

Background: Maintenance of cardiolipin molecular speciation by remodeling directly regulates mitochondrial bioenergetic efficiency.
Results: Transgenic expression of cardiolipin synthase accelerates cardiolipin remodeling, improves mitochondrial function, modulates mitochondrial signaling, and attenuates mitochondrial dysfunction during diabetes.
Conclusion: Cardiolipin synthase integrates multiple aspects of mitochondrial bioenergetic and signaling functions.
Significance: Cardiolipin synthase expression attenuates mitochondrial dysfunction in diabetic myocardium.
Keywords: Cardiolipin, Cardiomyopathy, Diabetic Cardiomyopathy, Lipids, Metabolism, Mitochondria, Lipidomics, Mitochondrial Dysfunction
Abstract
Lipidomic regulation of mitochondrial cardiolipin content and molecular species composition is a prominent regulator of bioenergetic efficiency. However, the mechanisms controlling cardiolipin metabolism during health or disease progression have remained elusive. Herein, we demonstrate that cardiac myocyte-specific transgenic expression of cardiolipin synthase results in accelerated cardiolipin lipidomic flux that impacts multiple aspects of mitochondrial bioenergetics and signaling. During the postnatal period, cardiolipin synthase transgene expression results in marked changes in the temporal maturation of cardiolipin molecular species during development. In adult myocardium, cardiolipin synthase transgene expression leads to a marked increase in symmetric tetra-18:2 molecular species without a change in total cardiolipin content. Mechanistic analysis demonstrated that these alterations result from increased cardiolipin remodeling by sequential phospholipase and transacylase/acyltransferase activities in conjunction with a decrease in phosphatidylglycerol content. Moreover, cardiolipin synthase transgene expression results in alterations in signaling metabolites, including a marked increase in the cardioprotective eicosanoid 14,15-epoxyeicosatrienoic acid. Examination of mitochondrial bioenergetic function by high resolution respirometry demonstrated that cardiolipin synthase transgene expression resulted in improved mitochondrial bioenergetic efficiency as evidenced by enhanced electron transport chain coupling using multiple substrates as well as by salutary changes in Complex III and IV activities. Furthermore, transgenic expression of cardiolipin synthase attenuated maladaptive cardiolipin remodeling and bioenergetic inefficiency in myocardium rendered diabetic by streptozotocin treatment. Collectively, these results demonstrate the unanticipated role of cardiolipin synthase in maintaining physiologic membrane structure and function even under metabolic stress, thereby identifying cardiolipin synthase as a novel therapeutic target to attenuate mitochondrial dysfunction in diabetic myocardium.
Introduction
Regulation of cardiac mitochondrial bioenergetic efficiency is a critical determinant of contractile function during hemodynamic stress (1). The principal determinant of myocardial bioenergetic efficiency is the functional capacity of cardiac mitochondria to mediate ATP generation, substrate utilization, calcium sequestration, apoptosis, and the cellular redox state (2). Pathologic alterations in mitochondrial function, such as those that occur in diabetic myocardium, result in inefficient utilization of substrate, promoting the progression of diastolic dysfunction and heart failure (3, 4). Thus, the identification of key regulatory mechanisms underlying cardiac bioenergetic dysfunction in the diabetic state is of fundamental importance to the development of novel pharmacologic targets for the treatment of diabetic cardiomyopathy.
Cardiolipin (CL)4 is a mitochondrial specific double negatively charged phospholipid that is intricately involved in regulating diverse mitochondrial activities, such as those of Complexes I, III, IV, and V of the electron transport chain (ETC) as well as state 3 respiration and the magnitude of uncoupling, which collectively determine myocardial bioenergetic efficiency (5). In addition, CL modulates adenine nucleotide translocase activity (6, 7), supercomplex formation (8, 9), acyl carnitine carrier activity (10), mitochondrial fission and fusion (11), phosphate carrier and pyruvate transporter activities (12), α-ketoglutarate dehydrogenase activities (13), and apoptosis (14–16). Understanding the chemical mechanisms underlying the multiple diverse functions of CL in mitochondrial bioenergetics and signaling will ultimately require the integration of genetic, lipidomic, and pharmacologic approaches to determine the interwoven relationships between CL content and molecular species composition with the pleiotropic roles of mitochondria in health and disease.
The coordinated regulation of CL content and composition is achieved through integrating CL de novo synthesis, remodeling, and catabolism (17–19). In eukaryotes, CL is synthesized by cardiolipin synthase (CLS) that catalyzes the condensation of phosphatidylglycerol and CDP-diacylglycerol in the mitochondrion (20, 21). Cardiolipin de novo synthesis results in the formation of nascent CL molecular species, which are largely comprised of 16:0- and 18:1-enriched CL molecular species. Nascent CL molecular species are subsequently remodeled by the sequential actions of phospholipase to form monolysocardiolipin followed by reacylation to form mature CL molecular species by remodeling enzymes, such as tafazzin, ALCAT1, or MLCAT (22–24). Although the molecular species compositions of CL vary between tissues, the predominant mature form of CL in myocardium is tetralinoleic (18:2) CL, which has been implicated in maximizing bioenergetic efficiency (25–28). Degradation of CL occurs through phospholipase A2-mediated hydrolysis to form monolysocardiolipin and dilysocardiolipin and through mitochondrial phospholipase D activation to form phosphatidic acid (5, 29). These processes are severely dysregulated in ischemia, Barth syndrome, cancer, hypothyroidism, diabetes, aging, and heart failure, leading to bioenergetic inefficiency, structural defects in mitochondria, and alterations in mitochondrial signaling (5, 24, 30–33). Thus, defining the key anabolic and catabolic regulatory steps in CL homeostasis that precipitate mitochondrial dysfunction during disease processes is of paramount importance.
Herein, we describe the generation of a transgenic cardiac myocyte-specific CLS mouse, which has enabled the identification of the chemical mechanisms mediating the regulation of CL metabolism that are dramatically altered in multiple disease states. Unexpectedly, the CLS transgenic mouse contained normal amounts of total CL but demonstrated increased cardiac CL remodeling and lipidomic flux that resulted in marked increases in tetralinoleic CL molecular species in conjunction with normal total CL content. These findings directly identify the molecular mechanisms integrating de novo synthesis, remodeling, and catabolism of CL in mitochondria to maintain optimal membrane surface charge, curvature, and molecular dynamics for integration of multiple mitochondrial functions. This novel discovery of CLS-mediated regulatory control of CL molecular species synthesis and flux was accompanied by mitochondrial functional alterations, including increased bioenergetic efficiency that was achieved through the coordinated regulation of multiple mitochondrial enzymatic activities. Remarkably, cardiac myocyte-specific transgenic expression of CLS attenuated streptozotocin-induced diabetic alterations in CL content and molecular species distribution and prevented mitochondrial bioenergetic dysfunction in diabetic myocardium. Thus, CLS represents a novel therapeutic target for attenuating myocardial mitochondrial bioenergetic dysfunction and the pathologic accumulation of lipids in diabetic myocardium.
EXPERIMENTAL PROCEDURES
Materials
Synthetic phospholipids used as internal standards in mass spectrometric analyses were purchased from Avanti Polar Lipids (Alabaster, AL). Western blot analyses of isolated mitochondria were performed as described previously (34). Polyclonal antibodies for Western blots were obtained from Protein Tech human CLS (14845-1), Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) (CDS2 (P-14) and tafazzin (JK-2)), Aviva Systems Biology (PGS1-ARP48895), MLCL AT (a generous gift from Dr. Grant Hatch, University of Manitoba), and Sigma-Aldrich (VDAC-V2139). Solvents for sample preparation and mass spectrometric analysis were purchased from Burdick and Jackson (Muskegon, MI) as well as Sigma-Aldrich.
Generation of Transgenic Mice Selectively Expressing Human CLS 1 (hCLS1) in Cardiomyocytes
Mice transgenically expressing hCLS1 were generated by exploiting the cardiac myocyte specificity of the α-myosin heavy chain (α-MHC) promoter. Briefly, we engineered SalI and HindIII sites at the 5′ and 3′ ends of the full-length 0.9-kb coding sequence of hCLS1. The SalI + HindIII-digested fragment was cloned into a SalI + HindIII-digested and alkaline phosphatase-treated α-MHC vector and sequenced in both directions. A HindIII fragment containing the α-MHC promoter in tandem with the hCLS1 sequence was utilized for microinjection of DNA directly into the pronuclei of mouse (B6CBAF1/J) zygotes, which resulted in integration of the transgene into the mouse germ line. Founder mice were identified by PCR analysis of mouse tail DNA and then bred with WT C57BL/6J mice (The Jackson Laboratories, Bar Harbor, ME) for at least seven generations to establish the transgenic line. The degree of hCLS1 expression was confirmed by quantitative PCR, microarray analysis, and its efficient translation and translocation into isolated mitochondria that was confirmed by Western blotting utilizing an antibody directed against hCLS1.
