

Background: Tissue plasminogen activator (tPA) modulates many physiological and pathological processes.
Results: tPA cleaves the NR2B subunit of the NMDA receptor.
Conclusion: Deletion of amino acid residues at the amino-terminal domain can alter pharmacological properties of NMDA receptors.
Significance: Identifying novel substrates for tPA may be crucial for stroke therapy and the design of therapeutic molecules targeted at the ATD of the NR2B subunit.
Keywords: Electrophysiology; Glutamate Receptors Ionotropic (AMPA, NMDA); Plasmin; Serine Protease; Tissue Plasminogen Activator (tPA); Amino-terminal Domain; Glycine; Ifenprodil; NMDA Receptor; NR2B
Abstract
Thrombolysis using tissue plasminogen activator (tPA) has been the key treatment for patients with acute ischemic stroke for the past decade. Recent studies, however, suggest that this clot-busting protease also plays various roles in brain physiological and pathophysiological glutamatergic-dependent processes, such as synaptic plasticity and neurodegeneration. In addition, increasing evidence implicates tPA as an important neuromodulator of the N-methyl-d-aspartate (NMDA) receptors. Here, we demonstrate that recombinant human tPA cleaves the NR2B subunit of NMDA receptor. Analysis of NR2B in rat brain lysates and cortical neurons treated with tPA revealed concentration- and time-dependent degradation of NR2B proteins. Peptide sequencing studies performed on the cleaved-off products obtained from the tPA treatment on a recombinant fusion protein of the amino-terminal domain of NR2B revealed that tPA-mediated cleavage occurred at arginine 67 (Arg67). This cleavage is tPA-specific, plasmin-independent, and removes a predicted ∼4-kDa fragment (Arg27-Arg67) from the amino-terminal domain of the NR2B protein. Site-directed mutagenesis of putative cleavage site Arg67 to Ala67 impeded tPA-mediated degradation of recombinant protein. This analysis revealed that NR2B is a novel substrate of tPA and suggested that an Arg27–Arg67-truncated NR2B-containing NMDA receptor could be formed. Heterologous expression of NR2B with Gln29–Arg67 deleted is functional but exhibits reduced ifenprodil inhibition and increased glycine EC50 with no change in glutamate EC50. Our results confirmed NR2B as a novel proteolytic substrate of tPA, where tPA may directly interact with NR2B subunits leading to a change in pharmacological properties of NR2B-containing NMDA receptors.
Introduction
Tissue plasminogen activator (tPA)3 is an important serine protease that has a wide range of functions in the body physiology. In intravascular space, tPA is well known for its thrombolytic ability to convert the zymogen plasminogen into the active plasmin (1). This proteolytic event leads to the degradation of fibrin and assists to dissolve blood clots (2). Hence, recombinant “clot-busting” tPA is to date the only drug approved by the Food and Drug Administration for the treatment of ischemic stroke (3–6). Albeit the desirable effect of injected tPA in restoring the cerebral blood flow through its fibrinolytic activity, ischemic stroke patients who are deemed eligible to undergo tPA therapy have a low risk of symptomatic intracerebral hemorrhage (3, 5, 7).
In the central nervous system (CNS), significant levels of tPA protein expression have been reported in the hippocampus, amygdala, hypothalamus, cerebellum, and cortex (8, 9). Endogenous tPA is synthesized by a variety of cells that include neurons, glia, and microglia (10, 11) and can be released into the extracellular space upon depolarizing stimulation (12–14). As an extracellular protease in the brain (11, 15), tPA is thought to have a very different function from thrombolysis; that is, promoting neurite outgrowth (16), motor learning (17), axonal regeneration (18), nerve regeneration (19), and long term potentiation including learning and memory (20, 21). Deleterious effects of tPA have been implicated in pathological events such as neuroexcitotoxicity, neurodegeneration, and neurovascular unit damage (10, 13, 22, 23).
In addition to endogenous sources of tPA in CNS, it is hypothesized that tPA from the vasculature, either endogenously expressed or from ischemic stroke treatment, together with other blood-derived proteases such as thrombin can enter brain tissues during head injuries, stroke, or ruptured blood vessels where the integrity of the blood-brain barrier is often compromised (24–26). In addition, tPA was reported to cross the intact blood-brain barrier through low density lipoprotein-related protein-mediated transcytosis (27).
Novel neurotoxic mechanisms mediated through the interactions between extracellular tPA and N-methyl-d-aspartate (NMDA) receptors have emerged in the past decade (13, 28–30). Although non-proteolytic mechanisms mediated by tPA involve extracellular-regulated kinase 1/2 activation (29, 30), proteolytic mechanisms implicate tPA through the cleavage of the amino-terminal domain (ATD) of NR1 subunits (13, 28). Furthermore, extracellular tPA has also been shown to directly interact non-proteolytically with another subunit of NMDA receptor, NR2B subunit, where this tPA-NR2B complex influences the regulation of NR2B-containing NMDA receptors during chronic ethanol administration, ethanol withdrawal-induced seizures, and acute stress (31, 32). tPA has also been proposed to indirectly potentiate NMDA-induced enhancement of intracellular Ca2+ level through the adaptor protein PSD95 upon its engagement of low density lipoprotein-related protein in neurons (33, 34). As a modulator of various surface receptors, tPA could also act on novel substrates that have yet to be identified.
In this study we demonstrated that tPA directly degrades rat brain NR2B protein through its proteolytic activity and not through plasmin activation. Using recombinant fusion NR2BATD protein, MS/MS spectroscopy, amino-terminal peptide sequencing analyses, and site-directed mutagenesis studies, we showed that tPA cleaves the extracellular domain of NR2B at arginine 67 (Arg67). We further demonstrated that the sensitivities of NR2B-containing NMDA receptor to glycine and ifenprodil are significantly reduced when the first 67 amino acids are truncated in NR2B using two-electrode voltage clamp recording on heterologously expressed NR1-1a/NR2B-ΔATD-R67 receptors. Taken together our data report the existence of a new tPA substrate, the NR2B subunit, and suggest plausible functional consequences on the tPA-cleaved NR2B-containing NMDA receptor. In addition, if the newly cleaved NR2B short peptide enters the bloodstream during tPA thrombolytic treatment, it could serve as a biomarker of central nervous system injury.
EXPERIMENTAL PROCEDURES
Animals
Male Sprague-Dawley rats (6–8 weeks old) were obtained from the National University of Singapore, Laboratory Animal Centre. Oocytes were isolated from Xenopus laevis as described previously (35). All procedures involving animals were approved by the National University of Singapore, Institutional Animal Care and Use Committee.
Preparation of Rat Brain Lysate
After decapitation, the rat brains were removed immediately and washed 3 times in ice-cold phosphate-buffered saline (PBS) (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 2 mm KH2PO4, pH 7.4). The brains were homogenized in ice-cold non-denaturing radioimmune precipitation assay buffer (50 mm Tris, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, pH 7.4) containing Complete Protease inhibitor mixture tablet (Roche Diagnostics). The mixture was subsequently centrifuged at 15,000 × g for 30 min, and the supernatant was retained. Protein concentration of the lysate was measured using Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL).
