

Background: The Gid ubiquitin ligase of yeast is composed of seven subunits that contain several remarkable motifs.
Results: Using a series of deletion mutants we performed co-immunoprecipitation studies to unravel the arrangement of the subunits.
Conclusion: An initial model of the topology of the Gid complex is created.
Significance: The model provides the basis to understand the functionality of the Gid complex.
Keywords: Carbohydrate Metabolism, E3 Ubiquitin Ligase, Gluconeogenesis, Ubiquitination, Yeast Metabolism
Abstract
In the yeast Saccharomyces cerevisiae, key regulatory enzymes of gluconeogenesis such as fructose-1,6-bisphosphatase are degraded via the ubiquitin proteasome system when cells are replenished with glucose. Polyubiquitination is carried out by the Gid complex, a multisubunit ubiquitin ligase that consists of seven different Gid (glucose-induced degradation-deficient) proteins. Under gluconeogenic conditions the E3 ligase is composed of six subunits (Gid1/Vid30, Gid2/Rmd5, Gid5/Vid28, Gid7, Gid8, and Gid9/Fyv10). Upon the addition of glucose the regulatory subunit Gid4/Vid24 appears, binds to the Gid complex, and triggers ubiquitination of fructose-1,6-bisphosphatase. All seven proteins are essential for this process; however, nothing is known about the arrangement of the subunits in the complex. Interestingly, each Gid protein possesses several remarkable motifs (e.g. SPRY, LisH, CTLH domains) that may play a role in protein-protein interaction. We, therefore, generated altered versions of individual Gid proteins by deleting or mutating these domains and performed co-immunoprecipitation experiments to analyze the interaction between distinct subunits. Thus, we were able to create an initial model of the topology of this unusual E3 ubiquitin ligase.
Introduction
The metabolic degradation of glucose via glycolysis provides energy as well as precursor molecules for a cell. When carbohydrates are missing, yeast cells are able to produce glucose from nonfermentable carbon sources such as ethanol or acetate via gluconeogenesis. Both metabolic pathways are controlled at different steps by reciprocally acting enzymes. One key enzyme of gluconeogenesis is fructose-1,6-bisphosphatase (FBPase)7 that is tightly regulated by different mechanisms. In Saccharomyces cerevisiae it is only expressed when cells are grown under gluconeogenic conditions, e.g. in medium with ethanol as the sole carbon source (1). After the addition of glucose, FBPase synthesis is repressed, and the enzyme is allosterically inhibited by AMP and fructose-2,6-bisphosphate (2) and inactivated by phosphorylation (3, 4). Furthermore, FBPase is ubiquitinated and rapidly degraded by the proteasome (5–8).
In general, ubiquitination first requires ATP-dependent activation of the 76-amino acid ubiquitin molecule by the ubiquitin activating enzyme (E1), resulting in a thioester bond between the C-terminal glycine of ubiquitin and the E1 enzyme. This activated ubiquitin is then transferred to the active site cysteine of the ubiquitin conjugating enzyme (E2). During the last step, which is catalyzed by the ubiquitin ligase (E3), the C-terminal carboxyl group of ubiquitin is covalently attached to a lysine residue of the target protein forming an isopeptide bond (9, 10). Two different types of E3 ligases exist: whereas HECT domain ligases first receive the ubiquitin from the E2 enzyme and afterward transfer it to the target protein, RING finger type ligases bind the E2 as well as the substrate and catalyze the direct transfer of ubiquitin from the E2 to the substrate (11, 12).
Polyubiquitination of FBPase is carried out by the Gid E3 ligase, a complex of about 600 kDa (13). It consists of seven different Gid (glucose-induced degradation-deficient) proteins of which two contain a degenerated RING domain providing ligase activity (14, 15). Under gluconeogenic conditions the complex is only composed of six members (Gid1/Vid30, Gid2/Rmd5, Gid5/Vid28, Gid7, Gid8, and Gid9/Fyv10). After the addition of glucose, Gid4/Vid24 appears, binds to the complex, and triggers ubiquitination of FBPase. Interestingly, Gid4 as well is rapidly degraded by the proteasome, and its elimination depends on the Gid complex (15). All seven Gid proteins are essential for the degradation of FBPase. Until now, nothing is known about the arrangement of the subunits in the complex. Computational protein-protein interaction prediction by PIPE analysis predicted a core complex consisting of Gid1, Gid8, and Gid5 and another yet unknown protein, Ydl176 (16). Gid2, Gid9, and Gid4 are supposed to be only weakly associated. Gid7, however, was not identified to be part of the complex.
All Gid proteins possess remarkable motifs that may play a role in protein-protein interaction (15). The LisH (lissencephaly type-1-like homology) motif, which is found in Gid1, Gid7, and Gid8, consists of an α-helical domain of about 30 amino acids originally identified in the LIS1 protein of human brains. Recent studies revealed that the LisH domain is necessary and sufficient for LIS1 dimerization and plays a role in determining protein half-life and intracellular localization (17). Adjacent to the LisH motif, often another α-helical region, the CTLH (C-terminal to LisH) motif, can be found. In S. cerevisiae, this domain is unique to Gid1, Gid2, Gid8, and Gid9. Gid1 and Gid8 are the only yeast proteins that possess the CRA (CT11-RanBPM) motif (18), another potential interaction domain. Gid1 furthermore contains a SPRY domain originally identified in SPla and ryanodine receptors (19). The E3 ligase activity of the Gid complex is contributed by Gid2 and Gid9, which both possess a degenerated RING finger domain (14, 15). Gid7 contains seven WD40 repeats, and in Gid5 only armadillo-like repeats are present. Apart from the RING domains and the WD40 repeats, no similarities to other known E3 subunits exist.
Interestingly, all Gid proteins, except Gid4, have mammalian orthologues that possess the same protein motifs (Fig. 1A) and are organized in the 20S CTLH complex (20). Until now, the CTLH complex has been linked to several different functions like regulation of cell morphology (21), proteasome-dependent degradation of non-ubiquitinated α-catenin (22), or modulation of endosome/lysosome-dependent degradation of ubiquitinated proteins via interaction with HRS (hepatocyte growth factor-regulated tyrosine kinase substrate) (23). Although some interaction studies were carried out between the components, the overall organization of the CTLH complex is still unclear.
FIGURE 1.
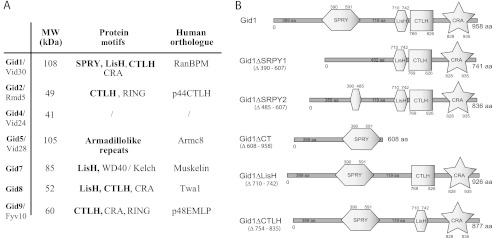
Protein motifs of the Gid proteins. A, Gid proteins and their human orthologues are shown. Motifs that are common to both proteins (according to SMART and UniProt) are in bold. The RING domains of Gid2 and Gid9 are described in Santt et al. (15) and Braun et al. (14). B, shown is a schematic overview of the protein domains of Gid1 and the Gid1 deletion mutants.
To elucidate the organization of the Gid E3 ligase, we deleted single domains of the predicted core proteins Gid1 and Gid8 and analyzed the interaction with other Gid proteins in vivo. Additionally, we knocked out certain GID genes and examined the effects on the complex composition. This enabled us to create an initial model of the topology of the Gid complex.
EXPERIMENTAL PROCEDURES
Media and Growth Conditions, Yeast Strains, and Plasmids
Standard methods were used for media preparation as well as genetic and molecular biological techniques (24, 25). Precultures were grown overnight at 30 °C in YP or synthetic complete medium containing 2% glucose, diluted 1:12.5 into the same medium, and grown for an additional 6–7 h. Thereafter, cells were transferred into YP or complete medium containing 2% ethanol and grown for 16 h to allow FBPase synthesis. To induce FBPase degradation, cells were shifted to YP or synthetic medium containing 2% glucose.