Induction of Diabetes by Streptozotocin Injection
A type I diabetic state was induced in male wild type (C57BL/6J) and CLS mice 4 months of age by a single intraperitoneal injection of streptozotocin (160 mg/kg body weight in 0.1 ml of 0.1 m citrate buffer, pH 4.5) as described previously (30, 35). Sham control mice received citrate buffer (0.1 ml) alone. Diabetes was confirmed within 48 h by glucose levels >300 mg/dl as measured by Chemstrip bG (Roche Applied Science). Mice were euthanized 45 days after diabetic induction, and cardiac tissue was collected. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Animal Studies Committee at Washington University School of Medicine.
Mitochondrial High Resolution Respirometry and ATP Synthesis Analysis
Mice used for experiments were sacrificed, and the hearts were immediately removed and dissected on ice (4 °C ambient temperature). Briefly, the dissected heart was placed in mitochondrial isolation buffer (0.21 m mannitol, 70 mm sucrose, 0.1 mm potassium-EDTA, 1 mm EGTA, 10 mm Tris-HCl, 0.5% BSA, pH 7.4) and homogenized using 12–15 passes with a Teflon homogenizer using a rotation speed of 120 revolutions/min. Next, the homogenate was centrifuged for 5 min at 850 × g, and the supernatant was collected and centrifuged for 7,200 × g for 10 min. The pellet was collected and resuspended in mitochondrial isolation buffer without BSA. Mitochondrial protein content was determined using a BCA protein assay (Thermo Fisher Scientific, San Jose, CA).
High resolution respirometry was performed using 50 μg of mitochondrial protein per 2-ml chamber with the substrate and inhibitor addition protocol described previously (36, 37). Additionally, for each assay, a 10-μl aliquot was collected from the respirometry chamber during state 3 respiration, saturated with DMSO, and stored at −80 °C for subsequent determination of ATP synthesis rate relative to ATP standards using the ENLITEN® detection system (Promega, Madison, WI) according to the manufacturer's instructions.
Multidimensional Mass Spectrometry-based Shotgun Lipidomic Analysis of the Cardiac Lipidome
Lipidomic analyses were performed as described previously (25, 36, 38). Individual lipid extracts were reconstituted with 1:1 (v/v) CHCl3/CH3OH, flushed with nitrogen, and stored at −20 °C prior to electrospray ionization-MS using a TSQ Quantum Ultra Plus triple-quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an automated nanospray apparatus (Advion Biosciences Ltd., Ithaca, NY) and customized sequence subroutine operated under Xcalibur software. Enhanced MDMS-SL analysis of cardiolipins were performed with a mass resolution setting of 0.3 Thomson as described previously in detail (39).
Measurement of Cardiolipin Synthase Activity
Cardiolipin synthase activity was measured as previously described in detail (40, 41) with minor modifications. Specifically, excised ventricular tissue was washed extensively with 5 mm Tris-HCl, pH 7.4, containing 0.25 m sucrose and 2 mm EDTA to remove blood prior to mincing the tissue in the same buffer (1.5 ml/heart). Next, the minced myocardial tissue was homogenized using 20 passes (rotation speed ∼150 revolutions/min) with a Teflon homogenizer and centrifuged at 500 × g to remove nuclei and myofibrils. The resultant supernatant was centrifuged at 12,000 × g to pellet mitochondria, which were resuspended at ∼5–10 mg protein/ml in the above buffer and briefly sonicated (five 1-s pulses). Mitochondrial homogenates (100 μg of protein/reaction) were added to buffer (35 mm Tris-HCl, pH 8.5, containing 0.25 mm EGTA) containing 0.6 mm 1,2-dihexadecanoyl-sn-glycero-3-(cytidine diphosphate) (CDP-diacylglycerol(di-16:0); Avanti Polar Lipids) and 100 μm 1-palmitoyl-2-stearoyl-[9,10-3H]phosphatidylglycerol (33 μCi/μmol; American Radiolabeled Chemicals) on ice and briefly sonicated (ten 1-s pulses). Reactions (100 μl) were initiated by the addition of 10 mm MgCl2 and incubated for 5–10 min at 37 °C. After termination of each reaction by the addition of 0.9 ml of 1% glacial acetic acid, 1 ml of methanol, 1 ml of chloroform followed by vigorous vortexing, lipid products extracted into the chloroform layer were dried under N2, dissolved in 75 μl of chloroform/methanol (2:1) containing 50 μg of cardiolipin standard, and resolved by thin layer chromatography (5/2.5/1/1.5 chloroform/acetone/methanol/acetic acid) using Whatman Partisil LK6D silica gel 60-Å plates. Lipids were visualized by iodine staining, the region corresponding to CL was scraped, and the amount of incorporated 3H was quantified by liquid scintillation counting.
Measurement of Cardiolipin Remodeling with 18:2 CoA
Mitochondria (1 mg of protein) were incubated in MiRO5 respiration buffer (with 20 μm palmitoyl-l-carnitine, 5 mm malate, and 1.25 mm ADP) with EGTA or with 0.5 mm CaCl2 in the presence of [1-14C]linoleoyl-CoA (55 mCi/mmol; 12 μm initial concentration; American Radiolabeled Chemicals, St. Louis, MO) to measure 18:2 specific CL remodeling. Aliquots of each reaction were removed at selected times over the course of 2 h, and lipids were twice extracted into chloroform by an acidified (1% glacial acetic acid) Bligh-Dyer method. After evaporating the chloroform layer under N2, cardiolipin was resolved from other lipid classes by thin layer chromatography (3/4/1/1/0.5 chloroform/acetone/methanol/acetic acid/water) and identified by iodine staining; the region of the plate corresponding to CL was scraped; and the amount of incorporated 14C label was determined by liquid scintillation counting. Measurements were calculated in the linear range of the reaction, and the -fold difference in reaction rate was determined.
Enzymatic Characterization of Electron Transport Chain and Functional Adenine Nucleotide Translocase Activities
Complex I
Complex I (NADH-ubiquinone oxidoreductase) activity was determined by measuring the decrease in the concentration of NADH at 340 nm and 37 °C as described previously (42, 43). The assay was performed in buffer containing 50 mm potassium phosphate (pH 7.4), 2 mm KCN, 5 mm MgCl2, 2.5 mg/ml BSA, 2 μm antimycin, 100 μm decylubiquinone, and 0.3 mm K2NADH. The reaction was initiated by adding purified mitochondria (5 μg). Enzyme activity was measured for 5 min, and values were recorded 30 s after the initiation of the reaction. Specific activities were determined by calculating the slope of the reaction in the linear range in the presence or absence of 1 μm rotenone (Complex I inhibitor).
Complex II
Complex II (succinate decylubiquinone 2,6-dichloroindophenol oxidoreductase) activity was determined by measuring the reduction of 2,6-dichloroindophenol at 600 nm as described previously (43, 44). The Complex II assay was performed in buffer containing 25 mm potassium phosphate (pH 7.4), 20 mm succinate, 2 mm KCN, 50 μm 2,6-dichloroindophenol, 2 μg/ml rotenone, and 2 μg/ml antimycin. Purified mitochondria (5 μg) were added prior to initiation of the reaction. The reaction was initiated by adding 56 μm decylubiquinone. Specific activities were determined by calculating the slope of the reaction in the linear range in the presence or absence of 0.5 mm thenoyltrifluoroacetone (Complex II inhibitor).
Complex III
Complex III (ubiquinol-cytochrome c reductase) activity was determined by measuring the reduction of cytochrome c at 550 nm and 30 °C. The Complex III assay was performed in buffer containing 25 mm potassium phosphate (pH 7.4), 1 mm EDTA, 1 mm KCN, 0.6 mm dodecyl maltoside, 32 μm oxidized cytochrome c, using purified mitochondria (1 μg). The reaction was initiated by adding 35 μm decylubiquinol. The reaction was measured following the linear slope for 1 min in the presence or absence of 2 μm antimycin (Complex III inhibitor). Decylubiquinol was made by dissolving decylubiquinone (10 mg) in 2 ml of acidified ethanol (pH 2) and using sodium dithionite as a reducing agent. Decylubiquinol was further purified by cyclohexane (42, 43, 45).
Complex IV
Complex IV (cytochrome c oxidase) activity was determined by measuring the oxidation of ferrocytochrome c at 550 nm and 25 °C. The Complex IV assay was performed in buffer containing 10 mm Tris-HCl and 120 mm KCl (pH 7.0), using purified mitochondria (2.5 μg). The reaction was initiated by adding 11 μm reduced ferrocytochrome c and monitoring the slope for 30 s in the presence or absence of 2.2 mm KCN (Complex IV inhibitor) (43, 46).
