

Background: Adenylyl cyclase 8 (AC8) expression is associated with pathological vascular smooth muscle cell (VSMC) trans-differentiation.
Results: The Notch-HRT1/3 pathway inhibits IL1β up-regulation of AC8 in VSMCs.
Conclusion: Notch3 pathway down-regulation (in the inflammatory context of pathological vascular remodeling) is a major event for sparking AC8 expression.
Significance: AC8 ranked as a marker of trans-differentiated VSMCs contributes to understanding the mechanisms involved in atherosclerosis and postangioplasty restenosis.
Keywords: Adenylate Cyclase, Atherosclerosis, Camp, Cardiovascular Disease, Vascular Smooth Muscle Cells, AC8, Notch Signaling Pathway, VSMC Trans-differentiation, Pathological Vascular Remodeling
Abstract
Vascular smooth muscle cell (VSMC) trans-differentiation, or their switch from a contractile/quiescent to a secretory/inflammatory/migratory state, is known to play an important role in pathological vascular remodeling including atherosclerosis and postangioplasty restenosis. Several reports have established the Notch pathway as tightly regulating VSMC response to various stress factors through growth, migration, apoptosis, and de-differentiation. More recently, we showed that alterations of the Notch pathway also govern VSMC acquisition of the inflammatory state, one of the major events accelerating atherosclerosis. We also evidenced that the inflammatory context of atherosclerosis triggers a de novo expression of adenylyl cyclase isoform 8 (AC8), associated with the properties developed by trans-differentiated VSMCs. As an initial approach to understanding the regulation of AC8 expression, we examined the role of the Notch pathway. Here we show that inhibiting the Notch pathway enhances the effect of IL1β on AC8 expression, amplifies its deleterious effects on the VSMC trans-differentiated phenotype, and decreases Notch target genes Hrt1 and Hrt3. Conversely, Notch activation resulted in blocking AC8 expression and up-regulated Hrt1 and Hrt3 expression. Furthermore, overexpressing Hrt1 and Hrt3 significantly decreased IL1β-induced AC8 expression. In agreement with these in vitro findings, the in vivo rat carotid balloon-injury model of restenosis evidenced that AC8 de novo expression coincided with down-regulation of the Notch3 pathway. These results, demonstrating that the Notch pathway attenuates IL1β-mediated AC8 up-regulation in trans-differentiated VSMCs, suggest that AC8 expression, besides being induced by the proinflammatory cytokine IL1β, is also dependent on down-regulation of the Notch pathway occurring in an inflammatory context.
Introduction
Atherosclerosis, a chronic disease of the arterial wall, and related cardiovascular incidents, are the number one causes of death throughout the world (World Health Organization). Trans-differentiation of vascular smooth muscle cells (VSMC),3 or their switch from a contractile/quiescent to a secretory/proliferative state, is known to play an important role in the development of atherosclerotic lesion development and/or postangioplasty neointimal formation. Therefore, many efforts are being made to determine the molecular mechanisms that are engaged in this process.
Over the past decade, several studies identified Notch as a critical player in controlling cell trans-differentiation by regulating intercellular communications and direct cell fate decisions (1, 2). In mammals, four Notch proteins have been discovered (Notch1–4), each being a large single-pass type 1 transmembrane receptor. Currently, five membrane-bound Notch ligands are known (Delta-like 1, 3, 4, and Jagged 1–2). Upon ligand binding, proteolytic cleavages (which involve extracellular proteases of ADAM/TACE, (a disintegrin and metalloprotease/tumor necrosis factor α converting enzyme, and the γ-secretase complex) trigger the release of the Notch intracellular domain (NICD) from the plasma membrane. Once translocated to the nucleus, NICD generally interacts with a transcriptional repressor called RBP-Jκ (recombinant signal-binding protein-1 for J-κ, also known as C promoter-binding factor 1 suppressor of hairless, Lag-1), which results in de-repression/activation of Notch-RBP-Jκ target genes (3). They include the transcriptional repressor factors HES (Hairy and Enhancer of Split) and HRT (Hairy Related Transcription factor), which both belong to the family of basal helix-loop-helix proteins. In mammals seven Hes (Hes1–Hes7) and three HRT (HRT1–HRT3, also known as Hey1,2,L, Hesr1,2,3, Herp1,2,3) genes have been described; HES1, HES5, HES7, and all three HRT proteins can be induced by the Notch pathway but HES2, HES3, and HES6 are seemingly independent of Notch signaling (4).