In Vitro Cleavage Treatments
Total rat brain lysates or MBP-ATD2B fusion proteins (36) were incubated with recombinant human tPA (human tPA (Calbiochem) or Actilyse® (Boehringer Ingelheim) in cleavage buffer (100 mm NaCl, 65 mm Tris, pH 7.5) at 37 °C. Reactions were stopped by adding 4× Laemmli buffer (50 mm dithiothreitol, 5% v/v glycerol, 1% w/v SDS, 30 mm Tris-HCl, pH 6.8, 0.0005% w/v bromphenol blue) and heated at 95–100 °C for 5 min. Cleavage experiments involving plasmin (Roche Diagnostics) and α2-antiplasmin (Calbiochem) were performed in a similar manner. Before the start of each cleavage experiment, recombinant tPA (Calbiochem and Actilyse®) activity was determined using Spectrozyme® tPA chromogenic substrate (methylsulfonyl-d-cyclohexyltyrosyl-glycyl-arginine para-nitroaniline acetate) (American Diagnostica Inc.) as described by the manufacturer. The rate of chromophore formation (measured spectrophotometrically at λ405 nm) is correlated back to the concentration of tPA. This is to ensure similar potency of tPA was used for each experiment. The concentrations of the synthetic tPA inhibitor tPA-STOPTM (American Diagnostica) used in experiments ranged from 500 to 600 μm unless otherwise stated.
Preparation of Cerebrocortical Neuronal Culture and Actilyse® Treatment
Primary cultures of rat cortical neurons were obtained as previously described (35). Neurons were seeded at densities of 17 × 106 cells/T75 flasks coated with 0.1 mg/ml poly-d-lysine, and cultured at 37 °C in a humidified 5% CO2 incubator. Day in vitro 10 cultures were treated with 10 μg/ml Actilyse® for 3 h. Neurons were washed in ice-cold PBS, harvested, and lysed with ice-cold non-denaturing radioimmune precipitation assay buffer containing protease inhibitors (2 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mg/ml pepstatin A, and 574 μm phenylmethylsulfonyl fluoride).
SDS-PAGE, Western Blot Analysis, and Coomassie Blue Gel Staining
Treated rat brain lysate samples and cortical neuronal lysate samples from Actilyse®-treated neurons were separated on 8.5% polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% w/v nonfat milk in Tris-buffered saline with 0.1% v/v Tween 20 (TBST) and probed with one or more of the following antibodies: mouse anti-NR2B (epitope 1–550 amino acids, 1:200, Zymed Laboratories Inc.), rabbit anti-NR2B (epitope amino acids 27–76, 1:200, Santa Cruz Biotechnology), and mouse anti-GAPDH antibody (1:50,000, Millipore). After primary antibodies incubation, washed membranes were incubated with goat anti-mouse IgG-horseradish peroxidase (HRP) (1:5,000, Santa Cruz Biotechnology), goat anti-rabbit IgG-HRP (1:20,000, Santa Cruz Biotechnology), or rabbit anti-goat IgG-HRP (1:15,000, Santa Cruz Biotechnology). Upon washing with TBST, detection was performed using either ECLTM or ECL PlusTM Western blotting detection reagents (Amersham Biosciences). Treated recombinant protein samples were separated on 15% polyacrylamide gels and transferred to PVDF membranes. Membranes were subjected to immunoblot analyses as described above but probed with rabbit anti-NR2B antibody (1:200) and mouse anti-MBP antibody (1:10,000, Santa Cruz Biotechnology). In addition to Western blot analyses, recombinant protein reaction samples were also separated on 15% polyacrylamide gel and stained with Coomassie Brilliant Blue R250 (Merck).
Mass Spectrometry and Amino-terminal Peptide Sequencing
Bands of interest on Coomassie Blue-stained gels or Coomassie Blue-stained PDVF membranes were excised and submitted for MALDI-TOF MS/MS peptide mass spectrometry analysis and amino-terminal peptide sequencing (Protein and Proteomics Centre, Department of Biological Sciences, National University of Singapore), respectively.
Site-directed Mutagenesis and Recombinant Protein Expression
The NR2BATD deletion constructs were generated by inserting BglII restriction enzyme sites between Ser28 and Ser31 and between Val65 and Val68 sequentially by QuikChange mutagenesis on pcDNA1 NR2B wild-type plasmid template as reported previously (37) to generate pcDNA1 NR2B-ΔATD-R67 construct (hereafter named as NR2B-ΔATD-R67). This construct retained the first 28 residues of NR2B (Met1–Ser28) coding for the putative signal peptide. The vector for NR1-1a (hereafter named as NR1) is pCIneo (35). Arginine at position 67 on MBP-ATD2B(Arg27–Arg393) recombinant fusion protein was mutated to alanine using the QuikChange site-directed method as described previously (38). Soluble recombinant wild-type and mutant MBP-ATD2B fusion proteins were expressed, purified, and concentrated as described previously (36).
Two-electrode Voltage Clamp Recording of Heterologously Expressed NR1/NR2B Receptors in Xenopus Oocytes
Briefly, stage V and VI oocytes were removed from X. laevis and defolliculated. Oocytes were maintained in Barth's solution (88 mm NaCl, 1 mm KCl, 24 mm NaHCO3, 10 mm HEPES, 0.82 mm MgSO4, 0.33 mm Ca(NO3)2, and 0.91 mm CaCl2) supplemented with 100 mg/ml gentamycin, 40 mg/ml streptomycin, and 50 mg/ml penicillin. Oocytes were injected with 5–15 ng of cRNAs that were synthesized in vitro from a linearized template cDNA using mMESSAGE mMACHINE® T7 kit (Ambion Inc, Applied Biosystems). The ratio of NR1 to NR2B cRNAs injected was 1:2. The injected oocytes were maintained in Barth's solution at 17 °C for up to 7 days. Two-electrode voltage clamp recordings on oocytes were described previously (35). Recordings were performed 2–3 days post-injection at room temperature using Warner OC-725C two-electrode voltage clamps (SDR Clinical Technology). The recording solution contained 90 mm NaCl, 1 mm KCl, 10 mm HEPES, 0.5 mm BaCl2 with the pH adjusted to 7.3 with 5 n NaOH. Solution exchange through an 8-modular valve positioner (Digital MVP Valve, NV) was controlled by the EasyOocyte software (a gift from Professor Stephen F. Traynelis, Emory University, Atlanta, GA). Voltage and current electrodes were filled with 0.3–3.0 m KCl, and current responses were recorded at a holding potential of −40 to −60 mV. 100 μm glutamate, 100 μm glycine, and 0.03–10 μm ifenprodil (Sigma) were used in all oocyte experiments. Only data from oocytes yielding current responses between 50 nA to 2 μA to glutamate (100 μm) and glycine (100 μm) were analyzed.