Complex V
Complex V (F1 ATPase) activity was determined using a coupled reaction measuring the decrease in NADH concentration at 340 nm and 37 °C as described previously (47–49). The Complex V assay was performed in buffer containing 50 mm Tris-HCl, 25 mm KCl, 5 mm MgCl2, 4 mm Mg-ATP, 200 μm K2NADH, 1.5 mm phospho(enol)pyruvate, 5 units of pyruvate kinase, 5 units of lactate dehydrogenase, 2.5 μm rotenone, 2 mm KCN, using purified mitochondria (10 μg). The reaction was initiated by the addition of mitochondria, and the reaction was monitored for 6 min. The slope in the linear range was used to calculate the reaction rate. 2.5 mg/ml oligomycin (Complex V inhibitor) was added to designated cuvettes to calculate the specific Complex V activity.
Functional Adenine Nucleotide Translocase (ANT) Activity
Measurement of functional ANT activity was performed using isolated mitochondria (50 μg) with high resolution respirometry. Briefly, isolated mitochondria were incubated with pyruvate (5 mm)/malate (5 mm), glutamate (10 mm)/malate (5 mm), palmitoyl-l-carnitine (20 μm)/malate (5 mm), or succinate (10 mm)/rotenone (1 μm) and then stimulated with ADP (1.25 mm) for state 3 respiration. Once maximal stimulated oxygen consumption was achieved, atractyloside was injected at sequential concentrations between 50 pmol and 20 nmol to determine the linear inhibition of ANT under control of the various respiratory substrates, as demonstrated previously (50–54).
Oxidized Lipid Metabolite Analysis
Heart Tissue Sample Preparations
Tissues (∼100 mg) were quickly washed with cold PBS (pH 7.4) solution, blotted, snap-frozen in liquid nitrogen, and stored at −80 °C until extraction. For extraction, 2 ml of ice-cold MeOH/CHCl3 (1:1 (v/v) with 1% acetic acid) and 2 μl of antioxidant mixture (0.2 mg/ml butylated hydroxyl-toluene, 0.2 mg/ml EDTA, 2 mg/triphenylphosphine, and 2 mg/ml indomethacin in a solution of 2:1:1 methanol/ethanol/H2O) were added to the tissue samples. Internal standards (250 pg each of TXB2-d4, PGE2-d4, LTB4-d4, 12-hydroxyeicosatetraenoic acid-d8, 13-hydroxyoctadecadienoic acid-d4, and 9,10-dihydroxyoctadecadienoic-d4 in 5 μl of acetonitrile) were also added at this step. The samples were immediately homogenized and subsequently vortexed several times during a 15-min incubation on ice. Then 1 ml of ice-cold H2O was added to the sample, which was briefly vortexed and centrifuged at 1,500 × g for 15 min. The CHCl3 layer was transferred to a new tube, and the remains were re-extracted and centrifuged at 1,500 × g for 15 min. The combined CHCl3 layers were dried down with N2 and reconstituted in 1 ml of 10% MeOH solution.
Solid Phase Extraction
The reconstituted solution was immediately applied to a Strata-X solid phase extraction cartridge that had been preconditioned with 1 ml of methanol followed by 1 ml of 10% methanol. The cartridge was then washed with 2 × 1 ml of 5% methanol, and additional solvent was pushed out with N2 at a pressure of 5 p.s.i. Eicosanoids were eluted with 1 ml of methanol containing 0.1% acetic acid. All cartridge steps were carried out using a vacuum manifold attached to a house vacuum line. After the organic solvent was evaporated with a SpeedVac, the residues were derivatized with AMPP.
Derivatization Reactions
The derivatization with AMPP was described in detail by Bollinger et al. Briefly, 12.5 μl of ice-cold acetonitrile, N,N-dimethylformamide (4:1, v/v) was added to the residue in the sample vial. Then 12.5 μl of ice-cold 640 mm (3-(dimethylamino)propyl)ethyl carbodiimide hydrochloride in HPLC grade water was added. The vial was briefly vortexed, and 25 μl of 5 mm N-hydroxybenzotriazole, 15 mm AMPP in acetonitrile was added. The vials were mixed briefly on a vortex and placed in a 60 °C water bath for 30 min.
LC/MS/MS Analysis
Arachidonic acid (AA), linoleic acid, and docosahexaenoic acid (DHA) metabolites were analyzed by LC/MS/MS. The metabolites were separated on a C18 reversed phase column (Ascentis Express, 2.7-μm particles, 150 × 2 mm), which was maintained at ambient temperature, using a mobile phase gradient (A, 0.1% glacial acetic acid in water; B, 0.1% glacial acetic acid in acetonitrile) at a flow rate of 0.2 ml/min. The solvent gradient program was 0–1.0 min, 5–20% B; 1.0–7.0 min, 20–25% B; 7.0–7.1 min, 25–40% B; 7.1–20 min, 40–60% B; 20–21 min, 60–100% B; 21–24 min, 100% B; 24–25 min, 100% B to 5% B. A 20-min column run was performed, followed by equilibration of the column at 5% B for next sample run.
Metabolites were analyzed using a hybrid tandem mass spectrometer (LTQ-Orbitrap, Thermo Scientific) via selected reaction monitoring in positive ion mode with sheath, auxiliary, and sweep gas flows of 30, 5, and 1 arbitrary units, respectively. The capillary temperature was set to 275 °C, and the electrospray voltage was 4.1 kV. Capillary voltage and tube lens were set to 2 and 100 V, respectively. Instrument control and data acquisition were performed using the Thermo Xcalibur version 2.1 software.
Statistical Analysis
Data were analyzed using a two-tailed unpaired Student's t test. Differences were regarded as significant at the p < 0.05 level (*) and p < 0.01 level (**). All data are reported as the means ± S.E. unless otherwise indicated.
RESULTS
Cardiac Myocyte Transgenic Expression of CLS Results in an Increased Content of Tetra-18:2 Cardiolipin Molecular Species
To investigate the metabolomic and functional sequelae of altering CL content in myocardial mitochondria, we generated a cardiac myocyte-specific (α-MHC) transgenic mouse expressing hCLS1. This approach allows the mechanistic dissection of the kinetic alterations that can be recruited to facilitate the regulation of CL content and molecular species distribution in mitochondria through genetic perturbation of the rate-limiting step in the formation of nascent cardiolipin molecular species. Moreover, through this approach, detailed insights into the downstream metabolomic and functional sequelae of alterations in cardiolipin metabolism on cardiac development, metabolism, signaling, and cellular bioenergetics can be examined to identify protective or maladaptive responses to metabolic stress (e.g. diabetic cardiomyopathy).
First, we generated mice expressing CLS in a cardiac myocyte-specific manner through utilization of the MHC promoter as described under “Experimental Procedures.” Human CLS transgene expression was confirmed by Western blot analysis by the appearance of a new band at the anticipated molecular mass of 61 kDa (Fig. 1A). Next, we determined that the expressed transgenic protein was catalytically active by quantifying the incorporation of radiolabeled phosphatidylglycerol into CL catalyzed by the transgenic CLS protein. Analysis of cardiolipin synthase activity revealed a 6-fold increase in activity in isolated cardiac mitochondria from the CLS compared with WT mice (Fig. 1B). Having established the expression and catalytic activity of the expressed transgene, we next focused on CLS transgene-mediated alterations in cardiolipin content and molecular species distribution in mature mice and during development.
FIGURE 1.

Mitochondrial translocation, enzymatic characterization, and MDMS-SL analysis of cardiolipin molecular species in wild type and cardiolipin synthase transgenic mice. A, Western blot analysis of isolated cardiac mitochondria revealed efficient translocation and expression of hCLS1 (hCLS) in the cardiac myocyte-specific cardiolipin synthase mouse as detected by a human cardiolipin synthase antibody. B, analysis of cardiac mitochondrial cardiolipin synthase activity revealed a 6-fold increase in the rate of CL synthesis compared with WT mice. Values represent the mean activity (pmol/h/mg protein) ± S.E. of four different mitochondrial isolates from each strain of mice per group. C, representative high resolution negative ion mode mass spectrum of anionic lipids for cardiolipin analysis from WT and CLS transgenic mouse myocardium. Cardiolipin molecular species were identified by the double charged isotopologue species as indicated with an asterisk. Spectra are representative of four different myocardium per group. All species were quantified with the isotopologue of the doubly charged tetra-14:0 cardiolipin standard (m/z 619.5). Tetra-18:2 cardiolipin species, the most predominant, is found at 723.5 m/z. *, p < 0.05.