The Notch pathway regulates some of the new features acquired by VSMCs in pathological vessels (for review, see Ref. 5). In particular, Notch1 and -3 regulate cell fate in VSMCs and control their growth and migration in response to growth factor stimulation following vascular injury (6, 7); Notch3 also promotes resistance to apoptosis of intimal smooth muscle cells, which is a prominent feature of the response to injury, and regulates the consequent formation of the neointima (6, 8); Notch signaling represses myocardin-dependent smooth muscle cell differentiation and maintenance of the contractile phenotype of VSMC (9). In addition, we showed that Notch3 and IL1β pathways exert opposite effects on the organization of the contractile apparatus of VSMCs and their transition toward an inflammatory/migratory state through the activation of NF-κB (10, 11). Consistently, cross-talk between the NF-κB and Notch pathways has been also shown to regulate the balance between VSMC proliferation and differentiation (12, 13).
Transformation of VSMCs into inflammatory/migratory cells results in part on the up-regulation of calcium/calmodulin positively regulated adenylyl cyclase isoform 8 (AC8) expression (14, 15). Indeed, AC8 emergence allows maximal secretion of phospholipase A2 type IIA, an inflammatory marker associated with an increased risk of coronary artery disease (14, 16); overexpression of AC8 in rat VSMCs triggered the re-colonization of the wounded zone, whereas blocking AC8 in IL1β-treated cells stopped the IL1β-induced migration. The cause to effect relationship between AC8 expression and the migratory (but not proliferative) response of the VSMCs translated, in the rat in vivo model of postangioplasty restenosis, into a pronounced transitory up-regulation of AC8 expression in VSMCs migrating to form the intimal layer (15). Of note: in mammals nine AC isoforms have been identified and characterized, each having a discrete tissue distribution and a specific pattern of regulation by G protein subunits, calcium-calmodulin and protein kinases (17); out of any inflammatory context, VSMCs mainly express AC isoforms 3, 5, and 6 (18), which, although all producing cAMP, have different functions and distinct membrane localizations. Here, we examine the role of the Notch pathway in regulating AC8 gene expression and provide evidence that the Notch pathway attenuates adenylyl cyclase 8 up-regulation, observed in the inflammatory state of trans-differentiated VSMCs.
EXPERIMENTAL PROCEDURES
Ethical Approval
All procedures were performed in accordance with European Community standards on the care and use of laboratory animals approved by the ethics committee for animal experimentation (Ile de France-Paris-Committee 3, Authorization 4270).
Balloon Injury of the Rat Carotid Artery
Adult male Wistar rats (Charles River, MA) weighing 400 g were anesthetized with pentobarbital (50 mg/kg intraperitoneally). Heparin (35 IU) was administered systemically by intraperitoneal injection. The left external carotid artery was injured using a 2F Fogarty embolectomy catheter (Baxter Healthcare Corp.) introduced into the common carotid artery through the external carotid, inflated to 2 atmospheres and withdrawn 3 times. The catheter was removed and the incision hole was ligated. Perfusion was restored in the common carotid and the neck incision was closed using 4-0 silk sutures. Carotids were collected 7 and 14 days postinjury, (after intraperitoneal injection of a lethal dose of pentobarbital) included in cryomatrix (OCT), and frozen at −80 °C. 12-μm Cross-sections were made from the entire length of the carotid and used for immunohistochemistry analysis.
Immunohistochemistry and Confocal Analysis
Tissue sections were fixed with acetone for 10 min at −20 °C. After blocking the nonspecific binding sites with normal goat serum, the tissue sections were incubated 1 h at 37 °C with rabbit anti-AC8 (Santa Cruz Biotechnology), rabbit anti-Notch3 (Abcam), mouse anti-PECAM1 (Chemicon International); rabbit anti-SM22 (Abcam); followed by incubation with the secondary antibody, Dylight 549 donkey anti-rabbit (Jackson ImmunoResearch), and Alexa 546 (Abcam). Tissue sections were examined with a Leica TCS4D confocal scanning laser microscope using Plan Apochromat objectives. Stacks of images were collected every 0.5 μm along the z axis. All settings were kept constant to allow comparison.
Immunocytochemistry
VSMCs were seeded directly onto glass coverslips placed in 12-well culture plates. After treatment (indicated in figure legends), cells were washed twice with PBS and fixed during 20 min at room temperature in 4% paraformaldehyde (neutralized with 50 mm of NH4Cl for 10 min), permeabilized in PBS + 0.2% Triton X-100, and incubated for 1 h in PBS + 10% FCS with mouse monoclonal α-actin antibody (DAKO) and rabbit polyclonal SM22 antibody (Abcam). Incubation with DyLight 488 goat anti-mouse secondary antibody and DyLight 546 donkey anti-rabbit secondary antibody was performed during 1 h followed by staining of cell nuclei with DAPI for 5 min. Coverslips were mounted with Dako fluorescent mounting medium (Dako, Carpinteria, CA). Cells were examined by confocal microscopy.