Densitometry, Curve-fitting, and Statistical Analyses
Densitometry analysis was performed using UN-SCAN-IT gelTM Version 6.1 (Silk Scientific Corp.). Quantified data are expressed as percentage (%) of control, where control is assigned 100%. All data are expressed as the mean ± standard error of the mean (S.E.). Plotting of bar charts and graphs, and all statistical analyses were performed using GraphPad PRISM® Version 4 (GraphPad Software Inc., CA). EC50 and IC50 curves were plotted and fitted to Equation 1 by constraining the maximum response and minimum response to 100 and 0, respectively, using GraphPad PRISM®,
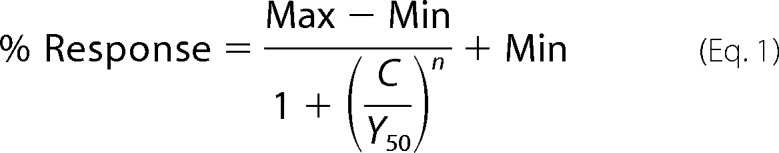 |
where n is the Hill slope, Y50 is agonist EC50 or antagonist IC50, C is agonist or antagonist concentration, Min is minimum response (%), and Max is maximum response (%). Error bars indicate S.E.
RESULTS
tPA Cleaves NR2B Protein
To determine whether tPA could cleave the NR2B subunit, we subjected rat brain lysates (40 μg) to increasing concentrations of tPA (ranging from 0.1 to 50 μg/ml). Using an anti-NR2B antibody that targets the extracellular domain of NR2B (epitope: amino acids 1–550), full-length NR2B protein immunopositive band (180 kDa) intensities were significantly decreased at 5 μg/ml (42.5 ± 5.5% of control, n = 4) to 50 μg/ml (6.1 ± 4.3% of control, n = 4) of tPA (p < 0.05, one-way ANOVA with Tukey's post hoc test) (Fig. 1, A and B). Despite the decreases in full-length NR2B immunoreactivity, no newly-generated fragments of NR2B were detected. The ability of tPA to induce cleavage of NR2B proteins was further validated using Actilyse®, the clinically applied human recombinant tPA. Actilyse® also degraded NR2B proteins in a similar concentration-dependent manner (supplemental Fig. 1). Moreover, Actilyse® degraded NR2B proteins, but not other glutamate receptor subunits NR1 (NMDA receptor subunit) and GluR2 (AMPA receptor subunit), in rat brain lysates prepared using non-denaturing radioimmune precipitation assay buffer as well as rat and mouse hippocampal lysates prepared using Tris-Triton-X buffer (supplemental Fig. 2). Hence, the NR2B subunit is a novel substrate of tPA.
FIGURE 1.

Representative immunoblots demonstrating the degradation of the NMDA receptor subunit NR2B upon recombinant tPA treatment. A, total rat brain lysate (RBL) (40 μg) was exposed to increasing concentrations of Calbiochem® tPA for 20 h at 37 °C. A Western blot was probed using anti-NR2B (Zymed Laboratories Inc.). No degradation of GAPDH was observed with increasing concentrations of tPA. B, shown is quantification of the blot seen in A. Data were normalized to control. n = 4; ** and *** denote p < 0.01 and p < 0.001, respectively (against control), one-way ANOVA with Tukey's post hoc test. The x axis was truncated to highlight the relative decrease in % NR2B immunoreactivity (≥1 μg/ml tPA). C, rat brain lysate (10 μg, ¼ of the amount used in A) was treated with 10 μg/ml Calbiochem® tPA for up to 3 h at 37 °C. A prominent decrease in NR2B immunoreactivity can be seen after tPA exposure for 30 min. Synthetic tPA inhibitor tPA-STOPTM prevented degradation of NR2B proteins. D, % NR2B immunoreactivity of full-length NR2B was reduced gradually with time upon incubation with tPA (based on quantification of C). NR2B proteins rapidly degraded within 30 min and reduced to ∼8% after 3 h. Data were normalized to untreated control of the corresponding treatment time. n = 4; ** and *** denote p < 0.01 and p < 0.001, respectively (against control), and # and φ denote p < 0.001 and p < 0.01 (against tPA-only treatment), one-way ANOVA with Tukey's post hoc test. E, a 3-h Actilyse® treatment of day in vitro 10 cortical neurons led to a decrease in NR2B protein levels. F, shown is quantification of the blot seen in E. Data were normalized to control. n = 6; * denote p < 0.05 (against control), two-tailed paired t test. A single blot was probed for NR2B and GAPDH for A, C, and E.
We next investigated the rate at which tPA degrades full-length NR2B over 3 h. 10 μg/ml tPA rapidly degraded NR2B proteins (10 μg) within 30 min and reduced the 180-kDa immunopositive band to ∼8% at the end of the 3-h treatment (Fig. 1, C and D). Likewise, no new NR2B-immunopositive fragments were detected by the anti-NR2B antibody (epitope: amino acids 1–550). Using a different anti-amino-terminal NR2B antibody (epitope: 27–76 amino acids, Santa Cruz Biotechnology), we observed a similar degree of full-length NR2B immunoreactivity reduction with no new NR2B band fragments (supplemental Fig. 3).
To investigate whether the cleavage of NR2B by tPA also occurs in native NMDA receptors, we treated cortical neurons in culture at days in vitro 10 with 10 μg/ml tPA for 3 h. tPA and culture medium were removed, and neurons were washed with 1× PBS and lysed. A statistically significant reduction in NR2B immunoreactivity was detected in the lysates obtained from tPA-treated neurons as compared with no tPA-treated neurons (Fig. 1, E and F, p < 0.05, two-tailed paired t test). These results suggest that NR2B is a possible proteolytic substrate for tPA.
tPA-mediated Degradation of NR2B Is Plasmin-independent
Physiologically, tPA converts plasminogen to plasmin. In vitro plasmin treatment of rat brain lysate led to the degradation of NR2B proteins that could be fully blocked by the specific plasmin inhibitor, α2-antiplasmin (Fig. 2A; also reported by Pawlak et al. (31)). Because plasminogen is found in the CNS (9, 10, 39), traces of plasminogen or plasmin could be present in the rat brain lysate preparations used for the cleavage experiments. To rule out the possibility that our observed NR2B cleavage in the presence of tPA was due to plasmin derived from plasminogen in our reaction mixture, α2-antiplasmin was co-incubated with tPA and rat brain lysate. The presence of α2-antiplasmin did not prevent NR2B degradation at 10 μg/ml tPA (n = 3, p > 0.05, one-way ANOVA with Tukey's post hoc test) (Fig. 2, B and C). On the contrary, in the presence of α2-antiplasmin, tPA continued to degrade NR2B to a similar degree as compared with tPA alone (n = 3, p > 0.05; one-way ANOVA with Tukey's post hoc test) (Fig. 2C). To validate that NR2B degradation is mediated by tPA, the specific synthetic tPA inhibitor (tPA-STOPTM) was co-incubated with tPA. tPA-STOPTM completely blocked the degradation of NR2B in the presence of tPA (n = 3, p > 0.05) (Fig. 2, B and C; see also Fig. 1, C and D).