Multidimensional mass spectrometry-based shotgun lipidomic (MDMS-SL) analyses of myocardial CL molecular species during cardiac development in WT and CLS mice revealed an accelerated rate of remodeling demonstrated by the selective enrichment of linoleic acid and decrease in docosahexaenoic acid (DHA)-containing CL molecular species in transgenic CLS mouse myocardium in comparison with WT littermates (Fig. 1C and supplemental Fig. 1). These specific alterations in the 18:2 fatty acyl content of CL were initially present at postnatal day 7 in the predominant molecular species that serve as intermediates in mature CL synthesis, such as 18:2–18:2–18:1–18:1 CL, 18:2–18:2–18:1–20:3 CL, and 18:1–18:2–18:2–20:2 CL. At postnatal day 14, species containing DHA were selectively and progressively decreased, and tetra-18:2 CL (the most abundant species in mature heart) was significantly increased in the transgenic CLS myocardium in comparison with WT littermates. These differences in CL remodeling are manifest through postnatal days 17–21, indicating accelerated CL remodeling in the CLS transgenic mouse during this time frame. The total CL content present in CLS myocardium was not statistically different in comparison with WT littermates and increased proportionally during development (Fig. 2A). By postnatal day 35, a mature CL profile was present in WT myocardium, whereas CLS myocardium exhibited a 40% increase in the predominant tetra-18:2 CL molecular species as identified by its doubly charged M + ½ isotopologue by MDMS-SL analysis (supplemental Fig. 1).
FIGURE 2.

Cardiac myocyte-specific cardiolipin synthase transgenic expression regulates myocardial lipidomic flux. A, total cardiolipin content of the cardiac myocyte-specific transgenic CLS mouse in comparison with wild type littermates. B, content of phosphatidylglycerol, the precursor for cardiolipin biosynthesis, was markedly depleted during development and remained at diminished levels in the CLS transgenic mouse. C, in contrast, tetra-18:2 cardiolipin content was dramatically increased starting at postnatal day 14 and maintained in the adult CLS transgenic mouse. Values represent the mean total lipid content or molecular species content (nmol/mg protein) ± S.E. (error bars) (n = 3–4 different hearts/group or isolated mitochondria). D, a 2-fold increase in 18:2-CoA incorporation into cardiolipin, indicative of cardiolipin remodeling, was present after calcium challenge in the CLS transgenic mouse. Black diamonds/bars, wild type mice; white squares/bars, CLS transgenic mice. *, p < 0.05 level; **, p < 0.01 level.
Transgenic Cardiac Myocyte CLS Expression Regulates Cardiolipin Flux and the Production of Oxidized Lipid Metabolites
Although the in vivo transgenic expression of CLS in myocardium was anticipated to result in an abundance of immature CL molecular species as well as the accumulation of CL mass, no increase in total CL mass was present. Rather, increased de novo synthesis was accompanied by an increase in phospholipase-mediated remodeling through accelerated phospholipase-mediated deacylation and subsequent acyltransferase/transacylation activities that served to regulate the overall content and composition of CL in the mitochondrial inner membrane. Thus, compensatory mechanisms (increased phospholipolysis) can be recruited to maintain the overall charge density and membrane curvature present in the mitochondrial inner membrane. A second prominent mechanism that is invoked to maintain mitochondrial membrane surface charge and membrane dynamics in the face of accelerated CL synthesis was the dramatic depletion of phosphatidylglycerol (PG), which serves as a co-substrate for CLS (Fig. 2B). Virtually all molecular species of PG were lower in the CLS mouse in comparison with WT littermates (supplemental Fig. 2A). However, based on the percentage distribution of PG molecular species, only the predominant 16:0–18:2 PG and 16:0–18:1 PG were proportionally decreased (supplemental Fig. 2B), indicating their major utilization in the synthesis of immature CL species. Interestingly, the content of phosphatidic acid, a precursor for PG synthesis, was unchanged in the CLS mouse relative to WT control mice (data not shown), indicating that CLS transgenic expression does not deplete additional distal upstream precursors.
More importantly, the effect of increased CLS activity not only resulted in increased catabolism of total CL species to balance net CL influx but also led to the selective remodeling of linoleic acid-enriched species, most notably tetra-18:2 CL (Fig. 2C). The increased remodeling rate of CL, however, was not the result of compensatory increases in the expression of upstream enzymes involved in CL biosynthesis (e.g. CDS2 and PGS1) or known CL remodeling enzymes, such as monolysocardiolipin acyltransferase or tafazzin (supplemental Fig. 3). To further investigate the rate of selective CL remodeling, we examined the incorporation of [14C]linoleoyl-CoA into CL in respiring mitochondria in the presence or absence of calcium ion. The results demonstrated a 2-fold increase in linoleic acid incorporation into CL in calcium-stimulated respiring mitochondria in the CLS transgenic mice versus their WT littermates (Fig. 2D). Thus, calcium serves as a modulator of CL content and composition, demonstrating a previously undescribed role for calcium in cellular bioenergetics through increased remodeling and resultant salutary alterations in mitochondrial bioenergetics.
To further investigate the sequelae of altering CL synthesis and remodeling on the composition of other myocardial lipids, we quantified the content and molecular species composition of choline and ethanolamine glycerophospholipids in WT and CLS transgenic hearts. The increased expression of CLS in transgenic mouse myocardium resulted in an increased content of specific choline and ethanolamine glycerophospholipid molecular species containing 18:2 and 20:4 acyl groups esterified at the sn-2 position accompanied by a corresponding decrease in the content of 22:6-containing molecular species (supplemental Figs. 4 and 5). We reasoned that the observed enrichment of linoleate and arachidonate in choline and ethanolamine glycerophospholipids augments the generation of lipid second messengers by phospholipase-mediated hydrolysis and subsequent oxidation. Thus, the CLS transgenic heart is presumably poised to produce biologically active signaling molecules that have previously been demonstrated to be potent regulators of cardiac contractility (55), bioenergetic efficiency, and vascular reactivity. Accordingly, we hypothesized that the altered composition of these molecular species reflected the intrinsic alterations in the production of lipid signaling molecules initiated by CLS transgene expression.
A mass spectrometric platform for measuring the diminutive amounts of these highly potent signaling metabolites in myocardium was developed using a charge switch methodology (56) in conjunction with a linear ion trap and accurate mass analysis of product ions using the LTQ-Orbitrap mass spectrometer. Mass spectrometric analysis of oxidized linoleic acid species revealed an ∼2-fold increase in 9-hydroxyoctadecadienoic acid, 13-oxo-octadecadienoic acid, 9-oxo-octadecadienoic acid, and 12(13)epoxy-9Z-octadecenoic acid (Fig. 3A). In contrast, other oxidized linoleic acid molecular species were unchanged. These results demonstrate the induction of specific signaling molecules that reflect adaptive metabolic programs initiated by the transgenic expression of CLS in murine myocardium. Similarly, analysis of 22:6 oxidized species demonstrated specific alterations in oxidized DHA molecular species after CLS transgenic expression. These included ∼2-fold decreases in 10,17-dihydroxy-4Z,7Z,11E,13Z,15E,19Z-docosahexaenoic acid, 19,20-dihydroxy-4Z,7Z,10Z,13Z,16Z-docosapentaenoic acid, and 7-hydroxy docosahexaenoic acid, whereas the majority of other DHA oxidized molecular species were unchanged in comparisons between CLS transgenic mice and their WT littermates despite a lower content of precursor 22:6 aliphatic chains in their phospholipid precursor pools (Fig. 3B). Additionally, analysis of oxidized AA metabolites revealed an overall increase in content of hydroxyeicosatetraenoic acids, epoxyeicosatrienoic acids, and prostanoids (Fig. 3C). Collectively, these results identify the unanticipated multiple pleiotropic regulatory roles of alterations in CL metabolic flux and remodeling on lipid second messenger generation in myocardium that can orchestrate multiple adaptive changes in cardiac bioenergetics and hemodynamic function.
FIGURE 3.

Myocardial generation of oxidized fatty acid metabolites is regulated by cardiac myocyte-specific cardiolipin synthase transgenic expression. A, oxidized linoleic acid (LA) metabolites; B, oxidized DHA metabolites; C, oxidized AA metabolites in wild type and CLS transgenic mouse hearts. Values represent the mean (pg/mg tissue) ± S.E. (error bars) (n = 6). Black bar, wild type mice; white bar, CLS transgenic mice. *, p < 0.05; **, p < 0.01.