All yeast strains used in this study are listed in Table 1. Epitope tagging of yeast genes was performed either by PCR-based modification (26) or by chromosomal integration of plasmids containing the corresponding gene fused to a tag. pWO1105 (GID1-MYC13-URA3) was constructed from pRM8 (GID1-HA3-URA3) by replacing the HA3 tag with a Myc tag. pRM8 consists of a SphI/KpnI fragment containing the GID1-HA3 gene followed by an ADH1 terminator and 640 nucleotides upstream including the promoter that was cloned into Yiplac211. pWO1105 served as a template for mutagenesis applying overlap extension PCR (27). Using primer pairs RM39/RM29 and RM30/RM40 (Table 2), a fragment was generated that was cloned with BseRI and BamHI into pWO1105, yielding a GID1 construct (pWO1106) in which the nucleotides encoding the SRPY domain (Fig. 1B) are deleted. In the same manner pWO1107 (GID1ΔSPRY2-MYC13), pWO1108 (GID1ΔCT-MYC13), and pWO1109 (GID1ΔCTLH-MYC13) were generated using primer pairs RM39/RM31 and RM32/RM40, RM39/RM33 and RM34/RM9, or RM39/RM35 and RM36/RM9. pWO1110 (GID1ΔLisH-MYC13) was constructed using JS1 and RM40. After digestion with PstI, pWO1105–1110 was transformed into YWO1246 (Δgid1), YWO1554 (GID2-HA3 Δgid1), YWO1249 (GID7-HA3 Δgid1), YWO1555 (GID8-HA3 Δgid1), and YWO1657 (GID5-HA3 Δgid1) to allow integration at the promoter region yielding YWO1530–1535, YWO1536-YWO1541, YWO1542-YWO1547, YWO1548-YWO1553, and YWO1658-YWO1663, respectively. To analyze the interaction between the Gid1 mutants and Myc-tagged Gid4 (YWO1872, YWO1924–1928) the sequence of the MYC13-epitope was replaced by the V5 epitope (28) in pWO1105–1110, and the resulting plasmids pWO1288 and pWO1295–1299 were integrated into YRM37 (MYC9-GID4 Δgid1).
TABLE 1.
Yeast strains used in this study
| Name | Genotype | Reference |
|---|---|---|
| W303-1B | Matα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 42 |
| YOS1 | W303-1B gid4::MYC9-GID4 | 14 |
| YRM37 | W303-1B gid4::MYC9-GID4 Δgid1::LEU2kl | This study |
| YRM49 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YRM76 | W303-1B gid1::GID1-V5-URA3 gid4::MYC9-GID4 Δgid5::his5+ LEU2kl | This study |
| YRM81 | W303-1B gid1::GID1-V5-URA3 gid4::MYC9-GID4 Δgid2::his5+ LEU2kl | This study |
| YRM82 | W303-1B gid1::GID1-V5-URA3 gid4::MYC9-GID4 Δgid7::his5+ LEU2kl | This study |
| YRM109 | W303-1B gid1::GID1-MYC13-URA3 Δgid2::GID2-V5-LEU2 Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YRM110 | W303-1B gid1::GID1-V5-URA3 GID1 | This study |
| YRM111 | W303-1B gid1::GID1-V5-URA3 GID1 Δgid2::kanMX | This study |
| YRM112 | W303-1B gid1::GID1-V5-URA3 GID1 Δgid5::kanMX | This study |
| YRM113 | W303-1B gid1::GID1-V5-URA3 gid1::GID1-HA3- his5+ Δgid7::LEU2kl | This study |
| YRM114 | W303-1B gid1::GID1-V5-URA3 GID1 Δgid8::kanMX | This study |
| YRM115 | W303-1B gid1::GID1-V5-URA3 GID1 Δgid9::kanMX | This study |
| YSA1 | W303-1B gid8::GID8-HA3-his5+ | 14 |
| YTP10 | W303-1B gid7::GID7-HA3-his5+ | 14 |
| YTP11 | W303-1B gid5::GID5-HA3-his5+ | 14 |
| YWO1246 | W303-1B Δgid1::kanMX | This study |
| YWO1249 | W303-1B gid7::GID7-HA3-his5+ Δgid1::LEU2kl | This study |
| YWO1530 | W303-1B gid1::GID1-MYC13-URA3 kanMX | This study |
| YWO1531 | W303-1B gid1::GID1ΔSPRY1-MYC13-URA3 kanMX | This study |
| YWO1532 | W303-1B gid1::GID1ΔSPRY2-MYC13-URA3 kanMX | This study |
| YWO1533 | W303-1B gid1::GID1ΔCT-MYC13-URA3 kanMX | This study |
| YWO1534 | W303-1B gid1::GID1ΔCTLH-MYC13-URA3 kanMX | This study |
| YWO1535 | W303-1B gid1::GID1ΔLisH-MYC13-URA3 kanMX | This study |
| YWO1536 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1537 | W303-1B gid1::GID1ΔSPRY1-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1538 | W303-1B gid1::GID1ΔSPRY2-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1539 | W303-1B gid1::GID1ΔCT-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1540 | W303-1B gid1::GID1ΔCTLH-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1541 | W303-1B gid1::GID1ΔLisH-MYC13-URA3 gid2::GID2-HA3-his5+ LEU2kl | This study |
| YWO1542 | W303-1B gid1::GID1-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1543 | W303-1B gid1::GID1ΔSPRY1-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1544 | W303-1B gid1::GID1ΔSPRY2-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1545 | W303-1B gid1::GID1ΔCT-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1546 | W303-1B gid1::GID1ΔCTLH-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1547 | W303-1B gid1::GID1ΔLisH-MYC13-URA3 gid7::GID7-HA3-his5+ LEU2kl | This study |
| YWO1548 | W303-1B gid1::GID1-MYC13-URA3 gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1549 | W303-1B gid1::GID1ΔSPRY1-MYC13-URA3 gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1550 | W303-1B gid1::GID1ΔSPRY2-MYC13-URA3 gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1551 | W303-1B gid1::GID1ΔCT-MYC13-URA3 gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1552 | W303-1B gid1::GID1ΔcTLH-MYC13-URA3gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1553 | W303-1B gid1::GID1ΔLisH-MYC13-URA3 gid8::GID8-HA3-his5+ LEU2kl | This study |
| YWO1554 | W303-1B Δgid1:: LEU2kl gid2::GID2-HA3-his5+ | This study |
| YWO1555 | W303-1B Δgid1:: LEU2kl gid8::GID8-HA3-his5+ | This study |
| YWO1657 | W303-1B Δgid1::kanMX gid5::GID5-HA3-his5+ | This study |
| YWO1658 | W303-1B gid1::GID1-MYC13-URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1659 | W303-1B gid1::GID1ΔSPRY1-MYC13 -URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1660 | W303-1B gid1::GID1ΔSPRY2-MYC13 -URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1661 | W303-1B gid1::GID1ΔCT-MYC13 -URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1662 | W303-1B gid1::GID1ΔLisH-MYC13 -URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1663 | W303-1B gid1::GID1ΔCTLH-MYC13 -URA3 gid5::GID5-HA3-his5+ kanMX | This study |
| YWO1672 | W303-1B gid2::GID2-V5 ura3::LEU2kl | This study |
| YWO1682 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-V5 ura3::LEU2kl kanMX | This study |
| YWO1850 | W303-1B gid1::loxP gid2::GID2-V5-HIS3 Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1852 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::MYC9-GID4-HIS3 Δgid5::loxP Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1853 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::loxP Δgid5::GID5-HA3-LEU2 Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1855 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::GID7-HA3-TRP1 Δgid8::loxP Δgid9::loxP | This study |