FIGURE 2.

Plasmin and tPA degrade NR2B proteins (10 μg). A, rat brain lysate was treated with 0.01 unit of plasmin for 1 h at 37 °C and probed with anti-NR2B (Zymed Laboratories Inc.). A representative immunoblot shows that NR2B proteins were completely degraded by plasmin. The plasmin-induced degradation could be prevented in the presence of α2-antiplasmin, a plasmin inhibitor (n = 3). B, rat brain lysate was treated with 10 μg/ml tPA in the presence of α2-antiplasmin for 3 h at 37 °C and probed with anti-NR2B (Zymed Laboratories Inc.). C, shown is quantification of B. Data were normalized to untreated control. n = 3; n.s. and *** denote not significant and p < 0.001, respectively (against control unless otherwise labeled), one-way ANOVA with Tukey's post hoc test. Single blots were probed for NR2B and GAPDH for A and B.
tPA Cleaves NR2B at Arg67
Because tPA is an extracellular protease (11, 15), extracellular regions of membrane proteins are plausible tPA substrates. It is thus noteworthy to examine whether tPA-induced degradation of NR2B (Figs. 1 and 2 and supplemental Figs. 1 and 2) could be due to tPA cleavage on extracellular domains of NR2B. The observations obtained using two different anti-amino-terminal NR2B antibodies (Figs. 1 and 2 and supplemental Fig. 3) suggest that (a) tPA could cleave NR2B at a site that obliterated the epitopes recognized by both anti-amino-terminal NR2B antibodies and/or (b) tPA might cleave within the region of NR2B proximal to amino acid 76 and generate a 5–6-kDa short fragment that cannot be resolved and detected by the experimental paradigm. We thus investigated these possibilities using a recombinant NR2BATD fusion construct that includes amino acids Arg27 to Arg393 (MBP-ATD2B) as the substrate for tPA (36).
tPA treatment degraded MBP-ATD2B (∼85 kDa) and generated four cleaved fragments, F1–F4 (Fig. 3A, arrowheads, Coomassie Blue-stained gel), and this degradation could be prevented in the presence of tPA-STOPTM. Western blot analyses using anti-MBP antibody (Fig. 3B, left panel) and anti-NR2B antibody (epitope: amino acids 27–76) (Fig. 3B, right panel) detected both the full-length MBP-ATD2B fusion protein and F1. F2 could only be detected by anti-MBP, whereas F3 was detected by anti-NR2B exclusively. Although Coomassie Blue staining of SDS-PAGE gel revealed additional faint band fragments in control conditions (Fig. 3, A and D, MBP-ATD2B only and MBP-ATD2B WT only, respectively), these band fragments are found inherently in the batch-purified recombinant proteins and were not detected in Western blot using MBP- and ATD2B-specific antibodies (Fig. 3B). Hence, fragments F1–F4 were the sole proteins fragments that resulted from tPA treatment of recombinant proteins. To verify the identity of the band fragments, F1 and F2 were subjected to MS/MS peptide sequencing, whereas F3 and F4 were subjected to amino-terminal peptide sequencing. MS/MS peptide sequencing identified F1 to contain MBP and NR2B sequences, whereas F2 contained only MBP sequences (Fig. 3C; predicted cleavages). F3 was identified to be a protein fragment cleaved by tPA at an arginine residue within the linker region of the MBP-ATD2B fusion protein. Amino-terminal peptide sequencing revealed that the first five amino acid residues of F4 to be VELVA, which matched that of the NR2B subunit at residues downstream of arginine 67 (Arg67). From these results, we deduce that tPA cleaves the ATD of NR2B at Arg67 (Fig. 3C).
FIGURE 3.

tPA cleaves the MBP-ATD2B fusion protein. A, a representative Coomassie Blue-stained SDS-PAGE gel shows that a 3-h tPA (8 μg/ml) treatment of MBP-ATD2B yielded cleaved fragments (F1–F4). † denotes a fragment sent for peptide identification by MS/MS peptide sequencing; †† denotes a fragment sent for amino-terminal peptide sequencing. The presence of the synthetic tPA inhibitor, tPA-STOPTM (250 μm), prevented the tPA-induced degradation of MBP-ATD2B. B, shown is a representative Western blot analysis of tPA treatment of MBP-ATD2B (shown in A) using anti-MBP (left) and anti-NR2B (Santa Cruz Biotechnology, Inc) (right). Fragments F2 and F3 were revealed by anti-MBP and anti-NR2B, respectively. C, shown is a schematic representation (not drawn to scale) of tPA-induced-cleavage sites in MBP-ATD2B. MS/MS peptide mass spectroscopy and amino-terminal sequencing revealed two cleavage sites in the fusion protein. tPA cleaves the ATD of NR2B at arginine 67. Met represents the amino acid methionine. D, a representative Coomassie Blue-stained SDS-PAGE gel showed that mutant MBP-ATD2B (R67A) was not susceptible to tPA cleavage. Both wild-type (WT) and mutant recombinant MBP-ATD2B were subjected to tPA treatment for 1 h at 37 °C. The lack of cleavage fragments F1 and F4 after tPA treatment showed that MBP-ATD2B (R67A) was not susceptible to tPA cleavage.
To further corroborate that Arg67 is the tPA cleavage site on ATD of NR2B, Arg67 was mutated to alanine on MBP-ATD2B and subjected to tPA cleavage. Although there was a decrease in the band intensity of the MBP-ATD2B(R67A) protein after tPA treatment, only F2 and F3 were readily detected (Fig. 3D, arrowheads). Our biochemical results suggest that NR2B is a plausible substrate for tPA, and Arg67 is the tPA cleavage site.
Truncation of NR2B ATD at Arg67 Forms Functional NR1/NR2B-ΔATD-R67 Receptors with Reduced Glycine and Ifenprodil Sensitivities
Proteolytic cleavage of NMDA receptors has often been reported with modifications of receptor function, and one possible explanation is due to structural or conformational change(s) upon cleavage (13, 28, 37, 40–43). A possible consequence of proteolytic cleavage of the NMDA receptor is the dissociation of cleavage fragments, which may result in the truncation of the full-length protein (37). The loss of any peptide fragment could lead to structural changes that may affect allosteric interactions, leading to a change in receptor property.