Enhanced Cardiolipin Flux and Remodeling Selectively Regulate Electron Transport Chain Flux, Adenine Nucleotide Translocase Activity, and Respiratory Control Modulating Bioenergetic Efficiency
To investigate the effects of increased CL turnover and enrichment of tetra-18:2 CL in the CLS mouse on mitochondrial bioenergetic function, multiple parameters were assessed. First, analysis of ETC activities in isolated cardiac mitochondria during development revealed a selective 30% increase in Complex III activity (initiated at 1 month of age) as well as a selective 40% decrease in Complex IV activity beginning at 14 days of age in CLS transgenic mice compared with their WT counterparts (Fig. 4A). These findings suggest that the extent of CL remodeling in the CLS mouse is initiated by changes in Complex IV activity that lead to further adaptive alterations in Complex III activity. Second, high resolution respirometry revealed changes in ANT activity utilizing palmitoyl-l-carnitine, glutamate, or succinate as substrate but not pyruvate, thereby demonstrating CL-induced substrate-selective alterations in bioenergetic efficiency in CLS transgenic mouse hearts (Fig. 4B). These results suggest that tetra-18:2 CL and increased CL flux control the efficiency of mitochondrial ATP synthesis by the coordinated regulation of ETC function and tricarboxylic acid cycle flux to drive ADP entry into mitochondria. Third, high resolution respirometric analysis of mitochondrial substrate utilization revealed substrate-selective regulation of respiration in the CLS mouse. Although cardiac mitochondria isolated from CLS transgenic and WT mice possessed nearly equivalent rates of pyruvate-stimulated state 3 respiration (Fig. 5A), administration of palmitoyl-l-carnitine, glutamate, pyruvate/glutamate, and succinate demonstrated lower rates of oxygen consumption in state 3 respiration in the CLS mouse in comparison with WT littermates (Fig. 5, B–D). Thus, these changes in the utilization of Complex I and Complex II substrates were not mediated by the selective control of the active complexes in the ETC (Fig. 5E), but rather by the specific metabolism of these substrates in the tricarboxylic acid cycle in conjunction with decreased oxygen consumption by the attenuation of Complex IV activity present in mitochondria isolated from myocardium of the CLS transgenic mouse in comparison with their WT littermates. This resulted in the vast majority of the lower respiratory activity when analyzed by substrate control ratios for CLS compared with WT mitochondria (Fig. 5F). Moreover, analysis of ATP production during respiration revealed that there were no differences in the relative amounts of ATP produced by each substrate, although less oxygen was consumed, demonstrating increased bioenergetic efficiency (Fig. 5G). Mechanistically, these results demonstrate the increased coupling of the ETC chain in the CLS transgenic mouse due to increased Complex III and decreased Complex IV activities and/or an increase in substrate level phosphorylation in the tricarboxylic acid cycle.
FIGURE 4.
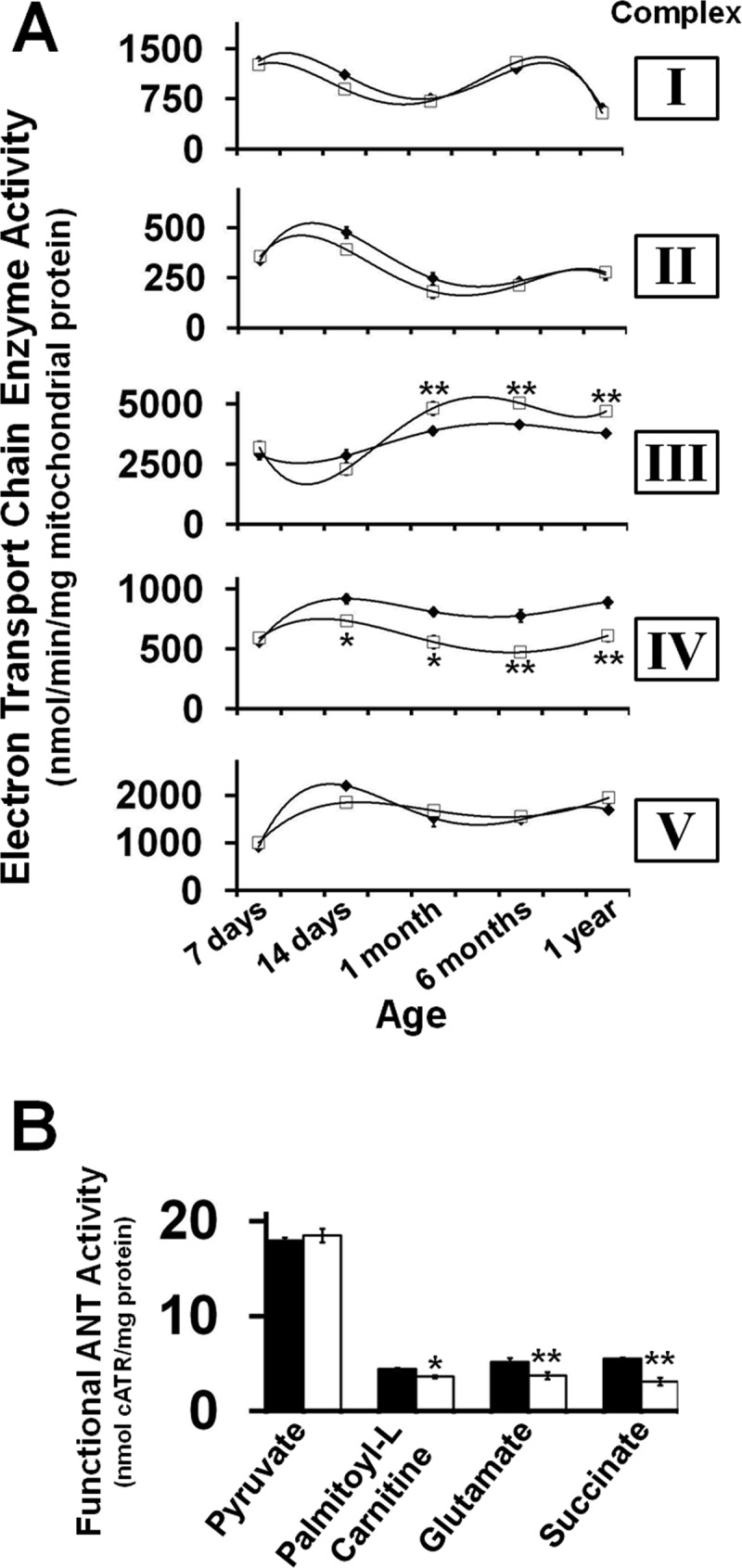
Developmental regulation of electron transport chain and adenine nucleotide translocase activities by cardiolipin synthase transgenic expression and enhanced cardiolipin remodeling. A, developmental effects of alterations in cardiac myocyte-specific CLS transgenic expression on the regulation of electron transport chain activities. B, functional adenine nucleotide translocase activity was decreased in a substrate-specific manner in the CLS transgenic mouse. Values represent the mean enzymatic activity (nmol/min/mg protein) ± S.E. (error bars) (n = 4–6 isolated mitochondrial preparations/group). Black diamonds/bars, wild type mice; white squares/bars, CLS transgenic mice. *, p < 0.05; **, p < 0.01.
FIGURE 5.

Cardiac mitochondrial substrate selectivity and bioenergetic efficiency are driven by cardiolipin synthase transgenic expression. High resolution respirometry demonstrated substrate-selective control of respiration utilizing pyruvate (Pyr) (A), palmitoyl-l-carnitine (Palm-Carn) (B), glutamate (Glut) (C), and pyruvate and glutamate (Pyr Glut) (D) through various respiratory and electron transport chain control states (E). F, comparative analysis of substrate control ratios revealed an overall decrease in substrate-stimulated respiration. However, cardiac mitochondria from CLS transgenic mice utilized less oxygen to generate an equivalent amount of ATP (G), demonstrating a high degree of coupling per molecule of oxygen consumed. FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; AA, antimycin A; Black bar, wild type mice; white bar, CLS transgenic mice; *, p < 0.05; **, p < 0.01. Error bars, S.E.
Cardiac Transgenic CLS Expression Attenuates Maladaptive Cardiolipin Remodeling and Bioenergetic Dysfunction in Mitochondria Isolated from Streptozotocin-induced Diabetic Mice
Previous work has demonstrated that streptozotocin (STZ)-induced diabetes leads to maladaptive CL remodeling, resulting in a decrease in tetra-18:2 CL molecular species and an increase in DHA-containing species (30, 33). Thus, we induced the diabetic state by STZ injection in both 4-month-old male CLS transgenic mice and their WT littermates and analyzed the resultant effects of CLS transgenic expression on diabetes-induced alterations in CL molecular species composition by the M + ½ isotopologue approach using MDMS-SL. Remarkably, we found that CLS transgenic expression attenuated the maladaptive remodeling of CL molecular species manifest in the diabetic state (Fig. 6).
FIGURE 6.

Cardiolipin synthase transgenic expression prevents maladaptive remodeling of 18:2- and 22:6-enriched species in streptozotocin-induced diabetes. Four-month-old wild type and CLS mice were injected with citrate (sham) or STZ to induce diabetes, and glucose levels were monitored to confirm the diabetic state. Following the induction of diabetes for 6 weeks, mice were sacrificed, and MDMS-SL cardiolipin molecular species analysis was performed. Analysis of 18:2-enriched (A) and 22:6-enriched cardiolipin molecular species (B) revealed maintenance of cardiolipin molecular speciation by cardiolipin synthase transgenic expression. Total analysis of cardiolipin molecular species can be found in supplemental Fig. 6. Values represent the mean molecular species content (nmol/mg protein) ± S.E. (error bars) (n = 4 different hearts/group). Black bar, wild type mice; white bar, CLS transgenic mice. *, p < 0.05; **, p < 0.01.