| YWO1858 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1863 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-V5-HIS3 Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1871 | W303-1B gid1::GID1-V5-URA3 Δgid2::loxP Δgid4::MYC9-GID4-HIS3 Δgid5::loxP Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1872 | W303-1B gid1::GID1-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1873 | W303-1B gid1::GID1-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::GID5-HA3-LEU2 Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1879 | W303-1B gid1::GID1-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::GID7-HA3-TRP1 Δgid8::loxP Δgid9::loxP | This study |
| YWO1880 | W303-1B gid1::GID1ΔCTLH-MYC13 -URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::GID7-HA3-TRP1 Δgid8::loxP Δgid9::loxP | This study |
| YWO1881 | W303-1B gid1::GID1ΔLisH-MYC13 -URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::GID7-HA3-TRP1 Δgid8::loxP Δgid9::loxP | This study |
| YWO1882 | W303-1B gid1::GID1-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1883 | W303-1B gid1::GID1ΔSPRY1-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1884 | W303-1B gid1::GID1ΔSPRY2-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1885 | W303-1B gid1::GID1ΔCT-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1886 | W303-1B gid1::GID1ΔCTLH-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1887 | W303-1B gid1::GID1ΔLisH-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1900 | W303-1B Δgid1::loxP Δgid2::GID2-V5-HIS3 Δgid4::loxP Δgid5::GID5-HA3-LEU2 Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1905 | W303-1B Δgid1::loxP Δgid2::loxP Δgid4::MYC9-GID4-HIS3 Δgid5::GID5-HA3-LEU2 Δgid7::loxP Δgid8::loxP Δgid9::loxP | This study |
| YWO1906 | W303-1B gid4::MYC9-GID4 gid5::GID5-HA3-LEU2 | This study |
| YWO1920 | W303-1B gid1::GID1-MYC13-URA3 Δgid2::GID2-V5-TRP1 Δgid4::loxP Δgid5::loxP Δgid7::loxP Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1923 | W303-1B Δgid1::GID1-MYC13-URA3 Δgid2::loxP Δgid4::loxP Δgid5::loxP Δgid7::GID7-HA3-TRP1 Δgid8::GID8-HA3-HIS3 Δgid9::loxP | This study |
| YWO1924 | W303-1B gid1::GID1ΔSPRY1-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1925 | W303-1B gid1::GID1ΔSPRY2-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1926 | W303-1B gid1::GID1ΔCT-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1927 | W303-1B gid1::GID1ΔCTLH-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1928 | W303-1B gid1::GID1ΔLisH-V5-URA3 gid4::MYC9-GID4 LEU2kl | This study |
| YWO1963 | W303-1B gid1::GID1-MYC13-URA3 Δgid8::LEU2kl kanMX | This study |
| YWO1964 | W303-1B gid1::GID1-MYC13-URA3 gid8::GID8-V5-HIS3 kanMX | This study |
| YWO1965 | W303-1B gid1::GID1-MYC13-URA3 gid8::GID8ΔLisH-V5-HIS3 kanMX | This study |
| YWO1966 | W303-1B gid1::GID1-MYC13-URA3 gid8::GID8CTLH*-V5-HIS3 kanMX | This study |
| YWO1967 | W303-1B gid1::GID1-MYC13-URA3 gid8::GID8CRA*-V5-HIS3 kanMX | This study |
| YWO1968 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-HA3-TRP1 gid8::GID8-V5-HIS3 kanMX | This study |
| YWO1969 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-HA3-TRP1 gid8::GID8ΔLisH-V5-HIS3 kanMX | This study |
| YWO1970 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-HA3-TRP1 gid8::GID8CTLH*-V5-HIS3 kanMX | This study |
| YWO1971 | W303-1B gid1::GID1-MYC13-URA3 gid2::GID2-HA3-TRP1 gid8::GID8CRA*-V5-HIS3 kanMX | This study |
| YWO1977 | W303-1B gid2::GID2-HA3-TRP1 gid8::GID8-V5-HIS3 | This study |
TABLE 2.
Primers used in this study
| JS1 | CAACTTAAGCGTGGATGACGGTTCAGTTAATGTCAATGGGCA ACATTC (AflII) |
| RM9 | ATTCGGTACCTCCATGTCGCTGGCCG (KpnI) |
| RM29 | CACGACAAATATGCTCATACGCCCAAGTGACTGCAAAGTCCAC |
| RM30 | GTGGACTTTGCAGTCACTTGGGCGTATGAGCATATTTGTCGTG |
| RM31 | CACGACAAATATGCTCATACGCCCCTTCATAAATATAATCACGCC |
| RM32 | GGCGTGATTATATTTATGAAGGGGCGTATGAGCATATTTGTCGTG |
| RM33 | CACCGTTAATTAACCCGGGGATCGCCAAACTCTTCCATTTATCTTG |
| RM34 | CAAGATAAATGGAAGAGTTTGGCGATCCCCGGGTTAATTAACGGTG |
| RM35 | CATTAGATAATTCTTGACCCTTCAAATCTTTACTTTCAGAATGTTGCCC |
| RM36 | GGGCAACATTCTGAAAGTAAAGATTTGAAGGGTCAAGAATTATCTAATG |
| RM39 | GAGAAGAAGGAAGTAGAGGAGC (BseRI) |
| RM40 | ATGTTAATTAACCCGGGGATCCG (BamHI) |
| RM74 | TATACTGCAGCTCGTTGCCGTATCTCAACG (PstI) |
| RM79 | AGAGGAGCTCGGCAGTGGCAAACGTCGAAG (SacI) |
| RM80 | GAGACCCGGGCCAGAGAAATTGGTACGGG |
| RM83 | TATAGTCGACCCGCTATCAGAGATGGACCG (SalI) |
| RM84 | TATAGCGGCCGCACTACAAAAAGCACCAAGGCC (NotI) |
The Δgid deletion strain YRM49 was constructed performing several rounds of PCR-based gene deletion using different markers that were afterward removed, applying the Cre/loxP recombination system (29). Expression of distinct Gid proteins was restored by chromosomal integration of tagged versions of the respective genes at the promoter region. For integration of wild type and mutated GID1, the above-described plasmids were used. pWO1283 contains the GID2-V5 coding sequence together with the promoter region and an ADH1 terminator amplified from YWO1672 (primers RM83 and RM84) and cloned into pRS303. pWO1282 contains the MYC9-GID4 coding sequence together with the promoter region and an ADH1 terminator amplified from YOS1 (primers RM79 and RM80) and cloned into pRS303. pWO1280 contains the GID5-HA3 coding sequence together with the promoter region and an ADH1 terminator amplified from YTP11 (primers RM9 and RM74) and cloned into Yiplac128. pWO1275 contains the GID8-HA3 coding sequence together with the promoter region and an ADH1 terminator amplified from YSA1 and cloned into pRS303. pWO1286 (GID8-V5) was constructed by replacing the HA3 tag with the V5 sequence. Using overlap extension PCR plasmids pWO1334 (deletion of the nucleotides corresponding to the entire LisH motif (amino acids 85–117)), pWO1335 (deletion of nucleotides corresponding to amino acids 193–206 of the CTLH motif), and pWO1336 (L225S and L256Q substitutions in the CRA domain of the expressed Gid8 protein) were generated. To analyze the interaction between Gid1 and the Gid8 mutants, the previously described plasmids were integrated into YWO1963 (GID1-MYC13 Δgid8) yielding YWO1964–1967. To further examine Gid2, these strains were transformed with pWO1338 (GID2-HA3) yielding YWO1968–1971 and YWO1977. GID9 was expressed from an ARS/Cen plasmid (pWO0747 (14)) and transformed into YWO1863, YWO1900, and YRM109.