To determine whether the removal of a short peptide proximal to Val68 (Gln29–Arg67 of mature NR2B protein) results in functionally modified NMDA receptors, a deletion construct of the ATD of NR2B up to Arg67 was generated (NR2B-ΔATD-R67) (Fig. 4A(ii)) to characterize the glutamate, glycine, and ifenprodil potencies by measuring the percentage of activation of NMDA receptor at maximum agonist-evoked steady state currents. The EC50 of glutamate measured in the presence of saturating concentration of glycine (100 μm) on NR1/NR2B-ΔATD-R67 (0.9 ± 0.2 μm, n = 6) did not differ from that of wild-type NR1/NR2B receptors (NR1/NR2BWT) (0.9 ± 0.1 μm, n = 7) (p > 0.05, two-tailed unpaired t test) (Fig. 4B). Interestingly, glycine potency was statistically significantly reduced for NR1/NR2B-ΔATD-R67 (1.4 ± 0.2 μm, n = 7) as compared with NR1/NR2BWT (0.28 ± 0.02 μm, n = 6) (p < 0.05, two-tailed unpaired t test) (Fig. 4C). In addition, the mean maximal current for the truncated NR1/NR2B-ΔATD-R67 (0.073 ± 0.017 μA, n = 16) was statistically significantly smaller than the wild-type NR1/NR2B receptors (0.674 ± 0.188 μA, n = 17, p < 0.0001; two-tailed Mann Whitney test) (supplemental Fig. 4).
FIGURE 4.

Electrophysiological characterization of heterologous NR1/NR2B-ΔATD-R67 receptors in Xenopus oocytes. A, (i) shown is a model of NR2B subunit consisting of ATD, agonist binding domain (S1S2), transmembrane domains, and the carboxyl terminus. Glutamate binds to the S1S2 domain, whereas critical amino acid residues affecting ifenprodil inhibition reside in the ATD. (ii) shown is a schematic representation of the full-length NR2B (NR2BWT) (upper panel) and truncated NR2B (deletion of Gln29–Arg67) (NR2B-ΔATD-R67) (lower panel) constructs. SP represents signal peptide, and M1, M3, and M4 represent transmembrane domain, whereas M2 represents the reentrant loop. B, the mean normalized concentration-response curve for glutamate in saturating concentrations of glycine (100 μm; pH 7.3) was obtained from oocytes expressing NR1/NR2BWT (open circles and solid line; n = 7) and NR1/NR2B-ΔATD-R67 (open triangle and dotted line; n = 6). Glutamate EC50 for NR1/NR2BWT and NR1/NR2B-ΔATD-R67 are 0.85 ± 0.12 and 0.94 ± 0.16 μm, respectively. p > 0.05, two-tailed unpaired t test. C, shown is a mean normalized concentration-response curve for glycine in saturating concentrations of glutamate (100 μm; pH 7.3) obtained from oocytes expressing NR1/NR2BWT (open circles and solid line; n = 6) and NR1/NR2B-ΔATD-R67 (open triangle and dotted line; n = 7). Glycine EC50 values for NR1/NR2BWT and NR1/NR2B-ΔATD-R67 are 0.28 ± 0.02 and 1.37 ± 0.19 μm, respectively, and are statistically significantly different. p < 0.001, two-tailed unpaired t test. D, shown is a mean normalized concentration-response curve for ifenprodil inhibition in saturating concentrations of glutamate and glycine (100 μm each; pH 7.3) obtained from oocytes expressing NR1/NR2BWT (open circles and solid line; n = 12) and NR1/NR2B-ΔATD-R67 (open triangle and dotted line; n = 4). Ifenprodil IC50 values for NR1/NR2BWT and NR2B-ΔATD-R67 are 0.35 ± 0.10 and 24.09. ± 18.15 μm, respectively, and are statistically significantly different. p < 0.05, two-tailed unpaired t test.
As the essential amino acids residues contributing to the sensitivity of ifenprodil shown by site-directed mutagenesis reside within the ATD of NR2B (38, 44) (Fig. 4A(i)), we further investigated the ifenprodil inhibition profile on the resultant NR2B-truncated NMDA receptors. Examination of ifenprodil concentration-response curves in the presence of maximum agonist-evoked steady state current revealed statistically significantly different IC50 values for NR1/NR2B-ΔATD-R67 (24.1 ± 18.2 μm, n = 4) and NR1/NR2B receptors (0.4 ± 0.1 μm, n = 12) (p < 0.001, two-tailed unpaired t test) (Fig. 4D).
DISCUSSION
In recent years proteases such as calpain, matrix metalloproteinase-3 and -7, thrombin, tPA, and plasmin have been reported to cleave NMDA receptor subunits and modify receptor function (13, 28, 34, 37, 40–43, 45). In particular, tPA and plasmin can modulate NMDA receptor through proteolytic cleavage of the ATDs of NR1 and NR2A, respectively (13, 28, 37). Moreover, we recently found that urokinase (or urokinase-type plasminogen activator), a serine protease, can also degrade NR2B.4 In this study, we report NR2B subunit of the NMDA receptor as a novel substrate for tPA. Using a recombinant fusion protein of the NR2B ATD, we identified the cleavage site at the peptide bond between Arg67 and Val68. Functionally, we characterized that the truncated form of NR2B (NR2B-ΔATD-R67) could lead to a reduction in ifenprodil inhibition and an increase in the EC50 of the co-agonist glycine.
Alignment of the sequence around our identified ATD cleavage site to the recognition sequences of known tPA substrates revealed several similarities (Table 1). Our identified tPA-mediated cleavage site on NR2B displayed conserved amino acids at the critical arginine (position denoted as P1) and valine (position denoted as P1′) positions when compared with plasminogen, the tPA natural endogenous substrate (Table 1). In addition, proteolytic profiling assay suggests that low hydrophobic amino acids (e.g. proline) and bulky hydrophobic amino acids (e.g. leucine, valine, phenylalanine, etc.) might be preferred at P2 and P3, respectively, for efficient tPA cleavage (46). This recognition motif coincides with our identified tPA cleavage site on NR2B (proline and valine at P2 and P3, respectively). In addition, the atomically resolved crystal structure of NR2BATD consists of two domains, R1 and R2, defined as residues 32–147, 287–359, and residues 148–286 and 360–394, respectively (47). Both apoNR2BATD and NR1ATD/NR2BATD in complex with ifenprodil revealed Arg67 is located at the tip of the R1 domain and not buried within the structure and is near the surface of the clamshell configuration of ATD (47, 48) (Fig. 5A). This suggests that Arg67 is potentially accessible to tPA in both the open and close states of the NR2BATD. However, external environmental conditions (e.g. redox status, pH, receptor conformation) could also influence the efficiency of cleavage.
TABLE 1.
Alignment of critical residues around the scissile peptide bonds of tPA substrates
Cleavage of tPA substrates occurs at the peptide bond between amino acid residues denoted by positions P1 and P1′. P2-P4 and P2′-P4′ refer to positions of amino acid residues flanking the left and right of the scissile peptide bond, respectively. Plasminogen activator inhibitor (PAI)-1 and -2 are the endogenous inhibitors of tPA.
FIGURE 5.