Analysis of the bioenergetic function in cardiac mitochondria revealed a decrease in pyruvate-stimulated, but not palmitoyl-l-carnitine-stimulated state 3 respiration in WT STZ mice compared with WT controls (Fig. 7, A and C). In contrast, no significant alterations were detected for either pyruvate- or palmitoyl-l-carnitine-stimulated state 3 respiration between CLS STZ and CLS mice (Fig. 7, B and D). Furthermore, measurements of ATP synthesis during respiration revealed a decrease in ATP production in WT STZ for both pyruvate- and palmitoyl-l-carnitine-stimulated respiration in comparison with WT injected animals. In sharp contrast, CLS transgenic mice rendered diabetic by STZ treatment maintained their ability to efficiently synthesize ATP despite lower rates of oxygen consumption, demonstrating preserved coupling efficiency (Fig. 7E). Enzymatic analysis of ETC activities in WT and CLS STZ mice revealed that STZ treatment of WT mice resulted in decreases in Complex I and Complex V function and increases in Complex II and Complex IV activities, probably leading to maladaptive compensatory alterations in bioenergetic capacity (Fig. 7F). In contrast, STZ treatment of CLS transgenic mice resulted only in a slight decrease in Complex V activity, which did not appear to affect total ATP synthesis because this was unaltered in the diabetic CLS transgenic mouse hearts (Fig. 7E). Collectively, these findings demonstrate that CLS transgenic expression can protect diabetic myocardium through its maintenance of physiologic CL molecular species, ETC activities, and bioenergetic efficiency in the diabetic state.
FIGURE 7.

Preservation of metabolic efficiency during streptozotocin-induced diabetes by cardiolipin synthase transgenic expression. High resolution respirometry using pyruvate (Pyr) with WT (A) or CLS transgenic mice (B) as well as palmitoyl-l-carnitine substrate (Palm-Carn) with WT (C) and CLS transgenic mice (D) stimulated respiration in sham-treated and STZ-induced diabetic mice. E, rate of ATP production driven by pyruvate or palmitoyl-l-carnitine. F, electron transport chain activities in diabetic and sham WT and CLS mice. Values represent the mean ± S.E. (error bars) (n = 5–8 different isolated mitochondria/group). *, p < 0.05; **, p < 0.01.
DISCUSSION
Dysfunctional regulation of CL content and/or molecular species distribution represents a membrane-mediated mechanism underlying mitochondrial bioenergetic inefficiency in multiple disease states (5, 24, 31, 33, 57, 58). However, due to the complex and interwoven nature of the multiple metabolic pathways involved in CL biosynthesis, remodeling, and metabolic flux, the mechanistic dissection of the roles of individual enzymes in the dysfunctional regulation of CL metabolism during the onset and progression of disease states has previously represented an intractable metabolomic problem (17, 26, 59, 60). In this study, we used cardiac myocyte-specific transgenic expression of CLS to identify the regulatory mechanisms that integrate multiple aspects of cardiolipin synthesis and remodeling to determine the functional sequelae of alterations in mitochondrial membrane composition, structure, and function. Through the detailed analysis of metabolomic, functional, and signaling alterations in the cardiac myocyte-specific CLS transgenic mouse, we provide compelling evidence that alterations in CLS activity coordinately regulate multiple downstream metabolic and operative processes, including CL remodeling, mitochondrial bioenergetic function, and the production of lipid second messengers of signal transduction. The results identify the previously unanticipated roles of CLS in 1) the regulation of CL molecular species distribution without changes in total CL content; 2) enhanced mitochondrial bioenergetic efficiency; 3) the regulation of biologically active lipid second messengers in myocardium; and 4) the ability of enhanced tetra-18:2 molecular species content in preventing pathologic alterations in cardiac mitochondrial lipidomic composition and bioenergetic function during streptozotozin-induced diabetes.
Alterations in CLS activity have previously been implicated in mitochondrial ETC complex assembly (19, 61) and mitochondrial fusion (62). The unique stereoelectronic relationships present in CL molecular species provide a powerful chemical palette that allows CL biosynthesis and remodeling to participate in the regulation of multiple mitochondrial processes, including protein import by Tam41 (63), fatty acid transport, and protein dynamics (64). Thus, alterations in CLS activity impact a multitude of mitochondrial processes that integrate mitochondrial structure, dynamics, and surface charge to facilitate adaptive changes that coordinately optimize mitochondrial bioenergetic and signaling functions.
Intense investigation has focused on identifying the molecular mechanisms that sculpt CL molecular species as well as regulate and maintain CL content in the mitochondrial membrane during development, health, and disease pathogenesis (18, 65). Alterations in CL content and molecular species have been demonstrated in heart failure, diabetes, Barth syndrome, myocardial ischemia, hyperthyroidism, oxidative stress, aging, and cancer (5, 24, 30, 31, 33, 66). In the heart, the predominant species of CL contains four linoleic acyl chains and is referred to as tetra-18:2 CL (67). Based on the structural symmetry and chiral centers present in tetra-18:2 CL as well as its predominance in highly oxidative tissues, such as myocardium, and its deficiency in Barth syndrome and other pathologic states, the maintenance of symmetric tetra-18:2 CL molecular species is evidently imperative for mitochondrial function (20, 28). Thus, modulation of tetra-18:2 CL content by the coordinated regulation of phospholipases and acyltransferases/transacylases during remodeling represents an upstream node for membrane-mediated mitochondrial bioenergetic and signaling functions. The selective increased content of tetra-18:2 CL (in the absence of an increase in total CL mass) in the CLS transgenic mouse provides a unique model system that has revealed the fundamental interrelationship between CL synthetic rates and phospholipase-initiated remodeling. Moreover, by perturbation of the metabolomic balance between CL synthesis and degradation, new insights into the fundamental mechanisms regulating CL content and molecular species composition have been uncovered.
In cardiac mitochondria isolated from the CLS transgenic mouse, we discovered that hyper-remodeling and increased tetra-18:2 CL content resulted in an increase in Complex III and a decrease in Complex IV activities in the CLS transgenic mouse compared with WT controls. In contrast, no differences were found in Complex I or V activities. Previously, analysis of fibroblasts from individuals with Barth syndrome, a disorder characterized by increased monolysocardiolipin content and decreased tetra-18:2 CL content, revealed a specific decrease in Complex III activity (68). Thus, it is evident that regulation of tetra-18:2 CL content in cardiac mitochondria coordinately regulates the activity of Complex III, thus influencing the coupling of proton and electron donation, through Complex I and II as well as the ubiquinone cycle, respectively, thereby increasing the potential driving force of the ETC. By selectively decreasing Complex IV activity, which is allosterically inhibited by ATP content and therefore controls the efficiency of ATP generation (69), the cytochrome c redox state in CLS cardiac mouse mitochondria is more efficiently coupled to the ETC, thereby utilizing less oxygen per molecule of ATP generated. Thus, selective control over specific components of the ETC complexes by CL content, molecular species composition, and lipid second messenger generation probably serves as an important regulatory mechanism underlying efficient mitochondrial bioenergetics function and signaling as well as its ability to circumvent maladaptive pathological diabetic insults (Fig. 8).
FIGURE 8.

Alterations in membrane structure, surface charge, and dynamics induced by cardiac myocyte-specific transgenic expression of cardiolipin synthase prevent mitochondrial dysfunction present in diabetes. Cardiolipin synthase transgenic expression regulates mitochondrial orchestrated lipidomic flux, substrate utilization for respiration, increased complex III activity and decreased complex IV activity affecting coupling capacity, and substrate connectivity with the adenine nucleotide translocase. These modulatory effects attenuate mitochondrial lipidomic alterations found with diabetes and prevent maladaptive mitochondrial dysfunction.
An unanticipated finding resulting from cardiac myocyte-specific transgenic expression of CLS was the increase in 18:2 and 20:4 fatty acyl and the decrease in 22:6 fatty acyl-containing choline and ethanolamine glycerophospholipid molecular species. This adaptation primed the generation of biologically potent oxidized fatty acid metabolites that are produced by downstream oxidative cascades. Previously, oxidized linoleic acid has been demonstrated to regulate intracellular calcium homeostasis, nitric oxide generation, and an antioxidant response (70–72). Additionally, AA metabolites, such as hydroxyeicosatetraenoic acids and epoxyeicosatrienoic acids, have demonstrated both vasoconstrictive and vasodilatory effects, respectively, on cardiovascular dynamics (73, 74). Thus, these results provide a novel mechanism through which alterations in mitochondrial bioenergetic state can be used to modulate blood flow and hence substrate delivery. Moreover, hydroxyeicosatetraenoic acids and epoxyeicosatrienoic acids have profound effects on cardiovascular hemodynamic function and ion channel kinetics (75, 76).