To analyze the interaction between Gid1 and Gid4 in single knock-out strains, GID2, GID5, and GID7 were replaced by the his5+ gene in YWO1872, yielding YRM81, YRM76, and YRM82. To further characterize the interaction between Gid1 and Gid5, pWO1288 (GID1-V5) was integrated into different strains containing deletions in single GID genes yielding YRM110–115.
Co-immunoprecipitation Experiments
For co-immunoprecipitation studies, cells were precultivated as described above and then grown overnight in medium containing 2% ethanol (gluconeogenic conditions). Cells (30 A600) were either harvested directly or, after the addition of 2% glucose for 30 min, washed with cold water, resuspended in buffer P3 (150 mm NaCl, 50 mm Tris/HCl, pH 7.5, 50 mm NaF, 5 mm EDTA, 0.1% Triton X-100) containing 1 mm PMSF and 1× ProteoBloc Protease Inhibitor Mixture (Fermentas), and lysed with glass beads at 8 °C for 20 min. After centrifugation for 20 min at 13,000 rpm, the supernatant was transferred to a new tube and incubated with the respective antibody for 2 h at 8 °C. After the addition of protein A-Sepharose CL-4B (GE Healthcare) and incubation for another 2 h or overnight, samples were centrifuged, and Sepharose beads were washed 3 times with buffer P3 and resuspended in urea buffer (200 mm Tris/HCl, pH 6.8, 8 m urea, 5% SDS, 0.1 mm EDTA, 1% 2-mercaptoethanol, 0.05% bromphenol blue). Bound proteins were released by incubation at 75 °C and analyzed via Western blotting.
Western Blotting and Antibodies
Western blotting was performed as described previously (7). For monitoring of FBPase degradation, 1.5 A600 of cells were harvested, and extracts were prepared via alkaline lysis. Proteins were precipitated with trichloroacetic acid and finally resuspended in urea buffer. Monoclonal HA antibody (clone 16B12, Covance), Myc antibody (clone 9E10, Santa Cruz Biotechnology), 3-phosphoglycerate kinase antibody (clone 22C5, Molecular Probes), and ubiquitin antibody (clone P4G7, Covance) were purchased. Polyclonal FBPase antiserum was raised against recombinant FBPase; Gid5 and Gid9 antisera were obtained after immunization of rabbits with peptides (Charles River, Germany).
Polyubiquitination Assay
Strains YWO1530–1535 and YWO1246 were transformed with a plasmid encoding TAP-tagged FBPase (15), and ubiquitination analysis was performed as described before (15) using IgG Sepharose.
RESULTS
Interaction Studies Utilizing Gid1 Deletion Mutants
Gid1 is the largest (108 kDa) protein of the Gid E3 ligase complex, and it contains four remarkable domains (SPRY, LisH, CTLH, CRA; Fig. 1B) that are thought to be involved in protein-protein interaction. PIPE analysis proposed that Gid1 might be a central component of the E3 ligase (16). We, therefore, generated various deletion mutants of Gid1 to learn more about the association of the proteins in the complex (comparable to (20)). We either removed the entire SPRY, LisH, or CTLH motif (GID1ΔSPRY1, GID1ΔLisH, GID1ΔCTLH) or, in case of the SPRY domain, just the part that is homologous to human RanBPM (GID1ΔSPRY2). Furthermore, we constructed a mutant in which the whole C-terminal part (GID1ΔCT) after the SPRY domain was eliminated. All constructs were integrated into the yeast genome replacing the wild type GID1 gene and were tested for their functionality. Because GID1 is a nonessential gene, the cells are viable. The mutant proteins are well expressed, although at different levels (see Fig. 3, Input). However, in every deletion mutant degradation of FBPase is impaired (Fig. 2A) due to a lack of ubiquitination (Fig. 2B) comparable to a GID1 deletion strain. This shows that any major change in Gid1 affects the functionality of the E3 ligase.
FIGURE 3.

The CTLH domain of Gid1 is necessary for interaction with Gid7, whereas the LisH domain is required for binding of Gid2, Gid8, Gid5, and Gid4. A, co-immunoprecipitation (IP) studies between the Gid1 mutants and Gid7 are shown. Cells were grown overnight in YPE and were then harvested either directly or 30 min after the addition of glucose. After glass bead lysis, Gid1 was precipitated using α-Myc-antibody and protein A-Sepharose. Precipitates were analyzed for the presence of Gid1 and Gid7 via Western blotting with α-Myc and α-HA antibodies. B, shown are co-IPs between Gid2 and the Gid1 mutants. In this experiment Gid2 was precipitated because HA3-tagged Gid2 has the same molecular weight as the heavy chain (HC) of the antibody. C–E, co-IPs between the Gid1 mutants and Gid8, Gid5, or Gid4, respectively, are shown. Because Gid4 only appears after the addition of glucose, samples were only taken 20 min after the shift. F, shown is a preliminary model of the Gid complex.
FIGURE 2.

Degradation of FBPase is impaired in all GID1 deletion mutants. A, cells were grown overnight in YP-ethanol (YPE), transferred to YPD, and harvested at the indicated time points. Cell extracts were prepared, and FBPase degradation was analyzed via Western blotting using FBPase or 3-phosphoglycerate kinase (Pgk) antibodies. 3-Phosphoglycerate kinase served as a loading control. B, shown is ubiquitination of FBPase in the GID1 mutants. Cells expressing FBPase-TAP from a plasmid were incubated overnight in YPE (sample E) and then transferred to YPD for 25 min to induce FBPase ubiquitination. After cell lysis, FBPase-TAP was pulled down using IgG-Sepharose beads. Ubiquitination was detected with ubiquitin antibodies after immunoblotting.
Next, we examined the interactions between these Gid1 mutant proteins and other subunits of the Gid complex performing co-immunoprecipitation studies. All interactions were analyzed in cells growing in ethanol-containing medium (gluconeogenic conditions) and after the addition of glucose (induction of FBPase degradation). Fig. 3A shows that in strains expressing HA-tagged Gid7 and Myc-tagged wild type Gid1, large amounts of Gid7 co-precipitate with Gid1 (IP, lanes 1 and 2). Gid7 comparably interacts with Gid1 when the LisH domain is missing (Gid1ΔLisH; IP, lane 11 and 12). Likewise, deletion of the SPRY domain or part of it allows binding of appropriate amounts of Gid7. Yet, the precipitated material of the Gid1ΔSPRY mutant protein is much lower, which consequently results in a weaker signal of co-precipitated Gid7. In contrast, removal of the entire C terminus of Gid1 (including the LisH and CTLH motif, lanes 7 and 8) or only of the CTLH domain (lane 9 and 10) abolishes binding of Gid7. This clearly demonstrates that the CTLH domain of Gid1 is necessary for its interaction with Gid7.
Subsequently, we analyzed the relationship between Gid1 and the remaining Gid proteins. Regarding the lysates (Input) in Fig. 3, B and C, it is apparent that the amounts of HA-tagged Gid2 and Gid8 are strongly reduced in all GID1 deletion mutants. Stability analysis after cycloheximide treatment revealed that both proteins are degraded rapidly in these cells in contrast to cells with full-length Gid1 (data not shown). This might indicate that the arrangement of the Gid complex is altered in the mutants, making Gid2 and Gid8 more accessible for breakdown. Nevertheless, we were able to analyze the interaction with Gid1. Because HA-tagged Gid2 has the same molecular weight as the heavy chain of the antibody, in this experiment we precipitated Gid2 and searched for binding of Gid1 (Fig. 3B). A strong interaction between Gid2 and wild type Gid1 is detectable (IP, lanes 1 and 2). Furthermore, deletion of the SPRY domain still allows co-precipitation of both proteins (lane 3–6). Comparable to Gid7, removal of the whole C terminus completely abolishes binding. However, Gid2 interacts with the Gid1ΔCTLH (lanes 9 and 10) mutant protein but not with Gid1ΔLisH. These findings indicate that for binding of Gid2, the LisH domain of Gid1 is crucial.