Crystal structure of apoNR2BATD. A, the strand representation of the crystal structure of apoNR2BATD (PDB code 3JPW) (47), which consists of two domains, R1 (light pink) and R2 (light purple) was created with Protein Workshop (60). Arg67 (R67; residue highlighted in red with balls and sticks) is situated at the tip of R1 (light pink) and thus is proposed to be exposed to the surrounding aqueous milieu. Left, shown is the front view of the NR2BATD crystal structure. Right, shown is a view of the crystal structure when the protein molecule is rotated as denoted. B, several structures that may be critical for the tertiary structure of the ATD upon the removal of Gln29–Arg67 are highlighted. The tPA cleavage site Arg67 (R67) is highlighted in red; Gly36–Val42 fragment (β1 sheet), Val68–Met73 (β2 sheet), and Val97–Asp101 (β3 sheet) are highlighted in dull yellow; Pro78–Asp91 (α1 helix) and Leu289–His311 (α8 helix) are highlighted in light green. The hypervariable loop (HVL) is highlighted in dark purple, and the two cysteine residues, Cys86 in α1 and Cys321 in HVL (highlighted in orange with balls and sticks), form a disulfide bond that helps to stabilize the ATD structure. The peptide upstream of Val68 can dissociate from the receptor after tPA treatment.
Mutations of critical amino acid residues residing between Thr76 and Val262 of the NR2B ATD have been shown to shift ifenprodil IC50 by more than 10-fold (44, 49). Moreover, truncation of the whole ATD of NR2B leads to an increase in the ifenprodil IC50 by up to 900-fold compared with the wild-type NR2B (50, 51). Our current electrophysiological functional data confirms that NR1/NR2B-ΔATD-R67 receptors exhibit statistically significantly lower sensitivity to ifenprodil-mediated inhibition compared with NR1/NR2B receptors. Noteworthy, Perin-Dureau et al. (44) had identified that the V42A mutation, which is proximal to the tPA cleavage site Arg67, led to an intermediate effect on ifenprodil-mediated inhibition (∼8-fold increase in IC50). The crystal structure of NR2BATD revealed that Gly36–Val42 portion of the tPA-cleaved fragment constitutes the β1 sheet, which is stacked in between β2 (Val68–Met73) and β3 (Val97–Asp101) sheets and flanked by α1 (Pro78–Asp91) and α8 helices (Leu289–His311) (47) (Fig. 5B). Although the R1 domain is partially organized by the Cys86 and Cys321 disulfide bond, the large change in the IC50 of ifenprodil upon removal of the peptide fragment proximal to Val68 suggests that the deletion of residues proximal to Val68 upon cleavage by tPA not only removes an important residue Val42 influencing ifenprodil sensitivity but may also disrupt the tertiary structure of NR2B-ATD. Such disruption may be significant enough to disrupt the ifenprodil sensitivity. However, it should be noted that upon cleavage at the ATD by tPA, it may also be possible that the cleaved fragment up to residue Arg67 may remain loosely associated with the tertiary receptor through various indirect attraction forces (e.g. van der Waals and hydrogen bonds).
In addition to affecting ifenprodil sensitivity, our electrophysiological results showed that NR2B-ΔATD-R67 containing NMDA receptors also resulted in decreased glycine affinity with a ∼5-fold increase in the EC50 of glycine as compared with NR1/NR2BWT receptors. On the contrary, no change in glutamate potency was observed. Although the binding site for the co-agonist glycine is located in the ligand binding domain of the NR1 subunit, the affinity of NMDA receptors for glycine depends on the identity of the NR2 subunit (52). In addition, the ATDs of the NR1 and NR2 subunits had been shown to play a critical role in NMDA receptor function (50, 53, 54). The removal of either the NR1-ATD or the NR2B-ATD will result in reduced glycine affinity, corresponding to a ∼17- and ∼5-fold reduction in glycine EC50, respectively (50, 54). Our results thus suggest that the amino acid residues proximal to Val68 of the ATD of NR2B may harbor potential molecular determinants that can allosterically affect glycine potency of NR2B-containing NMDA receptors.
The consequences of how a decrease in glycine sensitivity may affect NMDA receptor function in vivo are unclear. Endogenous glycine acts as a co-agonist on the glycine site situated in the ligand binding domain of the NR1 subunit. Together with the agonist glutamate, they activate NMDA receptors and allow for normal functioning of the NMDA receptors in various physiological processes such as synaptic transmission and synaptic plasticity (55, 56). Despite the high extracellular concentration of glycine found in the brain (with respect to the EC50 of glycine), various studies (both in vitro and in vivo) have shown that the glycine site in NMDA receptors are not saturated (for review, see Ref. 52). Thus, the non-saturation of the glycine sites coupled with a decrease in glycine potency on cleaved-NR2B-containing NMDA receptors could lead to lower levels of synaptic activity or hypofunction of the NMDA receptor. As the activation of NMDA receptors is critical during NMDA receptor-dependent long term potentiation (57), reduced NMDA receptor function due to tPA cleavage could potentially disrupt synaptic plasticity. This effect might be more pronounced in younger brains where NR2B-containing NMDA receptors are known to be the major NMDA receptor species (58, 59).
tPA can modulate NMDA receptor functions in many different ways, both proteolytically and non-proteolytically. However, it is difficult to decipher or understand which mode of NMDA receptor modulation by tPA is preferred in a given physiological/pathological condition and whether there is a temporal or spatial preference with respect to any particular pathways. More work will need to be done in this area to decode how tPA modulates NMDA receptor in physiological and pathophysiological situations. In summary, our current findings added NR2B subunit as a new proteolytic substrate to the few known endogenous ones that can be acted upon by tPA and augment existing knowledge on how tPA could modulate NMDA receptor functions. These results may have an impact in stroke therapy and future therapeutic molecules targeted at the amino-terminal domain of NR2B subunit.
Supplementary Material
Acknowledgments
We thank Dr. S. F. Heinemann for sharing NR1 and NR2B cDNAs. We are grateful to Dr. S. F. Traynelis for critical discussions and reading of the manuscript and L.-H. Lim, N. Anthony, and J.-T. Chen for contributions to initial recombinant protein work. We also thank Jeyapriya R. Sundaram and Noor Hazim Bin Sulaimee for technical assistance. We thank the National University Hospital-National University of Singapore Medical Publications Support Unit, Singapore, for assistance in the preparation of this article.
This work was supported by the Academic Research Fund Committee Ministry of Education, Singapore (to P. T.-H. W. and C.-M. L.), National University of Singapore Academic Research Funds, National Medical Research Council, and Biomedical Research Council grants (to C.-M. L).

This article contains supplemental Figs. 1–4.
K.-S. Ng, H.-W. Leung, P. T.-H. Wong, and C.-M. Low, unpublished data.
- tPA
- tissue plasminogen activator
- ATD
- amino-terminal domain
- NMDA
- N-methyl-d-aspartate
- MBP
- maltose-binding protein
- ANOVA
- analysis of variance.