Cell Culture and Treatment Conditions
All procedures were performed in accordance with the European Community Standards on the care and use of laboratory animals and conform to NIH Guidelines. Aortas were isolated from anesthetized 5-week-old male Wistar rats (180 g). After removing the fat tissue the aortas were subjected to enzymatic digestion at 37 °C under gentle shaking in Dulbecco's modified Eagle's medium (DMEM) containing glucose (4.5 g/liter) and a mixture of type I collagenase 45 min. The VSMC-rich media was separated from the adventitial layer with forceps, dilacerated in small rings, and re-incubated in the same mixture with elastase (1.5 g/liter) for 30–45 min to dissociate VSMCs. Cells were then collected by centrifugation (3 min at 500 × g) and re-suspended in complete medium containing DMEM supplemented with 10% fetal calf serum, glutamine (4 mm), penicillin (100 units/ml), and streptomycin (100 μg/ml), grown on calf skin type 1 collagen-coated dishes at 37 °C in a 5% CO2 environment. VSMCs were subcultured every 5 days in the same medium. All experiments were performed on cells at passages ranging from 2 to 6. Immunochemistry experiments visualizing α-actin show that among rat aorta vascular cell types, plated cells always display a well organized network of actin stress fibers in control untreated conditions. Twenty-four hours before any treatment, confluent cells were starved and experiments were continued in serum-free culture medium. Control cells were harvested at the same time as treated cells to prevent any effect of starvation. All interleukin-1β (IL1β) incubations were performed with human IL1β (PeproTech) dissolved in 2.5 μm BSA (bovine serum albumin, Sigma). Therefore, all incubations were performed in the presence of 2.5 μm BSA. When treated with Jagged1-Fc ((rat jagged 1, Ser32–Asp1068 (Gly57–Arg59del, D63T, R64L, and Val-Arg-P6Yins before Lys69) IEGMRD-Human IgG1 (Pro100–Lys330)-His6 tag; truncated at its C terminus and lacks the transmembrane as well as the intracellular domain), the ligand was clusterized with goat anti-human Fc antibodies for 1 h prior to IL1β treatment. N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) was dissolved in dimethyl sulfoxide (Sigma). The final concentration of dimethyl sulfoxide (0.01%) did not have any effect (as compared with control untreated cells) on the measured parameters (data not shown).
Adenoviral Infection
Rat VSMCs were infected with adenovirus expressing Ad-βGal, encoding β-galactosidase-GFP under the CMV promoter (19) Notch1 ICD-IRES-EGFP or Notch3 ICD-IRES-EGFP (20) (Ad-βgal, Ad-Notch1 ICD, Ad-Notch3 ICD, respectively) at a multiplicity of infection of 10 in complete medium for 36 h prior to serum starvation and subsequent treatment as indicated in the figure legends. Infection efficiency was controlled by counting GFP versus non-GFP positive cells.
RT-PCR
Total RNA of rat VSMCs and rat carotid arteries was extracted from VSMCs using the RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. After annealing oligo(dT) (1 μm) to template RNAs (0.5 μg) at 70 °C for 5 min, primer extension was initiated by adding the RT-Moloney murine leukemia virus enzyme plus 0.5 mm dNTP, 1 unit of RNAsin, and 10 mm dithiothreitol, and carried out for 45 min at 37 °C. Quantitative PCR was performed using the LightCycler LC480 (Roche Diagnostics). The PCR mixture included 5 μl of each reverse transcriptase (diluted 1:25) and 300 nm of each primer in 1× LightCycler DNA SYBR Green 1 Master Mix. Specific primers for complementary DNA (cDNA) were chosen with the LightCycler Probe Design2 program according to European Molecular Biology Laboratory accession numbers. The forward and reverse primers used to selectively amplify the cDNA encoding rat hypoxanthine phosphoribosyltransferase, AC3, AC6, AC8, HRT1–3, HES1, HES5, Notch1, Notch3, RBP-Jκ, α-actin, and SM22 are presented in Table 1. The PCRs were performed using the following thermal settings: denaturation and enzyme activation at 95 °C for 8 min, with cycling at 95 °C for 10 s, 64 °C for 10 s, and 72 °C for 8 s. Amplification was followed up online, and the PCRs were stopped after the logarithmic phase. Melting curve analyses were performed after PCR to check the reaction specificity. Controls and water blanks were included in each run; these were negative in all cases. Real-time quantitative PCR data were represented by the amount of each target messenger RNA (mRNA) relative to the amount of mRNA for a housekeeping reference gene, here, hypoxanthine phosphoribosyltransferase, estimated in the logarithmic phase of the PCR. Serial dilutions of reverse transcription products were used to determine the fit coefficients of the relative standard curve. When the PCR efficiencies of the targets were similar, individual cultures could be compared. If not, an internal calibrator was used to compare individual cultures. Results are expressed as fold over control (untreated) cells.