The rate-determining step in the synthesis of most biologically active eicosanoid metabolites is the release of AA from phospholipid molecular species by phospholipases A2. The released AA is subsequently oxidized by a rich repertoire of enzymes that generate specific eicosanoid metabolites in a spatio-temporal fashion that target discrete cellular functions through autocrine, paracrine, or endocrine pathways. Most of the intracellular phospholipases that participate in the release of AA are activated by negative membrane surface charge. Thus, CLS may directly participate in the production of biologically active oxidized lipid second messengers through phospholipid-mediated alterations in membrane surface charge and dynamics. Through the modulation of these distinct processes through increased CLS activity, the deleterious effects of the diabetic state on myocardial bioenergetics are attenuated, thereby preserving mitochondrial bioenergetic efficiency and metabolic capacity in diabetic myocardium.
In summary, our results demonstrate the critical importance of CLS in the coordination of bioenergetic capacity through CL flux, remodeling, and subsequent generation of bioactive oxidized lipid products in myocardium. Regulation of CLS activity thus represents a key metabolic and physiologic node that provides a novel therapeutic target for the treatment of bioenergetic dysfunction in diabetic myocardium and other disorders precipitated by mitochondrial dysfunction.
Supplementary Material
Acknowledgments
We thank Annie Nguyen and Susie Grathwohl for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant 5PO1HL57278. This work was also supported by the Barth Syndrome Foundation. X. H. has a financial relationship with LipoSpectrum. R. W. G. has financial relationships with LipoSpectrum and Platomics.
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE33451).

This article contains supplemental Figs. 1–6.
- CL
- cardiolipin
- ETC
- electron transport chain
- CLS
- cardiolipin synthase
- hCLS1
- human cardiolipin synthase 1
- ANT
- adenine nucleotide translocase
- MDMS-SL
- multidimensional mass spectrometry-based shotgun lipidomics
- PG
- phosphatidylglycerol
- AA
- arachidonic acid
- DHA
- docosahexaenoic acid
- STZ
- streptozotocin
- AMPP
- N-(4-aminomethylphenyl) pyridinium.
REFERENCES
- 1. Frohman M. A. (2010) Mitochondria as integrators of signal transduction and energy production in cardiac physiology and disease. J. Mol. Med. 88, 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brookes P. S., Yoon Y., Robotham J. L., Anders M. W., Sheu S. S. (2004) Calcium, ATP, and ROS. A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833 [DOI] [PubMed] [Google Scholar]
- 3. Bugger H., Abel E. D. (2010) Mitochondria in the diabetic heart. Cardiovasc. Res. 88, 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosca M. G., Hoppel C. L. (2010) Mitochondria in heart failure. Cardiovasc. Res. 88, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chicco A. J., Sparagna G. C. (2007) Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 292, C33–C44 [DOI] [PubMed] [Google Scholar]
- 6. Beyer K., Klingenberg M. (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24, 3821–3826 [DOI] [PubMed] [Google Scholar]
- 7. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M., Mileykovskaya E., Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 9. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 10. Noël H., Pande S. V. (1986) An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur. J. Biochem. 155, 99–102 [DOI] [PubMed] [Google Scholar]
- 11. Choi S. Y., Huang P., Jenkins G. M., Chan D. C., Schiller J., Frohman M. A. (2006) A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 8, 1255–1262 [DOI] [PubMed] [Google Scholar]
- 12. Klingenberg M. (2009) Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 1788, 2048–2058 [DOI] [PubMed] [Google Scholar]
- 13. van Gestel R. A., Rijken P. J., Surinova S., O'Flaherty M., Heck A. J., Killian J. A., de Kroon A. I., Slijper M. (2010) The influence of the acyl chain composition of cardiolipin on the stability of mitochondrial complexes. An unexpected effect of cardiolipin in α-ketoglutarate dehydrogenase and prohibitin complexes. J. Proteomics 73, 806–814 [DOI] [PubMed] [Google Scholar]
- 14. Schug Z. T., Gottlieb E. (2009) Cardiolipin acts as a mitochondrial signaling platform to launch apoptosis. Biochim. Biophys. Acta 1788, 2022–2031 [DOI] [PubMed] [Google Scholar]
- 15. Tyurin V. A., Tyurina Y. Y., Osipov A. N., Belikova N. A., Basova L. V., Kapralov A. A., Bayir H., Kagan V. E. (2007) Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid. Conflict or assistance in apoptosis. Cell Death Differ. 14, 872–875 [DOI] [PubMed] [Google Scholar]
- 16. Kuwana T., Mackey M. R., Perkins G., Ellisman M. H., Latterich M., Schneiter R., Green D. R., Newmeyer D. D. (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342 [DOI] [PubMed] [Google Scholar]
- 17. Hatch G. M. (1998) Cardiolipin. Biosynthesis, remodeling, and trafficking in the heart and mammalian cells (Review). Int. J. Mol. Med. 1, 33–41 [DOI] [PubMed] [Google Scholar]
- 18. Houtkooper R. H., Vaz F. M. (2008) Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. 65, 2493–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schlame M., Rua D., Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 [DOI] [PubMed] [Google Scholar]
- 20. Schlame M. (2008) Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 49, 1607–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Houtkooper R. H., Akbari H., van Lenthe H., Kulik W., Wanders R. J., Frentzen M., Vaz F. M. (2006) Identification and characterization of human cardiolipin synthase. FEBS Lett. 580, 3059–3064 [DOI] [PubMed] [Google Scholar]
- 22. Cao J., Liu Y., Lockwood J., Burn P., Shi Y. (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734 [DOI] [PubMed] [Google Scholar]
- 23. Taylor W. A., Hatch G. M. (2009) Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J. Biol. Chem. 284, 30360–30371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vreken P., Valianpour F., Nijtmans L. G., Grivell L. A., Plecko B., Wanders R. J., Barth P. G. (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 [DOI] [PubMed] [Google Scholar]
- 25. Kiebish M. A., Bell R., Yang K., Phan T., Zhao Z., Ames W., Seyfried T. N., Gross R. W., Chuang J. H., Han X. (2010) Dynamic simulation of cardiolipin remodeling. Greasing the wheels for an interpretative approach to lipidomics. J. Lipid Res. 51, 2153–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Houtkooper R. H., Turkenburg M., Poll-The B. T., Karall D., Pérez-Cerdá C., Morrone A., Malvagia S., Wanders R. J., Kulik W., Vaz F. M. (2009) The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 1788, 2003–2014 [DOI] [PubMed] [Google Scholar]
- 27. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. (2002) Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 28. Schlame M., Ren M., Xu Y., Greenberg M. L., Haller I. (2005) Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 [DOI] [PubMed] [Google Scholar]
- 29. Pope S., Land J. M., Heales S. J. (2008) Oxidative stress and mitochondrial dysfunction in neurodegeneration. Cardiolipin a critical target? Biochim. Biophys. Acta 1777, 794–799 [DOI] [PubMed] [Google Scholar]
- 30. Han X., Yang J., Yang K., Zhao Z., Abendschein D. R., Gross R. W. (2007) Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes. A shotgun lipidomics study. Biochemistry 46, 6417–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kiebish M. A., Han X., Cheng H., Chuang J. H., Seyfried T. N. (2008) Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria. Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 49, 2545–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saini-Chohan H. K., Holmes M. G., Chicco A. J., Taylor W. A., Moore R. L., McCune S. A., Hickson-Bick D. L., Hatch G. M., Sparagna G. C. (2009) Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 50, 1600–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han X., Yang J., Cheng H., Yang K., Abendschein D. R., Gross R. W. (2005) Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44, 16684–16694 [DOI] [PubMed] [Google Scholar]
- 34. Kiebish M. A., Han X., Cheng H., Lunceford A., Clarke C. F., Moon H., Chuang J. H., Seyfried T. N. (2008) Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 106, 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han X., Abendschein D. R., Kelley J. G., Gross R. W. (2000) Diabetes-induced changes in specific lipid molecular species in rat myocardium. Biochem. J. 352, 79–89 [PMC free article] [PubMed] [Google Scholar]
