, and function. Curr. Opin. Cell Biol. 19, 584–592 [DOI] [PubMed] [Google Scholar]
- 22. Wu Q., Maniatis T. (1999) A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790 [DOI] [PubMed] [Google Scholar]
- 23. Wu Q., Zhang T., Cheng J. F., Kim Y., Grimwood J., Schmutz J., Dickson M., Noonan J. P., Zhang M. Q., Myers R. M., Maniatis T. (2001) Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res. 11, 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu Q., Maniatis T. (2000) Large exons encoding multiple ectodomains are a characteristic feature of protocadherin genes. Proc. Natl. Acad. Sci. U.S.A. 97, 3124–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu J. S., Koujak S., Nagase S., Li C. M., Su T., Wang X., Keniry M., Memeo L., Rojtman A., Mansukhani M., Hibshoosh H., Tycko B., Parsons R. (2008) PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene 27, 4657–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Novak P., Jensen T., Oshiro M. M., Watts G. S., Kim C. J., Futscher B. W. (2008) Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 68, 8616–8625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dallosso A. R., Hancock A. L., Szemes M., Moorwood K., Chilukamarri L., Tsai H. H., Sarkar A., Barasch J., Vuononvirta R., Jones C., Pritchard-Jones K., Royer-Pokora B., Lee S. B., Owen C., Malik S., Feng Y., Frank M., Ward A., Brown K. W., Malik K. (2009) Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms' tumor. PLoS Genet. 5, e1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu J., Cheng Y. Y., Tao Q., Cheung K. F., Lam C. N., Geng H., Tian L. W., Wong Y. P., Tong J. H., Ying J. M., Jin H., To K. F., Chan F. K., Sung J. J. (2009) Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology 136, 640–651.e1 [DOI] [PubMed] [Google Scholar]
- 29. Nairn A. V., Kinoshita-Toyoda A., Toyoda H., Xie J., Harris K., Dalton S., Kulik M., Pierce J. M., Toida T., Moremen K. W., Linhardt R. J. (2007) Glycomics of proteoglycan biosynthesis in murine embryonic stem cell differentiation. J. Proteome Res. 6, 4374–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharbati-Tehrani S., Kutz-Lohroff B., Bergbauer R., Scholven J., Einspanier R. (2008) miR-Q: a novel quantitative RT-PCR approach for the expression profiling of small RNA molecules such as miRNAs in a complex sample. BMC Mol. Biol. 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gürlevik E., Woller N., Schache P., Malek N. P., Wirth T. C., Zender L., Manns M. P., Kubicka S., Kühnel F. (2009) p53-dependent antiviral RNA-interference facilitates tumor-selective viral replication. Nucleic Acids Res. 37, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ginestier C., Hur M. H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C. G., Liu S., Schott A., Hayes D., Birnbaum D., Wicha M. S., Dontu G. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fiegl H., Millinger S., Goebel G., Müller-Holzner E., Marth C., Laird P. W., Widschwendter M. (2006) Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 66, 29–33 [DOI] [PubMed] [Google Scholar]
- 34. Terada K., Okochi-Takada E., Akashi-Tanaka S., Miyamoto K., Taniyama K., Tsuda H., Asada K., Kaminishi M., Ushijima T. (2009) Association between frequent CpG island methylation and HER2 amplification in human breast cancers. Carcinogenesis 30, 466–471 [DOI] [PubMed] [Google Scholar]
- 35. Hoque M. O., Prencipe M., Poeta M. L., Barbano R., Valori V. M., Copetti M., Gallo A. P., Brait M., Maiello E., Apicella A., Rossiello R., Zito F., Stefania T., Paradiso A., Carella M., Dallapiccola B., Murgo R., Carosi I., Bisceglia M., Fazio V. M., Sidransky D., Parrella P. (2009) Changes in CpG islands promoter methylation patterns during ductal breast carcinoma progression. Cancer Epidemiol. Biomarkers Prev. 18, 2694–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang T. H., Wu F., Loeb G. B., Hsu R., Heidersbach A., Brincat A., Horiuchi D., Lebbink R. J., Mo Y. Y., Goga A., McManus M. T. (2009) Up-regulation of miR-21 by HER2/neu signaling promotes cell invasion. J. Biol. Chem. 284, 18515–18524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujita S., Ito T., Mizutani T., Minoguchi S., Yamamichi N., Sakurai K., Iba H. (2008) miR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 378, 492–504 [DOI] [PubMed] [Google Scholar]