TABLE 1.
PCR primer sequences
| Target gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| HPRT | aggacctctcgaagtgt | atccctgaagtgctcattata |
| AC8 | attgcctcagtggtggtgacta | caaactcctcgggct |
| AC3 | ccctcctgtctagttaagtcaatg | attgcactttctgctctc |
| AC6 | agaagtattcacggaaagtagac | cggcataaatcccgagtatc |
| HES1 | gacggccaatttgctttc | gacactgcgttaggaccc |
| HES5 | accgcatcaacagcagcatt | aggctttgctgtgcttcaggt |
| HRT1 | gaagcgccgacgagaccgaatcaa | cagggcgtgcgcgtcaaaataacc |
| HRT2 | cgacgtggggagcgagaacaat | ggcaagagcatgggcatcaaagta |
| HRT3 | agaccgcatcaacagca | caagtgatccacggtcat |
| RBP-Jκ | atccattatggacagactgtca | atccagtaatgctgtctgc |
| α-Actin | acccagattatgtttgagacc | cagagtccagcacaatacca |
| SM22 | tatggcagcagtgcagag | ctttcttcataaaccagttggga |
Transfection
Smooth muscle cells were transfected by electroporation in the Amaxa electroporation device using the D-33 program. Briefly, 1 million cells were re-suspended in 100 μl of Amaxa electroporation transfection solution and 4 μg of each vector (RBP-Jκ-DN, IκBα(32–36), HRT1-myc, HRT2-myc, HRT3-myc) were added. After electroporation, transfected cells were plated in two wells of a six-well plate, each containing 1 ml of cell culture medium. 24 h after plasmid transfection, the cells were starved overnight in serum-free medium then treated as indicated in the figure legends.
Protein Extraction and Western Blot Analysis
Cells were washed with ice-cold phosphate-buffered saline and re-suspended in lysis buffer (20 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1% Triton, 1% sodium deoxycholate) plus Complete protease inhibitor mixture (Roche Diagnostics) for 10 min on ice. The lysate was centrifuged at 13,000 × g for 10 min at 4 °C. Cell lysates (20–40 μg) were analyzed by SDS-PAGE on a 12% resolving gel followed by transfer to nitrocellulose membranes (NuPAGE system). The free binding sites on membranes were blocked with Tris-buffered saline containing 0.1% Tween 20 (TBST) and 5% fat-free milk for 1 h at room temperature. The membranes were then incubated overnight with primary antibody c-myc (Santa Cruz) in TBST with 5% milk overnight at 4 °C. Next, the membranes were washed in TBST and incubated with horseradish peroxidase-conjugated secondary antibody (P.A.R.I.S. Ltd., France) for 1 h at room temperature followed by repeated washes with TBST. Signals were detected with the ECL detection system and exposed to Fujifilm LAS-300 (Fujifim Medical Systems).
Flow Cytometry
To detect Jagged1-Fc binding, the cells were re-suspended in PBS with 1% BSA and treated for 1 h with 1 μg/ml of Jagged1-Fc at 4 °C. Jagged1 binding was revealed by an anti-human Fc goat polyclonal antibody followed by a secondary 549 Dylight donkey anti-goat antibody (Jackson ImmunoResearch). Endogenous Jagged1 expression was examined as follows: VSMCs were starved for 24 h and treated with or without 1.5 ng/ml of IL1β in serum-free culture medium for 48 h. Subsequently the cells were re-suspended in phosphate-buffered saline (PBS) containing 1% BSA. Cells were stained with a mouse anti-Jagged1 polyclonal antibody (Santa Cruz Biotechnology) followed by 488 Dylight goat anti-mouse secondary antibody (1/400 Jackson ImmunoResearch); Delta was detected with an anti-Delta directly coupled to APC (allophycocyanine; Biolegend). In all cases, stained cells were washed with PBS containing 1% BSA and analyzed immediately on a MoFlow XL4 cytometer (Beckman-Coulter) with Submitt Software.