Concerning Gid8 (Fig. 3C), after precipitation of the Gid1 mutant proteins a similar picture emerges; the interaction between both proteins depends on the LisH domain. Interestingly, Gid5 and also Gid4 show the same binding patterns (Fig. 3, D and E). Unfortunately, we were not able to perform these studies with Gid9 because tagging of the protein disturbs the functionality of the Gid complex, and the polyclonal antiserum raised against a Gid9 peptide is not able to detect the reduced amounts of endogenous Gid9 present in the GID1 mutants.
The fact that removal of the CTLH domain of Gid1 specifically inhibits binding of Gid7 but still allows interaction with Gid2, Gid4, Gid5, and Gid8 indicates that the deletion does not disturb the overall structure of the residual protein. Vice versa, deletion of the nearby LisH domain prevents binding of Gid2, Gid4, Gid5, and Gid8 but has no influence on the interaction with Gid7. Interestingly, removal of the large SPRY domain (217 amino acids) retains binding of all other Gid proteins. However, after the addition of glucose to the GID1ΔSPRY mutant cells, less Gid7 and Gid8 seem to co-precipitate with Gid1. This might indicate that a conformational change occurs in the complex that weakens the interaction between the proteins. However, the amount of precipitated Gid1ΔSPRY is also lower in these samples, and in case of Gid8 significantly less protein is present in the lysates. These small alterations are, therefore, difficult to interpret. Based on the data presented above, we generated a first simple model of the Gid complex (Fig. 3F).
Gid1 Binds Directly to Gid7 and Gid8
The LisH motif only consists of about 30 amino acids. For this reason it is hard to imagine that four proteins (Gid2, Gid4, Gid5, and Gid8) can simultaneously attach to this small domain. It is, however, possible that one subunit connects the others to Gid1. To determine whether the interaction between two Gid proteins is direct or mediated by other members of the complex, we first expressed each Gid protein separately in Escherichia coli. However, most of the expressed material aggregated and was found in inclusion bodies. This is not surprising, as in yeast all Gid proteins are part of a large complex and deletion of single Gid proteins or only domains of e.g. Gid1 results in severe instability of the remaining Gid proteins (Fig. 3, B and C).8 Moreover, at least Gid1 and Gid7 are phosphorylated in vivo (data not shown); this posttranslational modification will be absent in the recombinant material. Taken further into account that no activity assay exists for single Gid proteins, we are not able to prove whether the subunits are folded properly. Therefore, we decided to establish an in vivo system and created a yeast knock-out strain in which all seven GID genes are deleted (Δgid). We then restored the expression of particular, tagged Gid proteins and again performed co-immunoprecipitation studies. This strain is most suitable for our purpose because all proteins are expressed from their endogenous promoter and in their natural environment, allowing posttranslational modification and proper folding (30). Furthermore, simultaneous expression of different combinations of several proteins is easy. The possibility that the interaction of the Gid proteins is mediated by a yet unknown protein is very unlikely as several different approaches only identified one additional protein as part of the Gid complex, the uncharacterized protein Ydl176 (31, 32), which is not necessary for degradation of FBPase and only binds to the complex when cells are grown in YPD.9
Fig. 4, A–C, illustrates that neither Gid2, Gid4, nor Gid5 was able to bind to Gid1 in the absence of all other Gid proteins (Δgid) even though they co-precipitate well in the wild type background (WT). This indicates that their interaction is mediated by other subunits of the complex.
FIGURE 4.

Only Gid7 and Gid8 bind directly to Gid1. A–C, shown are co-IPs between Gid1 and Gid2, Gid4, or Gid5, respectively, as described in Fig. 3. Studies were performed in the wild type background (WT) and in a Δgid strain in which only the indicated Gid proteins were expressed endogenously. Cells were grown overnight in YPE and were harvested directly (A and C) or 20 min after shift to glucose (B). D–E, co-IPs between Gid1 mutant proteins and Gid7 or Gid8 are shown. Cells were grown overnight in YPE. F, shown is a preliminary model of the Gid complex. HC, heavy chain.
Gid7, on the other hand, binds directly to Gid1 (Fig. 4D). The amount of co-precipitated material, however, is quite low compared with cells with wild type background. Nevertheless, the interaction is specific as Gid7 does not bind any more when the CTLH domain of Gid1 is deleted, consistent with the data from wild type cells (Fig. 3A, see model B in Fig. 4F).
The only strong binding partner of Gid1 is Gid8 (Fig. 4E). In the Δgid deletion strain both proteins interact to the same extent as in the wild type background, and binding depends on the LisH domain as shown before (Fig. 3C, see model B in Fig. 4F).
Gid5 Is the Adaptor Molecule for the Regulatory Subunit Gid4
Furthermore, we tested the interaction between all other combinations of Gid proteins in our Δgid strain. One additional interaction we could identify occurs between Gid4 and Gid5 (Fig. 5A), which was verified from both directions. Because Gid4 is not a permanent component of the E3 ligase but only appears and binds after the addition of glucose, it is likely that Gid5 serves as an adaptor molecule for Gid4. We, therefore, examined the interaction between Gid1 and Gid4 in a strain where just Gid5 was eliminated. Fig. 5B illustrates that deletion of GID5 completely prevents binding of Gid4 to the ligase complex, whereas removal of GID2 or GID7 has no effect. This clearly shows that Gid5 connects the regulatory subunit Gid4 to the complex.
FIGURE 5.
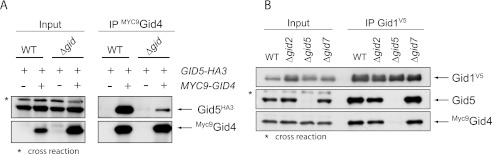
Gid5 is the adaptor molecule for Gid4. A, shown are co-IPs between Gid4 and Gid5 in the wild type background (WT) and the Δgid strain as described in Fig. 3. Cells were grown overnight in YPE and harvested 20 min after the addition of glucose. B, shown are co-IPs between Gid1 and Gid4 in distinct GID deletion strains. Cells were grown overnight in YPE and harvested 20 min after the addition of glucose. Gid5 was detected with polyclonal antiserum.
Gid8 Recruits Gid2, Gid9, and Gid5 to the Complex
Gid2 contains a degenerated RING domain providing ubiquitin ligase activity to the Gid complex (15). RING ligases often form homo- or heterodimers via their RING domain or adjacent motifs (12). Recently, we showed that Gid9 likewise possesses a RING motif and that it interacts with Gid2 in the Δgid strain (14). Subsequently, we analyzed how this Gid2/Gid9 dimer is attached to the complex. As mentioned above, the interaction between Gid1 and Gid8 is very strong, indicating that they might build the central part of the E3 ligase. This offers the possibility that both proteins together form a binding site for the remaining subunits. We, therefore, performed immunoprecipitation experiments between Gid2, Gid9, Gid1, and Gid8. Fig. 6A shows that co-expression of Gid8 significantly enhances binding of Gid2 to Gid1 in the Δgid strain. Unfortunately, the amounts of Gid9 in this experiment were below the detection limit of our antibody. However, in strains that do not harbor the GID9 expression plasmid (right panel), Gid2 does not bind to the core complex (consisting of Gid1 and Gid8), indicating that dimer formation between Gid2 and Gid9 is necessary for the interaction. Moreover, dimerization appears to stabilize Gid2 because the amounts of the protein in the cell extract are always higher when Gid9 is present.