- 38. Pierce J. M. (2009) in Handbook of Glycomics (Cummings R. D., Pierce J. M., eds) 1st Ed., pp. 399–429, Elsevier, London [Google Scholar]
- 39. Obata S., Sago H., Mori N., Rochelle J. M., Seldin M. F., Davidson M., St John T., Taketani S., Suzuki S. T. (1995) Protocadherin Pcdh2 shows properties similar to, but distinct from, those of classical cadherins. J. Cell Sci. 108, 3765–3773 [DOI] [PubMed] [Google Scholar]
- 40. Morishita H., Umitsu M., Murata Y., Shibata N., Udaka K., Higuchi Y., Akutsu H., Yamaguchi T., Yagi T., Ikegami T. (2006) Structure of the cadherin-related neuronal receptor/protocadherin-α first extracellular cadherin domain reveals diversity across cadherin families. J. Biol. Chem. 281, 33650–33663 [DOI] [PubMed] [Google Scholar]
- 41. Hambsch B., Grinevich V., Seeburg P. H., Schwarz M. K. (2005) γ-Protocadherins, presenilin-mediated release of C-terminal fragment promotes locus expression. J. Biol. Chem. 280, 15888–15897 [DOI] [PubMed] [Google Scholar]
- 42. Buchanan S. M., Schalm S. S., Maniatis T. (2010) Proteolytic processing of protocadherin proteins requires endocytosis. Proc. Natl. Acad. Sci. U.S.A. 107, 17774–17779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones P. A., Baylin S. B. (2007) The epigenomics of cancer. Cell 128, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Widschwendter M., Jones P. A. (2002) DNA methylation and breast carcinogenesis. Oncogene 21, 5462–5482 [DOI] [PubMed] [Google Scholar]
- 45. Wu L., Fan J., Belasco J. G. (2006) MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. U.S.A. 103, 4034–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eulalio A., Huntzinger E., Izaurralde E. (2008) Getting to the root of miRNA-mediated gene silencing. Cell 132, 9–14 [DOI] [PubMed] [Google Scholar]
- 47. Lu J., Getz G., Miska E. A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B. L., Mak R. H., Ferrando A. A., Downing J. R., Jacks T., Horvitz H. R., Golub T. R. (2005) MicroRNA expression profiles classify human cancers. Nature 435, 834–838 [DOI] [PubMed] [Google Scholar]
- 48. Calin G. A., Croce C. M. (2006) MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866 [DOI] [PubMed] [Google Scholar]
- 49. Selcuklu S. D., Donoghue M. T., Spillane C. (2009) miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 37, 918–925 [DOI] [PubMed] [Google Scholar]
- 50. Volinia S., Calin G. A., Liu C. G., Ambs S., Cimmino A., Petrocca F., Visone R., Iorio M., Roldo C., Ferracin M., Prueitt R. L., Yanaihara N., Lanza G., Scarpa A., Vecchione A., Negrini M., Harris C. C., Croce C. M. (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Si M. L., Zhu S., Wu H., Lu Z., Wu F., Mo Y. Y. (2007) miR-21-mediated tumor growth. Oncogene 26, 2799–2803 [DOI] [PubMed] [Google Scholar]
- 52. Moreira F., Kiehl T. R., So K., Ajeawung N. F., Honculada C., Gould P., Pieper R. O., Kamnasaran D. (2011) NPAS3 demonstrates features of a tumor suppressive role in driving the progression of astrocytomas. Am. J. Pathol. 179, 462–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayashi T., Koyama N., Azuma Y., Kashimata M. (2011) Mesenchymal miR-21 regulates branching morphogenesis in murine submandibular gland in vitro. Dev. Biol. 352, 299–307 [DOI] [PubMed] [Google Scholar]
- 54. Pasquale E. B. (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frankel L. B., Christoffersen N. R., Jacobsen A., Lindow M., Krogh A., Lund A. H. (2008) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 56. Pan W., Zhu S., Yuan M., Cui H., Wang L., Luo X., Li J., Zhou H., Tang Y., Shen N. (2010) MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 184, 6773–6781 [DOI] [PubMed] [Google Scholar]
- 57. Adams B. D., Furneaux H., White B. A. (2007) The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-α (ERα) and represses ERα messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 21, 1132–1147 [DOI] [PubMed] [Google Scholar]
- 58. Yokoe S., Takahashi M., Asahi M., Lee S. H., Li W., Osumi D., Miyoshi E., Taniguchi N. (2007) The Asn418-linked N-glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res. 67, 1935–1942 [DOI] [PubMed] [Google Scholar]
- 59. Guo H. B., Johnson H., Randolph M., Lee I., Pierce M. (2009) Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 19, 547–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.