Statistical Analysis
Data are reported as mean ± S.E. The numbers of independent experiments are reported in the figure legends. One-way analysis of variance Tukey-Kramer Multiple Comparisons Test was used to compare the mean values between groups using GraphPad InStat (GraphPad Software, San Diego, CA).
RESULTS
Notch 3 Expression Opposes AC8 Expression
IL1β induces the trans-differentiation of VSMCs and stimulates the expression of AC8 (15). As an initial approach to determine whether there is any correlation between AC8 and Notch expressions in VSMCs, we analyzed their transcript expressions when treated with various concentrations of IL1β. As shown in Fig. 1A, IL1β-induced AC8 expression and Notch3 down-regulation are dose-dependent. Because AC8 transcript induction displayed an EC50 value of ∼1.5 ng/ml, we chose to treat VSMCs with this concentration of IL1β (which was used for all the in vitro experiments reported in this article). We next measured the kinetics of AC8 and Notch3 receptor mRNA expression over 48 h of IL1β treatment. The analysis of the kinetics revealed that AC8 mRNA expression was significant after 16 h of treatment (compared with untreated cells; see the legend to Fig. 1 for details), whereas a strong (∼60%) down-regulation of Notch3 was evident as soon as 6 h (Fig. 1B). Notch1 mRNA levels, although displaying a tendency of inhibition at 16 h, did not change significantly (data not shown); similar results were obtained for Notch2 (data not shown). Altogether, this data indicates that IL1β-induced Notch3 inhibition preceded AC8 up-regulation.
FIGURE 1.

Notch3 expression opposes AC8 expression in trans-differentiated VSMC. A, dose-response effect of IL1β on AC8 expression in VSMC. Rat VSMCs were treated with various concentrations of IL1β as indicated for 48 h. Total RNA was extracted and AC8 and Notch3 gene expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of nontreated (control) cells. Data represent the mean ± S.E. of 4 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (versus control); ##, p < 0.01 (between IL1β-treated cells). B, rat VSMCs were treated with 1.5 ng/ml of human IL1β at the indicated time points. Total RNA was extracted and AC8 and Notch3 gene expression was analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of un-treated (control) cells (data not shown) for each time point to avoid any effect of the starvation. Data represent the mean ± S.E. of 4 independent experiments. **, p < 0.01; ***, p < 0.001 (versus control); ##, p < 0.01; ###, p < 0.001 (between IL1β-treated cells).
Inhibition of the Notch Pathway Reinforces the Effect of IL1β on AC8 Expression
To determine the involvement of the Notch signaling pathway in AC8 expression, we coincubated VSMCs with IL1β (1.5 ng/ml) and/or DAPT (referred to as D), a potent inhibitor of the γ-secretase complex activity, for 48 h. (The γ-secretase complex generates the intracellular active domain of Notch, as mentioned under the Introduction.) As illustrated in Fig. 2A, Notch inhibition using DAPT (2.5 μm) doubled the effect of IL1β on AC8 mRNA induction; DAPT alone had no effect on AC8 transcript expression when compared with vehicle-treated (referred to as control, Ctl) cells. DAPT treatment did not have any effect on AC3 and AC6, which are two main isoforms of adenylyl cyclases present in VSMCs, although AC3 transcripts were decreased in IL1β-treated VSMCs (Fig. 2B). Lower (0.5 μm) or higher (10 μm) concentrations of DAPT provided similar results (data not shown). Notch inhibition by DAPT treatment was confirmed by attesting its inhibitory effect on Notch target genes expression (HRT1–3, HES1, and HES5) (Fig. 2C).
FIGURE 2.

γ-Secretase complex inhibitor DAPT reinforces the effect of IL1β on AC8 expression. Rat VSMCs were treated with 1.5 ng/ml of human IL1β and/or 2.5 μm DAPT for 48 h. Total RNA was extracted and AC8 (A), AC3 and AC6 (B), and Notch target genes HRT1, HRT2, HRT3, HES1, and HES5 (C) expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of untreated (control) cells and represent the mean ± S.E. of 4 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (versus control); ##, p < 0.01 (between IL1β-treated cells).