FIGURE 6.

Gid8 recruits the Gid2/Gid9 dimer and thus Gid5 to the complex. A, co-expression of Gid8 enhances binding of Gid2 to Gid1 in the Δgid strain. In the absence of Gid9, no interaction is detectable. Cells were grown in complete medium. HC, heavy chain. B, co-expression of Gid8 does not enhance binding between Gid7 and Gid1 in the Δgid deletion strain. C, shown is a preliminary model of the Gid complex.
As mentioned above, binding of Gid7 to Gid1 in the Δgid strain is very weak (Fig. 4D). We, therefore, examined whether co-expression of Gid8 enhances the interaction between the two proteins. As is shown in Fig. 6B, this is not the case. The new data are depicted in Model C (Fig. 6C).
The LisH Domain of Gid8 Mediates Binding to Gid1, Whereas the CTLH and CRA Domains Are Necessary for Interaction with Gid2 and Gid5
Gid8 is one of the smaller subunits (52 kDa) and contains a LisH, CTLH, and CRA motif. Using deletion or point mutants, we examined which domain of Gid8 is involved in attachment to Gid1 in the wild type background. Interestingly, each mutation inhibits degradation of FBPase (data not shown). Fig. 7A reveals that deletion of the LisH motif of Gid8 completely abolishes binding to Gid1, whereas small deletions in the nearby CTLH domain (14 amino acids) or amino acid substitution of two conserved residues in the CRA domain (L255S/L246Q) still allows interaction comparable to wild type Gid8. These findings clearly demonstrate that Gid8 and Gid1 interact via their LisH domains.
FIGURE 7.
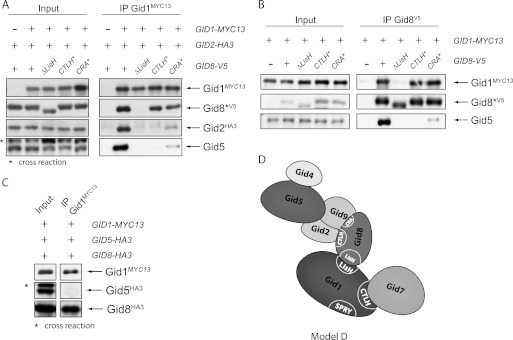
The LisH domain of Gid8 is responsible for binding to Gid1, whereas the CTLH and CRA domain mediates contact to Gid2 and Gid5. A, shown are immunoprecipitations of Gid1 in the wild type background. The precipitates were analyzed for the presence of Gid8, Gid2, and Gid5. Gid5 was detected with polyclonal Gid5 antiserum. B, shown are immunoprecipitations of the Gid8 mutant proteins in the wild type background. C, shown is a co-IP of Gid1, Gid5, and Gid8 in the Δgid strain. D, shown is a preliminary model of the Gid complex.
Additionally, we examined the binding of Gid2 to Gid1 in these strains. Disruption of the interaction between Gid1 and Gid8 totally impairs co-precipitation of Gid2 with Gid1 (Fig. 7A). This is consistent with our previous data showing that Gid8 mediates binding of the Gid2/Gid9 dimer to Gid1 (Fig. 6, A and C). Furthermore, mutations in the CTLH or CRA domain of Gid8 also affect the attachment of Gid2, indicating that these regions are necessary for the interaction between Gid8 and Gid2.
Regarding Gid5, a similar picture arises. Interestingly, when we precipitated Gid8, no binding of Gid5 could be detected when Gid8 did not interact with Gid1, revealing that Gid8 on its own is not able to bind Gid5 but needs contact to Gid1 (Fig. 7B). Interaction studies in the Δgid strain exhibit that even Gid1 and Gid8 together are not sufficient to bind Gid5, implying that the proteins are linked by another Gid protein (Fig. 7C). To analyze how Gid5 is incorporated into the complex in relationship to the Gid2/Gid9 dimer, we precipitated the core component Gid1 in strains in which just a single GID gene was deleted and analyzed the precipitates for the presence of Gid5 and also Gid9 with our antisera (Fig. 8). Binding of Gid9 is unimpaired by deletion of GID7 and GID5, whereas deletion of GID8 totally prevents it, confirming that Gid8 connects Gid9 with Gid1. Deletion of GID2 only weakens the interaction, implying that Gid9 links the Gid2/Gid9 dimer to the complex, although the contact is more stable in the presence of Gid2. This view is supported by the data presented in Fig. 6A showing that Gid2 only interacts with Gid1 and Gid8 when Gid9 is co-expressed.
FIGURE 8.

Binding of Gid9 to the core complex depends on Gid8, whereas binding of Gid5 additionally requires Gid8 and Gid9. Gid1 was precipitated in different GID deletion strains, and the interaction to Gid5 and Gid9 was analyzed with polyclonal antisera.
Binding of Gid5 likewise is independent of Gid7. However, besides Gid8, Gid9 is also necessary for the interaction with Gid1, showing that Gid9 connects Gid5 to the ligase complex. Deletion of GID2 consequently only weakens the interaction. Our present model of the Gid complex is illustrated in Fig. 9.
FIGURE 9.

Model of the topology of the Gid ubiquitin ligase complex.
DISCUSSION
The Gid E3 ligase complex of S. cerevisiae consists of seven different Gid proteins that are all essential for ubiquitination of gluconeogenic enzymes. Each Gid protein possesses several potential protein-protein interaction motifs; however, their relevance for the organization of the complex as well as the general arrangement of the subunits is unknown. We, therefore, deleted some of these motifs in two Gid proteins, and performed a series of co-immunoprecipitation studies in the wild type background. Furthermore, we examined the binding between individual subunits selectively co-expressed in a Δgid deletion strain. Altogether our studies enabled us to create an initial model of the topology of the Gid complex (Fig. 9). We identified Gid1 and Gid8 as the central components of the E3 ligase. In the absence of all other Gid subunits, both proteins strongly interact (Fig. 4E). Even though these studies were not performed in vitro but in the yeast background, it is very unlikely that the interaction is mediated by a yet unknown protein. The only additional component of the complex identified in four different approaches (13, 16, 31, 32) is the uncharacterized protein Ydl176. Studies in our laboratory show that it is not involved in degradation of FBPase and only binds under glycolytic conditions.9 Nevertheless, even if an unknown protein would link Gid1 and Gid8, our data clearly show that their interaction depends on their LisH domains (Figs. 4E and 8) fitting data from mammalian cells which show that this motif mediates homo- or heterodimerization of proteins (33, 34).
Another direct binding partner of Gid1 is Gid7 (Fig. 4D). This interaction depends on the CTLH domain of Gid1. Because it is much weaker in the Δgid deletion strain than in the wild type background, further components seem to be necessary to stabilize the contact between both proteins. Gid8 would be a good candidate for this purpose as the LisH and CTLH motifs of Gid1 are adjacent to each other. However, Gid8 does not enhance binding of Gid7 to Gid1 (Fig. 6C), revealing that the nearby LisH and CTLH motifs independently contact their binding partners. Furthermore, we identified Gid5 as the adaptor molecule for the regulatory subunit Gid4, which only appears after the shift from gluconeogenic to glycolytic growth conditions and acts as a trigger for ubiquitination of FBPase.
Recent data show that Gid2 and Gid9, which both contain a degenerated RING domain, form a heterodimer (14). Our analysis reveals that Gid9 establishes contact to the complex and that binding requires intact CTLH and CRA domains of Gid8 (Figs. 7A and 8). Attachment of Gid5 depends on the presence of Gid9 and consequently on Gid8 (Fig. 8). Interestingly, Gid8 is necessary but not sufficient for the recruitment of the Gid2/Gid9 dimer and Gid5 as the interaction between Gid1 and Gid8 is also required.