REFERENCES
- 1. Vassalli J. D., Sappino A. P., Belin D. (1991) The plasminogen activator/plasmin system. J. Clin. Invest. 88, 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Collen D. (1999) The plasminogen (fibrinolytic) system. Thromb. Haemost. 82, 259–270 [PubMed] [Google Scholar]
- 3. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group (1995) Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N. Engl. J. Med. 333, 1581–1587 [DOI] [PubMed] [Google Scholar]
- 4. Adams H. P., Jr., del Zoppo G., Alberts M. J., Bhatt D. L., Brass L., Furlan A., Grubb R. L., Higashida R. T., Jauch E. C., Kidwell C., Lyden P. D., Morgenstern L. B., Qureshi A. I., Rosenwasser R. H., Scott P. A., Wijdicks E. F., American Heart Association, American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups (2007) Guidelines for the early management of adults with ischemic stroke. A guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups. The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke 38, 1655–1711 [DOI] [PubMed] [Google Scholar]
- 5. Goldstein L. B. (2007) Acute ischemic stroke treatment in 2007. Circulation 116, 1504–1514 [DOI] [PubMed] [Google Scholar]
- 6. Meyers P. M., Schumacher H. C., Higashida R. T., Barnwell S. L., Creager M. A., Gupta R., McDougall C. G., Pandey D. K., Sacks D., Wechsler L. R. (2009) Indications for the performance of intracranial endovascular neurointerventional procedures. A scientific statement from the American Heart Association Council on Cardiovascular Radiology and Intervention, Stroke Council, Council on Cardiovascular Surgery and Anesthesia, Interdisciplinary Council on Peripheral Vascular Disease, and Interdisciplinary Council on Quality of Care and Outcomes Research. Circulation 119, 2235–2249 [DOI] [PubMed] [Google Scholar]
- 7. Adibhatla R. M., Hatcher J. F. (2008) Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke. Therapeutic strategies. CNS Neurol. Disord. Drug Targets 7, 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sappino A. P., Madani R., Huarte J., Belin D., Kiss J. Z., Wohlwend A., Vassalli J. D. (1993) Extracellular proteolysis in the adult murine brain. J. Clin. Invest. 92, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salles F. J., Strickland S. (2002) Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J. Neurosci. 22, 2125–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsirka S. E., Rogove A. D., Bugge T. H., Degen J. L., Strickland S. (1997) An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J. Neurosci. 17, 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benchenane K., López-Atalaya J. P., Fernández-Monreal M., Touzani O., Vivien D. (2004) Equivocal roles of tissue-type plasminogen activator in stroke-induced injury. Trends Neurosci. 27, 155–160 [DOI] [PubMed] [Google Scholar]
- 12. Gualandris A., Jones T. E., Strickland S., Tsirka S. E. (1996) Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J. Neurosci. 16, 2220–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicole O., Docagne F., Ali C., Margaill I., Carmeliet P., MacKenzie E. T., Vivien D., Buisson A. (2001) The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat. Med. 7, 59–64 [DOI] [PubMed] [Google Scholar]
- 14. Bruno M. A., Cuello A. C. (2006) Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 6735–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samson A. L., Medcalf R. L. (2006) Tissue-type plasminogen activator. A multifaceted modulator of neurotransmission and synaptic plasticity. Neuron 50, 673–678 [DOI] [PubMed] [Google Scholar]
- 16. Seeds N. W., Siconolfi L. B., Haffke S. P. (1997) Neuronal extracellular proteases facilitate cell migration, axonal growth, and pathfinding. Cell Tissue Res. 290, 367–370 [DOI] [PubMed] [Google Scholar]
- 17. Seeds N. W., Williams B. L., Bickford P. C. (1995) Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science 270, 1992–1994 [DOI] [PubMed] [Google Scholar]
- 18. Minor K., Phillips J., Seeds N. W. (2009) Tissue plasminogen activator promotes axonal outgrowth on CNS myelin after conditioned injury. J. Neurochem. 109, 706–715 [DOI] [PubMed] [Google Scholar]
- 19. Zou T., Ling C., Xiao Y., Tao X., Ma D., Chen Z. L., Strickland S., Song H. (2006) Exogenous tissue plasminogen activator enhances peripheral nerve regeneration and functional recovery after injury in mice. J. Neuropathol. Exp. Neurol. 65, 78–86 [DOI] [PubMed] [Google Scholar]
- 20. Madani R., Hulo S., Toni N., Madani H., Steimer T., Muller D., Vassalli J. D. (1999) Enhanced hippocampal long term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 18, 3007–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pang P. T., Teng H. K., Zaitsev E., Woo N. T., Sakata K., Zhen S., Teng K. K., Yung W. H., Hempstead B. L., Lu B. (2004) Cleavage of proBDNF by tPA/plasmin is essential for long term hippocampal plasticity. Science 306, 487–491 [DOI] [PubMed] [Google Scholar]
- 22. Wang Y. F., Tsirka S. E., Strickland S., Stieg P. E., Soriano S. G., Lipton S. A. (1998) Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat. Med. 4, 228–231 [DOI] [PubMed] [Google Scholar]
- 23. Yepes M., Roussel B. D., Ali C., Vivien D. (2009) Tissue-type plasminogen activator in the ischemic brain. More than a thrombolytic. Trends Neurosci. 32, 48–55 [DOI] [PubMed] [Google Scholar]
- 24. Nagy Z., Kolev K., Csonka E., Vastag M., Machovich R. (1998) Perturbation of the integrity of the blood-brain barrier by fibrinolytic enzymes. Blood Coagul. Fibrinolysis 9, 471–478 [DOI] [PubMed] [Google Scholar]
- 25. Gingrich M. B., Traynelis S. F. (2000) Serine proteases and brain damage. Is there a link? Trends Neurosci. 23, 399–407 [DOI] [PubMed] [Google Scholar]
- 26. Fujimoto S., Katsuki H., Ohnishi M., Takagi M., Kume T., Akaike A. (2008) Plasminogen potentiates thrombin cytotoxicity and contributes to pathology of intracerebral hemorrhage in rats. J. Cereb. Blood Flow Metab. 28, 506–515 [DOI] [PubMed] [Google Scholar]
- 27. Benchenane K., Berezowski V., Ali C., Fernández-Monreal M., López-Atalaya J. P., Brillault J., Chuquet J., Nouvelot A., MacKenzie E. T., Bu G., Cecchelli R., Touzani O., Vivien D. (2005) Tissue-type plasminogen activator crosses the intact blood-brain barrier by low density lipoprotein receptor-related protein-mediated transcytosis. Circulation 111, 2241–2249 [DOI] [PubMed] [Google Scholar]
- 28. Fernández-Monreal M., López-Atalaya J. P., Benchenane K., Cacquevel M., Dulin F., Le Caer J. P., Rossier J., Jarrige A. C., Mackenzie E. T., Colloc'h N., Ali C., Vivien D. (2004) Arginine 260 of the amino-terminal domain of NR1 subunit is critical for tissue-type plasminogen activator-mediated enhancement of N-methyl-d-aspartate receptor signaling. J. Biol. Chem. 279, 50850–50856 [DOI] [PubMed] [Google Scholar]
- 29. Medina M. G., Ledesma M. D., Domínguez J. E., Medina M., Zafra D., Alameda F., Dotti C. G., Navarro P. (2005) Tissue plasminogen activator mediates amyloid-induced neurotoxicity via Erk1/2 activation. EMBO J. 24, 1706–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baron A., Montagne A., Cassé F., Launay S., Maubert E., Ali C., Vivien D. (2010) NR2D-containing NMDA receptors mediate tissue plasminogen activator-promoted neuronal excitotoxicity. Cell Death Differ. 17, 860–871 [DOI] [PubMed] [Google Scholar]