Apart from generating NICD, the γ-secretase complex is also responsible for intramembrane processing of over 60 substrates (21). Thus, to confirm that inhibition of the Notch signaling pathway increases IL1β-induced AC8 expression, we used the soluble human recombinant Jagged1-Fc fusion protein, reported to have an antagonistic effect on Notch signaling (22, 23) and assayed AC8 transcript expression. The main reason for choosing Jagged1 rather than Delta-like Notch ligands is that Jagged1-but not Delta-like 4-induced Notch signaling regulates VSMC differentiation (24). Jagged1-Fc binding efficiency was assessed by flow cytometry using an anti-Fc antibody on Jagged1-Fc-treated VSMC (Fig. 3A, left panel). As shown in Fig. 3A, right panel, Jagged1-Fc (1 μg/ml) treatment doubled the effect of IL1β on AC8 mRNA expression. Similar results were obtained when using a concentration of 2.5 μg/ml of Jagged1-Fc (data not shown). As expected for an antagonist, this recombinant protein significantly decreased the expression of Notch targets genes, HRT1 and -3 (Fig. 3B). Although not significant, HRT2 expression also tended to decrease, whereas HES1 and HES5 were unaffected.
FIGURE 3.

Notch pathway inhibition increases the effects of IL1β on AC8 expression. Rat VSMCs were treated with 1.5 ng/ml of IL1β and/or 1 μg/ml of clusterized Jagged1-Fc for 48 h. A, left panel, Jagged1 binding was assessed using an anti-Fc antibody and subsequently analyzed by flow cytometry (A, right panel). B, total RNA was extracted and AC8 expression and Notch target genes HRT1, HRT2, HRT3, HES1, and HES5 expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of nontreated cells and represent the mean ± S.E. of 3 independent experiments. *, p < 0.05; ***, p < 0.001 (versus control); ###, p < 0.001 (between IL1β-treated cells). C, rat VSMCs were transfected with RBP-Jκ-DN (dominant-negative, a mutated form of RBP-Jκ), IκBα(32–36) (a nonphosphorylatable form of NF-κB), or empty (mock) plasmids and treated for 48 h with IL-1β (1.5 ng/ml). Total RNA was extracted and AC8 (left panel) and RBP-Jκ (right panel) gene expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of nontreated cells and represent the mean ± S.E. of 3 (left panel) or 2 (right panel) independent experiments. *, p < 0.05 (versus control); #, p < 0.05 (between IL1β-treated cells).
Once in the nucleus, NICD interacts with transcriptional repressor RBP-Jκ (recombinant signal-binding protein 1 for J-κ, also known as C promoter binding factor 1 suppressor of hairless, Lag-1), which results in activating Notch-RBP-Jκ target genes (3). As evidenced in Fig. 3C, left panel, overexpressing RBP-Jκ dominant-negative (RBP-Jκ-DN, shown by real time PCR in Fig. 3C, right panel) increased the levels of IL1β-induced AC8 expression, similar to DAPT and Jagged1-Fc treatment (Figs. 2A and 3A, right panel). Of note, RBP-J-DN associates with Notch ICD but lacks its DNA binding capability, therefore altering the transcription of Notch target genes in response to Notch activation (25, 26).
Notch Pathway Inhibition Amplifies the Deleterious Effects of IL1β on VSMC Phenotype
To examine effects of the Notch pathway inhibition on VSMC morphology and trans-differentiation, we inhibited the Notch pathway with DAPT or Jagged1-Fc and evaluated the expression of α-actin and SM22, both markers of differentiated, contractile VSMCs. As demonstrated by RT quantitative PCR analysis, α-actin and SM22 transcripts were decreased when VSMCs were treated with IL1β (1.5 ng/ml for 48 h). This down-regulation was further enhanced when Notch was inhibited whether using DAPT (Fig. 4A) or Jagged1-Fc (Fig. 4C). As visualized by immunostaining in Fig. 4, B and D, the intensity and number of cells positive for α-actin and SM22 labeling were much lower in cells treated with Notch inhibitors (DAPT or Jagged1-Fc) and IL1β versus IL1β alone after 5 days of treatment, demonstrating that Notch inhibition exacerbates IL1βs effect on VSMC trans-differentiation. When observing cell morphology at a higher magnification (×63), α-Actin and SM22 networks are less dense, disorganized, and display a tangled arrangement in IL1β + DAPT or Jagged1-Fc-treated cells that still expressed these markers (supplemental Fig. S4). Altogether these experiments clearly demonstrate that morphology and differentiation markers (α-actin and SM22) are drastically modified along with AC8 mRNA potentiation (Figs. 2A and 3A, right panel) when Notch is inhibited in IL1β-treated cells.
FIGURE 4.