Some time ago the Gid complex was identified by bioinformatics methods using PIPE, a novel protein-protein interaction prediction engine (16). Depending on the score values of the protein pairs, the study suggests a model of the internal architecture of the complex, which places Gid1, Gid5, Gid8, and the protein Ydl176 at the center. The strongest interaction by far was predicted between Gid1 and Gid5. As described above, our detailed biochemical analysis only partly confirms this model, revealing the limitation of the computational prediction method. Especially, the absence of Gid7, which we and other groups (31) identified as a stable binding partner, is surprising.
Our present model of the topology of the Gid complex helps to understand the function of this unusual E3 ligase. Gid1 and Gid8 act as a scaffold for the other subunits. Gid1 is extremely important for the architecture of the complex as every alteration in this protein disturbs the whole assembly, leading to instability of Gid8 and Gid2 (Fig. 3) and thus complete loss of ligase activity. Ligase activity is contributed by Gid2 and Gid9, which are bound at the periphery of the complex. This arrangement might facilitate the contact to the ubiquitin-conjugating enzyme (E2) and the substrate. Furthermore, our data elucidate how Gid4 can act as a trigger. Binding of Gid4 to Gid5 may induce a conformational shift in the adaptor molecule that is directly passed on to the Gid2/Gid9 dimer, resulting in activation of the ligase.
Until now no comparable E3 ligase was found in yeast, yet an orthologous complex called the CTLH complex was found in mammalian cells. It contains the mammalian counterparts of all Gid proteins except Gid4 (20, 35), and several fundamental similarities exist between both complexes. Recently, it has been shown that the orthologue of Gid5, ARMc8, likewise functions as an adaptor molecule and associates with α-catenin, a component of the cadherin-catenin complex that forms adherens junctions (22). Binding of ARMc8 up-regulates the proteasome-dependent degradation of α-catenin, which, however, seems to be independent of ubiquitination. ARMc8, just as Gid5, is a peripheral protein (22), indicating that the basic topologies of the Gid and the CTLH complex are very similar.
RanBPM, the human orthologue of Gid1, as well acts as a scaffold and is associated with a wide range of functions (36). It is expressed ubiquitously in mammalian tissues and has been found to bind to microtubules, to adhesion molecules of the immune response or nervous system, and to numerous kinases. Furthermore, it has been shown that RanBPM interacts with the SUMO E2 enzyme Ubc9 and promotes sumoylation of the viral transcription factor Rta (37). Moreover, it is an activator of the mitochondrial apoptotic pathway (38). Similarly, yeast Gid1 has been associated with further functions besides ubiquitination of gluconeogenic enzymes such as transcription of nitrogen-regulated genes (39). At least Gid1, Gid2, and Gid5 are involved in rapamycin-induced internalization of the high affinity hexose transporter Hxt7 and its degradation via the endocytic pathway (40). Furthermore, most of the yeast Gid proteins are necessary for the vacuolar-dependent degradation of FBPase, which occurs when cells are replenished with glucose after prolonged starvation (41). In mammalian cells, ARMc8 was found to interact with the endosomal protein HRS hepatocyte growth factor regulated tyrosine kinase substrate, which plays an important role in endosomal trafficking of membrane proteins (23). HRS recognizes monoubiquitinated proteins via an ubiquitin interacting motif and regulates their endosome/lysosome-dependent degradation. Taken together, these data demonstrate that both complexes are members of different degradation pathways of the cell. Our results concerning the topology of the Gid E3 ligase provide a basis to understand the multifunctionality of the complex.
Acknowledgments
We thank Lise Barbin and Corinna Schelzel for providing plasmids.
This work was supported by the Deutsche Forschungsgemeinschaft, Bonn.
R. Menssen, J. Schweiggert, J. Schreiner, D. Kušević, J. Reuther, B. Braun, and D. H. Wolf, unpublished results.
R. Menssen and F. Wartlick, unpublished results.
- FBPase
- fructose-1,6-bisphosphatase
- CRA
- CT11-RanBPM
- CTLH
- C-terminal to LisH
- Gid
- glucose-induced degradation-deficient
- LisH
- lissencephaly type-1-like homology
- YPD
- yeast extract/peptone/dextrose
- YPE
- yeast extract/peptone/ethanol
- IP
- immunoprecipitation.
REFERENCES
- 1. Gancedo C., Salas M. L., Giner A., Sols A. (1965) Reciprocal effects of carbon sources on the levels of an AMP-sensitive fructose-1,6-diphosphatase and phosphofructokinase in yeast. Biochem. Biophys. Res. Commun. 20, 15–20 [DOI] [PubMed] [Google Scholar]
- 2. von Herrath M., Holzer H. (1988) Sensitivity of fructose-1,6-biphosphatase from yeast, liver, and skeletal muscle to fructose-2,6-biphosphate and 5'-adenosine monophosphate. Z. Lebensm.-Unters.-Forsch. 186, 427–430 [DOI] [PubMed] [Google Scholar]
- 3. Mazón M. J., Gancedo J. M., Gancedo C. (1982) Inactivation of yeast fructose-1,6-bisphosphatase. In vivo phosphorylation of the enzyme. J. Biol. Chem. 257, 1128–1130 [PubMed] [Google Scholar]
- 4. Müller M., Müller H., Holzer H. (1981) Immunochemical studies on catabolite inactivation of phosphoenolpyruvate carboxykinase in Saccharomyces cerevisiae. J. Biol. Chem. 256, 723–727 [PubMed] [Google Scholar]
- 5. Schork S. M., Bee G., Thumm M., Wolf D. H. (1994) Site of catabolite inactivation. Nature 369, 283–284 [DOI] [PubMed] [Google Scholar]
- 6. Schork S. M., Bee G., Thumm M., Wolf D. H. (1994) Catabolite inactivation of fructose-1,6-bisphosphatase in yeast is mediated by the proteasome. FEBS Lett. 349, 270–274 [DOI] [PubMed] [Google Scholar]
- 7. Schork S. M., Thumm M., Wolf D. H. (1995) Catabolite inactivation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. Degradation occurs via the ubiquitin pathway. J. Biol. Chem. 270, 26446–26450 [DOI] [PubMed] [Google Scholar]
- 8. Hämmerle M., Bauer J., Rose M., Szallies A., Thumm M., Düsterhus S., Mecke D., Entian K. D., Wolf D. H. (1998) Proteins of newly isolated mutants and the amino-terminal proline are essential for ubiquitin-proteasome-catalyzed catabolite degradation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. J. Biol. Chem. 273, 25000–25005 [DOI] [PubMed] [Google Scholar]
- 9. Glickman M. H., Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway. Destruction for the sake of construction. Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 10. Wolf D. H. (2011) The ubiquitin clan. A protein family essential for life. FEBS Lett. 585, 2769–2771 [DOI] [PubMed] [Google Scholar]
- 11. Fang S., Weissman A. M. (2004) A field guide to ubiquitylation. Cell. Mol. Life Sci. 61, 1546–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 13. Regelmann J., Schüle T., Josupeit F. S., Horak J., Rose M., Entian K. D., Thumm M., Wolf D. H. (2003) Catabolite degradation of fructose-1,6-bisphosphatase in the yeast Saccharomyces cerevisiae. A genome-wide screen identifies eight novel GID genes and indicates the existence of two degradation pathways. Mol. Biol. Cell 14, 1652–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braun B., Pfirrmann T., Menssen R., Hofmann K., Scheel H., Wolf D. H. (2011) Gid9, a second RING finger protein contributes to the ubiquitin ligase activity of the Gid complex required for catabolite degradation. FEBS Lett. 585, 3856–3861 [DOI] [PubMed] [Google Scholar]