- 31. Pawlak R., Melchor J. P., Matys T., Skrzypiec A. E., Strickland S. (2005) Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc. Natl. Acad. Sci. U.S.A. 102, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Norris E. H., Strickland S. (2007) Modulation of NR2B-regulated contextual fear in the hippocampus by the tissue plasminogen activator system. Proc. Natl. Acad. Sci. U.S.A. 104, 13473–13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin A. M., Kuhlmann C., Trossbach S., Jaeger S., Waldron E., Roebroek A., Luhmann H. J., Laatsch A., Weggen S., Lessmann V., Pietrzik C. U. (2008) The functional role of the second NPXY motif of the LRP1 β-chain in tissue-type plasminogen activator-mediated activation of N-methyl-d-aspartate receptors. J. Biol. Chem. 283, 12004–12013 [DOI] [PubMed] [Google Scholar]
- 34. Samson A. L., Nevin S. T., Croucher D., Niego B., Daniel P. B., Weiss T. W., Moreno E., Monard D., Lawrence D. A., Medcalf R. L. (2008) Tissue-type plasminogen activator requires a co-receptor to enhance NMDA receptor function. J. Neurochem. 107, 1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wee X. K., Ng K. S., Leung H. W., Cheong Y. P., Kong K. H., Ng F. M., Soh W., Lam Y., Low C. M. (2010) Mapping the high affinity binding domain of 5-substituted benzimidazoles to the proximal N terminus of the GluN2B subunit of the NMDA receptor. Br. J. Pharmacol. 159, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ng F. M., Soh W., Geballe M. T., Low C. M. (2007) Improving solubility of NR2B amino-terminal domain of N-methyl-d-aspartate receptor expressed in Escherichia coli. Biochem. Biophys. Res. Commun. 362, 69–74 [DOI] [PubMed] [Google Scholar]
- 37. Yuan H., Vance K. M., Junge C. E., Geballe M. T., Snyder J. P., Hepler J. R., Yepes M., Low C. M., Traynelis S. F. (2009) The serine protease plasmin cleaves the amino-terminal domain of the NR2A subunit to relieve zinc inhibition of the N-methyl-d-aspartate receptors. J. Biol. Chem. 284, 12862–12873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ng F. M., Geballe M. T, Snyder J. P., Traynelis S. F., Low C. M. (2008) Structural insights into phenylethanolamines high affinity binding site in NR2B from binding and molecular modeling studies. Mol. Brain 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Basham M. E., Seeds N. W. (2001) Plasminogen expression in the neonatal and adult mouse brain. J. Neurochem. 77, 318–325 [DOI] [PubMed] [Google Scholar]
- 40. Dong Y. N., Wu H. Y., Hsu F. C., Coulter D. A., Lynch D. R. (2006) Developmental and cell-selective variations in N-methyl-d-aspartate receptor degradation by calpain. J. Neurochem. 99, 206–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gingrich M. B., Junge C. E., Lyuboslavsky P., Traynelis S. F. (2000) Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 20, 4582–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guttmann R. P., Baker D. L., Seifert K. M., Cohen A. S., Coulter D. A., Lynch D. R. (2001) Specific proteolysis of the NR2 subunit at multiple sites by calpain. J. Neurochem. 78, 1083–1093 [DOI] [PubMed] [Google Scholar]
- 43. Szklarczyk A., Ewaleifoh O., Beique J. C., Wang Y., Knorr D., Haughey N., Malpica T., Mattson M. P., Huganir R., Conant K. (2008) MMP-7 cleaves the NR1 NMDA receptor subunit and modifies NMDA receptor function. FASEB J. 22, 3757–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perin-Dureau F., Rachline J., Neyton J., Paoletti P. (2002) Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J. Neurosci. 22, 5955–5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pauly T., Ratliff M., Pietrowski E., Neugebauer R., Schlicksupp A., Kirsch J., Kuhse J. (2008) Activity-dependent shedding of the NMDA receptor glycine binding site by matrix metalloproteinase 3. A PUTATIVE mechanism of postsynaptic plasticity. PLoS ONE 3, e2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Butenas S., Kalafatis M., Mann K. G. (1997) Analysis of tissue plasminogen activator specificity using peptidyl fluorogenic substrates. Biochemistry 36, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 47. Karakas E., Simorowski N., Furukawa H. (2009) Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 28, 3910–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karakas E., Simorowski N., Furukawa H. (2011) Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mony L., Kew J. N., Gunthorpe M. J., Paoletti P. (2009) Allosteric modulators of NR2B-containing NMDA receptors. Molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 157, 1301–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Madry C., Mesic I., Betz H., Laube B. (2007) The N-terminal domains of both NR1 and NR2 subunits determine allosteric Zn2+ inhibition and glycine affinity of N-methyl-d-aspartate receptors. Mol. Pharmacol. 72, 1535–1544 [DOI] [PubMed] [Google Scholar]
- 51. Mony L., Krzaczkowski L., Leonetti M., Le Goff A., Alarcon K., Neyton J., Bertrand H. O., Acher F., Paoletti P. (2009) Structural basis of NR2B-selective antagonist recognition by N-methyl-d-aspartate receptors. Mol. Pharmacol. 75, 60–74 [DOI] [PubMed] [Google Scholar]
- 52. Yang C. R., Svensson K. A. (2008) Allosteric modulation of NMDA receptor via elevation of brain glycine and D-serine. The therapeutic potentials for schizophrenia. Pharmacol. Ther. 120, 317–332 [DOI] [PubMed] [Google Scholar]
- 53. Gielen M., Siegler Retchless B., Mony L., Johnson J. W., Paoletti P. (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459, 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yuan H., Hansen K. B., Vance K. M., Ogden K. K., Traynelis S. F. (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 29, 12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dingledine R., Borges K., Bowie D., Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–62 [PubMed] [Google Scholar]
- 56. Papadia S., Soriano F. X., Léveillé F., Martel M. A., Dakin K. A., Hansen H. H., Kaindl A., Sifringer M., Fowler J., Stefovska V., McKenzie G., Craigon M., Corriveau R., Ghazal P., Horsburgh K., Yankner B. A., Wyllie D. J., Ikonomidou C., Hardingham G. E. (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 11, 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhuo M. (2009) Plasticity of NMDA receptor NR2B subunit in memory and chronic pain. Mol. Brain 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kew J. N., Richards J. G., Mutel V., Kemp J. A. (1998) Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J. Neurosci. 18, 1935–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X. B., Murray K. D., Jones E. G. (2004) Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J. Neurosci. 24, 8885–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moreland J. L., Gramada A., Buzko O. V., Zhang Q., Bourne P. E. (2005) The Molecular Biology Toolkit (MBT). A modular platform for developing molecular visualization applications. BMC Bioinformatics 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.