Inhibition of Notch pathway amplifies the deleterious effects of IL1β on VSMC phenotype. Rat VSMCs were treated for 48 h (A and C) or 5 days (B and D) with 1.5 ng/ml of human IL1β and/or with 2.5 μm DAPT (A and B) or 1 μg/ml of clusterized Jagged1-FC (C and D). A and C, rat VSMCs were treated for 48 h and total RNA was extracted. α-Actin (left panel) and SM22 (right panel) expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of untreated (control) cells and represent the mean ± S.E. of 4 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B and D, rat VSMCs were treated for 5 days and immunostaining was performed on paraformaldehyde-fixed cells using primary antibodies against α-actin and SM22 and secondary antibodies coupled to FITC (green) and Dylight 549 (red), respectively. Cell nuclei were stained with DAPI and analyzed using confocal microscopy (×40).
Notch3-ICD and/or Notch1-ICD Prevent the Effects of IL1β on AC8 Expression through HRT1 and/or HRT3
We next evaluated the effects of Notch activation on IL1β-induced AC8 expression. Thus, we infected our cell model with an adenovirus expressing either the Notch3 or the Notch1 constitutively active intracellular domains (Notch1-ICD-IRES-EGFP or Notch3-ICD-IRES-EGFP). An adenovirus expressing β-Gal was used as a control. Infection efficiency (estimated by counting GFP positive cells) was around 80% in all conditions and maintained upon IL1β treatment (data not shown). As evidenced in Fig. 5A, left panel, Notch1-ICD and Notch3-ICD blocked the induction from IL1β of AC8 by 70%. Both Notch1-ICD and Notch3-ICD induced significant up-regulation of the Notch downstream effector transcripts HRT1 and HRT3 but not HRT2, HES1, and HES5 (Fig. 5B). Although N1-ICD had no effect on AC3 and AC6 expression whether in control or IL1β-treated cells, N3-ICD infection increased the expression of both of these cyclases (supplemental Fig. S1). Of note: IL1β-induced AC3 down-regulation (Fig. 2B) is maintained in N3-ICD-infected VSMCs (supplemental Fig. S1). This set of experiments demonstrates that Notch1 and -3 activities prevent the expression of AC8 induced by IL1β.
FIGURE 5.

Overexpression of Notch1 and Notch3 ICD prevent IL1β-induced AC8 gene expression. Rat VSMCs were infected with adenoviruses (Ad) containing β-gal-EGFP, Notch1-ICD-EGFP, or Notch3-ICD-EGFP for 36 h. Cells were starved in serum-free medium for 24 h, then treated with 1.5 ng/ml of IL1β for 24 h. Total RNA was extracted and AC8 (A, left panel) and Notch target genes HRT1, HRT2, HRT3, HES1, and HES5. B, gene expression levels were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of nontreated cells and represent the mean ± S.E. of 5 independent experiments. *, p < 0.05; **, p < 0.01 (versus control); #, p < 0.05; ##, p < 0.01 (between IL1β-treated cells). A, right panel, Ad-β-gal-EGFP, Ad-Notch1-ICD-EGFP, and Ad-Notch3-ICD-EGFP protein expression of VSMCs after 36 h of infection (×20).
Regarding the systematic modifications of HRT1 and -3 gene expressions when the Notch pathway is modulated (Figs. 2C, 3B, and 5B) we next addressed the hypothesis of whether they contribute to repressing AC8 expression. To accomplish this, we overexpressed HRT1, HRT2, and HRT3 in IL1β-treated rat VSMCs and analyzed the effects on AC8 expression. Transfection efficiency was controlled by Western blot with an anti-c-myc because HRTs constructs are c-myc tagged (Fig. 6B). As shown in Fig. 6A, HRT1 and HRT3 overexpression, in contrast to HRT2, significantly decreased AC8 mRNA transcript expression. Collectively, these results suggest that in cultured VSMCs Notch1 and -3 intracellular active domains prevent the ability of IL1β to induce AC8 expression through enhancing the expression of transcriptional repressors HRT1 and/or HRT3.
FIGURE 6.