- 15. Santt O., Pfirrmann T., Braun B., Juretschke J., Kimmig P., Scheel H., Hofmann K., Thumm M., Wolf D. H. (2008) The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol. Biol. Cell 19, 3323–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pitre S., Dehne F., Chan A., Cheetham J., Duong A., Emili A., Gebbia M., Greenblatt J., Jessulat M., Krogan N., Luo X., Golshani A. (2006) PIPE. A protein-protein interaction prediction engine based on the reoccurring short polypeptide sequences between known interacting protein pairs. BMC Bioinformatics 7, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerlitz G., Darhin E., Giorgio G., Franco B., Reiner O. (2005) Novel functional features of the Lis-H domain. Role in protein dimerization, half-life, and cellular localization. Cell Cycle 4, 1632–1640 [DOI] [PubMed] [Google Scholar]
- 18. Menon R. P., Gibson T. J., Pastore A. (2004) The C terminus of fragile X mental retardation protein interacts with the multidomain Ran-binding protein in the microtubule-organizing center. J. Mol. Biol. 343, 43–53 [DOI] [PubMed] [Google Scholar]
- 19. Woo J. S., Imm J. H., Min C. K., Kim K. J., Cha S. S., Oh B. H. (2006) Structural and functional insights into the B30.2/SPRY domain. EMBO J. 25, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi N., Yang J., Ueda A., Suzuki T., Tomaru K., Takeno M., Okuda K., Ishigatsubo Y. (2007) RanBPM, Muskelin, p48EMLP, p44CTLH, and the armadillo-repeat proteins ARMC8α and ARMC8β are components of the CTLH complex. Gene 396, 236–247 [DOI] [PubMed] [Google Scholar]
- 21. Valiyaveettil M., Bentley A. A., Gursahaney P., Hussien R., Chakravarti R., Kureishy N., Prag S., Adams J. C. (2008) Novel role of the muskelin-RanBP9 complex as a nucleocytoplasmic mediator of cell morphology regulation. J. Cell Biol. 182, 727–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki T., Ueda A., Kobayashi N., Yang J., Tomaru K., Yamamoto M., Takeno M., Ishigatsubo Y. (2008) Proteasome-dependent degradation of α-catenin is regulated by interaction with ARMc8α. Biochem. J. 411, 581–591 [DOI] [PubMed] [Google Scholar]
- 23. Tomaru K., Ueda A., Suzuki T., Kobayashi N., Yang J., Yamamoto M., Takeno M., Kaneko T., Ishigatsubo Y. (2010) Armadillo repeat containing 8α binds to HRS and promotes HRS interaction with ubiquitinated proteins. Open Biochem. J. 4, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ausubel F. M., Kingston R. E., Seidman F. G., Struhl K., Moore D. D., Brent R., Smith F. A. (eds.) (1992) in Current Protocols in Molecular Biology (Greene, ed.) Greene, New York [Google Scholar]
- 25. Guthrie C., Fink G. R. (1991) Guide to Yeast Genetics and Molecular Biology, Academic Press, Inc., Vol. 194, San Diego, CA [Google Scholar]
- 26. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 27. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 28. Funakoshi M., Hochstrasser M. (2009) Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 26, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gueldener U., Heinisch J., Koehler G. J., Voss D., Hegemann J. H. (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Betts S., King J. (1999) There's a right way and a wrong way. In vivo and in vitro folding, misfolding, and subunit assembly of the P22 tailspike. Structure 7, R131–R139 [DOI] [PubMed] [Google Scholar]
- 31. Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., Yang L., Wolting C., Donaldson I., Schandorff S., Shewnarane J., Vo M., Taggart J., Goudreault M., Muskat B., Alfarano C., Dewar D., Lin Z., Michalickova K., Willems A. R., Sassi H., Nielsen P. A., Rasmussen K. J., Andersen J. R., Johansen L. E., Hansen L. H., Jespersen H., Podtelejnikov A., Nielsen E., Crawford J., Poulsen V., Sørensen B. D., Matthiesen J., Hendrickson R. C., Gleeson F., Pawson T., Moran M. F., Durocher D., Mann M., Hogue C. W., Figeys D., Tyers M. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 32. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., Robinson M. D., Paccanaro A., Bray J. E., Sheung A., Beattie B., Richards D. P., Canadien V., Lalev A., Mena F., Wong P., Starostine A., Canete M. M., Vlasblom J., Wu S., Orsi C., Collins S. R., Chandran S., Haw R., Rilstone J. J., Gandi K., Thompson N. J., Musso G., St Onge P., Ghanny S., Lam M. H., Butland G., Altaf-Ul A. M., Kanaya S., Shilatifard A., O'Shea E., Weissman J. S., Ingles C. J., Hughes T. R., Parkinson J., Gerstein M., Wodak S. J., Emili A., Greenblatt J. F. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 33. Mateja A., Cierpicki T., Paduch M., Derewenda Z. S., Otlewski J. (2006) The dimerization mechanism of LIS1 and its implication for proteins containing the LisH motif. J. Mol. Biol. 357, 621–631 [DOI] [PubMed] [Google Scholar]
- 34. Ahn J., Novince Z., Concel J., Byeon C. H., Makhov A. M., Byeon I. J., Zhang P., Gronenborn A. M. (2011) The Cullin-RING E3 ubiquitin ligase CRL4-DCAF1 complex dimerizes via a short helical region in DCAF1. Biochemistry 50, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Umeda M., Nishitani H., Nishimoto T. (2003) A novel nuclear protein, Twa1, and Muskelin comprise a complex with RanBPM. Gene 303, 47–54 [DOI] [PubMed] [Google Scholar]
- 36. Murrin L. C., Talbot J. N. (2007) RanBPM, a scaffolding protein in the immune and nervous systems. J. Neuroimmune Pharmacol. 2, 290–295 [DOI] [PubMed] [Google Scholar]
- 37. Chang L. K., Liu S. T., Kuo C. W., Wang W. H., Chuang J. Y., Bianchi E., Hong Y. R. (2008) Enhancement of transactivation activity of Rta of Epstein-Barr virus by RanBPM. J. Mol. Biol. 379, 231–242 [DOI] [PubMed] [Google Scholar]
- 38. Atabakhsh E., Bryce D. M., Lefebvre K. J., Schild-Poulter C. (2009) RanBPM has proapoptotic activities that regulate cell death pathways in response to DNA damage. Mol. Cancer Res. 7, 1962–1972 [DOI] [PubMed] [Google Scholar]
- 39. van der Merwe G. K., Cooper T. G., van Vuuren H. J. (2001) Ammonia regulates VID30 expression, and Vid30p function shifts nitrogen metabolism toward glutamate formation especially when Saccharomyces cerevisiae are grown in low concentrations of ammonia. J. Biol. Chem. 276, 28659–28666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Snowdon C., Hlynialuk C., van der Merwe G. (2008) Components of the Vid30c are needed for the rapamycin-induced degradation of the high affinity hexose transporter Hxt7p in Saccharomyces cerevisiae. FEMS Yeast Res. 8, 204–216 [DOI] [PubMed] [Google Scholar]
- 41. Hung G. C., Brown C. R., Wolfe A. B., Liu J., Chiang H. L. (2004) Degradation of the gluconeogenic enzymes fructose-1,6-bisphosphatase and malate dehydrogenase is mediated by distinct proteolytic pathways and signaling events. J. Biol. Chem. 279, 49138–49150 [DOI] [PubMed] [Google Scholar]
- 42. Chiang M. C., Chiang H. L. (1998) Vid24p, a novel protein localized to the fructose-1,6-bisphosphatase-containing vesicles, regulates targeting of fructose-1,6-bisphosphatase from the vesicles to the vacuole for degradation. J. Cell. Biol. 140, 1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]