- 36. Mancuso D. J., Sims H. F., Yang K., Kiebish M. A., Su X., Jenkins C. M., Guan S., Moon S. H., Pietka T., Nassir F., Schappe T., Moore K., Han X., Abumrad N. A., Gross R. W. (2010) Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 285, 36495–36510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jüllig M., Hickey A. J., Chai C. C., Skea G. L., Middleditch M. J., Costa S., Choong S. Y., Philips A. R., Cooper G. J. (2008) Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics 8, 2556–2572 [DOI] [PubMed] [Google Scholar]
- 38. Yang K., Cheng H., Gross R. W., Han X. (2009) Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 81, 4356–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han X., Yang K., Yang J., Cheng H., Gross R. W. (2006) Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 47, 864–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hatch G. M., McClarty G. (1996) Regulation of cardiolipin biosynthesis in H9c2 cardiac myoblasts by cytidine 5′-triphosphate. J. Biol. Chem. 271, 25810–25816 [DOI] [PubMed] [Google Scholar]
- 41. Schlame M., Hostetler K. Y. (1991) Solubilization, purification, and characterization of cardiolipin synthase from rat liver mitochondria. Demonstration of its phospholipid requirement. J. Biol. Chem. 266, 22398–22403 [PubMed] [Google Scholar]
- 42. Birch-Machin M. A., Turnbull D. M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97–117 [DOI] [PubMed] [Google Scholar]
- 43. Ellis C. E., Murphy E. J., Mitchell D. C., Golovko M. Y., Scaglia F., Barceló-Coblijn G. C., Nussbaum R. L. (2005) Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking α-synuclein. Mol. Cell Biol. 25, 10190–10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. King T. E. (1967) Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. Methods Enzymol. 10, 322–331 [Google Scholar]
- 45. Degli Esposti M. (2001) Assessing functional integrity of mitochondria in vitro and in vivo. Methods Cell Biol. 65, 75–96 [DOI] [PubMed] [Google Scholar]
- 46. Yonetan T. (ed) (1967) Cytochrome oxidase. Beef heart. Methods Enzymol. 10, 332–335 [Google Scholar]
- 47. Bosetti F., Yu G., Zucchi R., Ronca-Testoni S., Solaini G. (2000) Myocardial ischemic preconditioning and mitochondrial F1F0-ATPase activity. Mol. Cell Biochem. 215, 31–37 [DOI] [PubMed] [Google Scholar]
- 48. Kipp J. L., Ramirez V. D. (2001) Effect of estradiol, diethylstilbestrol, and resveratrol on F0F1-ATPase activity from mitochondrial preparations of rat heart, liver, and brain. Endocrine 15, 165–175 [DOI] [PubMed] [Google Scholar]
- 49. Zheng J., Ramirez V. D. (1999) Rapid inhibition of rat brain mitochondrial proton F0F1-ATPase activity by estrogens. Comparison with Na+,K+-ATPase of porcine cortex. Eur. J. Pharmacol. 368, 95–102 [DOI] [PubMed] [Google Scholar]
- 50. Roussel D., Chainier F., Rouanet J., Barré H. (2000) Increase in the adenine nucleotide translocase content of duckling subsarcolemmal mitochondria during cold acclimation. FEBS Lett. 477, 141–144 [DOI] [PubMed] [Google Scholar]
- 51. Oliveira P. J., Wallace K. B. (2006) Depletion of adenine nucleotide translocator protein in heart mitochondria from doxorubicin-treated rats. Relevance for mitochondrial dysfunction. Toxicology 220, 160–168 [DOI] [PubMed] [Google Scholar]
- 52. Gellerich F. N., Kunz W. S., Bohnensack R. (1990) Estimation of flux control coefficients from inhibitor titrations by non-linear regression. FEBS Lett. 274, 167–170 [DOI] [PubMed] [Google Scholar]
- 53. Guillet V., Gueguen N., Verny C., Ferre M., Homedan C., Loiseau D., Procaccio V., Amati-Bonneau P., Bonneau D., Reynier P., Chevrollier A. (2010) Adenine nucleotide translocase is involved in a mitochondrial coupling defect in MFN2-related Charcot-Marie-Tooth type 2A disease. Neurogenetics 11, 127–133 [DOI] [PubMed] [Google Scholar]
- 54. Brown J. C., Gerson A. R., Staples J. F. (2007) Mitochondrial metabolism during daily torpor in the dwarf Siberian hamster. Role of active regulated changes and passive thermal effects. Am. J. Physiol. 293, R1833–R1845 [DOI] [PubMed] [Google Scholar]
- 55. Dzau V. J., Packer M., Lilly L. S., Swartz S. L., Hollenberg N. K., Williams G. H. (1984) Prostaglandins in severe congestive heart failure. Relation to activation of the renin-angiotensin system and hyponatremia. N. Engl. J. Med. 310, 347–352 [DOI] [PubMed] [Google Scholar]
- 56. Bollinger J. G., Thompson W., Lai Y., Oslund R. C., Hallstrand T. S., Sadilek M., Turecek F., Gelb M. H. (2010) Improved sensitivity mass spectrometric detection of eicosanoids by charge reversal derivatization. Anal. Chem. 82, 6790–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoch F. L. (1992) Cardiolipins and biomembrane function. Biochim. Biophys. Acta 1113, 71–133 [DOI] [PubMed] [Google Scholar]
- 58. Li J., Romestaing C., Han X., Li Y., Hao X., Wu Y., Sun C., Liu X., Jefferson L. S., Xiong J., Lanoue K. F., Chang Z., Lynch C. J., Wang H., Shi Y. (2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hauff K. D., Hatch G. M. (2006) Cardiolipin metabolism and Barth Syndrome. Prog. Lipid Res. 45, 91–101 [DOI] [PubMed] [Google Scholar]
- 60. Osman C., Voelker D. R., Langer T. (2011) Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhao M., Schlame M., Rua D., Greenberg M. L. (1998) Cardiolipin synthase is associated with a large complex in yeast mitochondria. J. Biol. Chem. 273, 2402–2408 [DOI] [PubMed] [Google Scholar]
- 62. Xu F. Y., McBride H., Acehan D., Vaz F. M., Houtkooper R. H., Lee R. M., Mowat M. A., Hatch G. M. (2010) The dynamics of cardiolipin synthesis post-mitochondrial fusion. Biochim. Biophys. Acta 1798, 1577–1585 [DOI] [PubMed] [Google Scholar]
- 63. Kutik S., Rissler M., Guan X. L., Guiard B., Shui G., Gebert N., Heacock P. N., Rehling P., Dowhan W., Wenk M. R., Pfanner N., Wiedemann N. (2008) The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J. Cell Biol. 183, 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mitchell R. W., Hatch G. M. (2009) Regulation of cardiolipin biosynthesis by fatty acid transport protein-1 in HEK 293 cells. Biochim. Biophys. Acta 1788, 2015–2021 [DOI] [PubMed] [Google Scholar]
- 65. Sparagna G. C., Lesnefsky E. J. (2009) Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53, 290–301 [DOI] [PubMed] [Google Scholar]
- 66. Sparagna G. C., Chicco A. J., Murphy R. C., Bristow M. R., Johnson C. A., Rees M. L., Maxey M. L., McCune S. A., Moore R. L. (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 48, 1559–1570 [DOI] [PubMed] [Google Scholar]
- 67. Schlame M., Horvàth L., Vìgh L. (1990) Relationship between lipid saturation and lipid-protein interaction in liver mitochondria modified by catalytic hydrogenation with reference to cardiolipin molecular species. Biochem. J. 265, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barth P. G., Van den Bogert C., Bolhuis P. A., Scholte H. R., van Gennip A. H., Schutgens R. B., Ketel A. G. (1996) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome). Respiratory chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 19, 157–160 [DOI] [PubMed] [Google Scholar]
- 69. Arnold S., Kadenbach B. (1997) Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome c oxidase. Eur. J. Biochem. 249, 350–354 [DOI] [PubMed] [Google Scholar]
- 70. Wang R., Kern J. T., Goodfriend T. L., Ball D. L., Luesch H. (2009) Activation of the antioxidant response element by specific oxidized metabolites of linoleic acid. Prostaglandins Leukot. Essent. Fatty acids 81, 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Searles C. D. (2006) Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 291, C803–C816 [DOI] [PubMed] [Google Scholar]
- 72. Obinata H., Izumi T. (2009) G2A as a receptor for oxidized free fatty acids. Prostaglandins Other Lipid Mediat. 89, 66–72 [DOI] [PubMed] [Google Scholar]
- 73. Li N., Liu J. Y., Qiu H., Harris T. R., Sirish P., Hammock B. D., Chiamvimonvat N. (2011) Use of metabolomic profiling in the study of arachidonic acid metabolism in cardiovascular disease. Congest. Heart Fail. 17, 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sudhahar V., Shaw S., Imig J. D. (2010) Epoxyeicosatrienoic acid analogs and vascular function. Curr. Med. Chem. 17, 1181–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grosser T., Yu Y., Fitzgerald G. A. (2010) Emotion recollected in tranquility. Lessons learned from the COX-2 saga. Annu. Rev. Med. 61, 17–33 [DOI] [PubMed] [Google Scholar]
- 76. Wang D., Patel V. V., Ricciotti E., Zhou R., Levin M. D., Gao E., Yu Z., Ferrari V. A., Lu M. M., Xu J., Zhang H., Hui Y., Cheng Y., Petrenko N., Yu Y., FitzGerald G. A. (2009) Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc. Natl. Acad. Sci. U.S.A. 106, 7548–7552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.