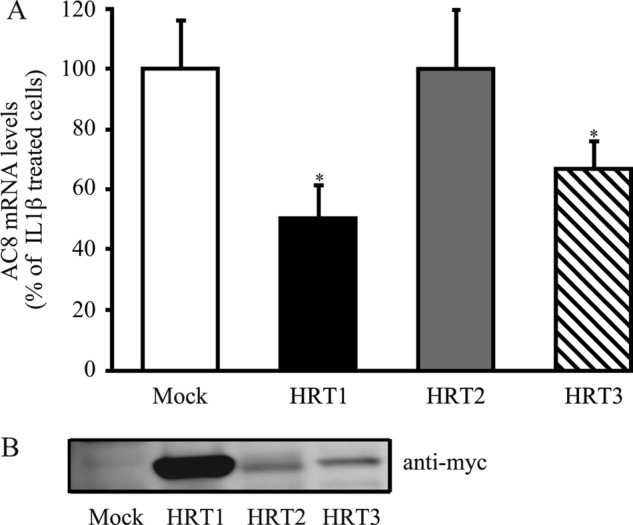
HRT1 and HRT3 overexpression decrease IL1β-induced AC8 gene expression. Rat VSMCs were transfected with empty (mock), HRT1, HRT2, or HRT3 plasmids and treated for 48 h with IL-1β (1.5 ng/ml). A, total RNA was extracted and AC8 gene expression levels were analyzed using real time PCR. Results are expressed as a percentage of IL1β-treated cells and represent the mean ± S.E. of three independent experiments. *, p < 0.05. B, HRT1, HRT2, and HRT3 protein expressions were analyzed by Western blot. Anti-myc was used to detect myc-tagged HRTs proteins.
Notch 3 and AC8 Expression Are Reciprocally Related in Vivo
As an initial approach to evaluate the concordance of this in vitro finding with in vivo VSMC remodeling toward the inflammatory/migratory/proliferating state, we used the rat carotid balloon injury model of restenosis (Fig. 7). The differentiation status of medial cells as well as their identity was attested by SM22 labeling (Fig. 7A). Efficiency of the balloon injury was attested by post-intervention endothelial denudation, visualized here with altered CD31 expression (endothelial cell marker). Immunofluorescence staining revealed the presence of Notch3 in medial/contractile VSMCs of control non-injured vessels, whereas AC8 expression was not detectable. At day 7 post-injury, expression of Notch3 and SM22, a marker of the contractile phenotype, were no longer detectable in medial or neointimal VSMCs, whereas AC8 expression was up-regulated. Fourteen days postinjury, Notch3 expression was restored in the neointima, but also among the (medial) elastin fibers. A similar tendency was observed for SM22 expression although its expression still seemed low at this time. This was consistent with the completion of neointimal formation usually occurring between 14 and 21 days postinjury when neointimal as well as medial VSMCs regain a differentiated state (15). In accordance with the in vitro findings mentioned above, AC8 expression opposed that of Notch3. In particular, one might notice that the expression of AC8 was absent in medial/differentiated VSMCs of non-injured vessels, but was highly present in the media and neointima 7 days after balloon injury and disappeared 14 days postintervention.
FIGURE 7.

Notch3 and AC8 expression are reciprocally related in vivo. A, expression of AC8, Notch3, SM22, and CD31 in rat carotid arteries after balloon injury. Confocal immunofluorescence (red) of noninjured and 7- or 14-day post-balloon injury rat carotid arteries using anti-AC8, Notch3, SM22, or CD31 primary antibodies. Autofluorescence of elastin is observed in green. The images are representative of independent experiments performed on 3 to 6 rats. a, adventitia; m, media; ni, neo-intima; L, lumen; ec, endothelial cells. B, total RNA was extracted from rat carotid arteries after balloon injury at the indicated time points; AC8 and Notch3 gene expressions were analyzed using real time PCR. Results are expressed as a fold-induction of the mRNA level of noninjured arteries. Data represent the mean ± S.E. of 3–7 rats depending on the time point. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
These observations were confirmed by real-time RT-PCR analysis of AC8 and Notch3 mRNA levels in post-balloon-injured rat carotids (analyzed from 2 to 30 days) (Fig. 7B). Indeed, Notch3 expression dramatically decreases 2 days postinjury prior to AC8 expression, which was maximal at day 4. AC8 up-regulation was maintained until 10 days postinjury and returned to basal levels at 30 days postinjury; simultaneously, relative expression of Notch3 was progressively restored during this same period. Furthermore, the expression of Notch ligands Delta1 and Jagged1, and target genes HRT1, HRT2, HRT3, and Hes1 were also down-regulated in rat carotid arteries 2 days postinjury, (supplemental Fig. S3). Therefore, whether in vitro or in vivo, the inhibition of the Notch3 signaling pathway precedes AC8 expression up-regulation and the two are reciprocally related.
Taken together, this data demonstrated that AC8 expression is dependent on Notch pathway down-regulation in inflammatory, trans-differentiating VSMCs and strongly support that AC8 gene silencing occurring in a noninflammatory context potentially relies on Notch-induced expression of HRT1 and/or HRT3 transcriptional repressors.
DISCUSSION
The expansion of atherosclerotic lesions and postangioplasty restenosis is due, in part, to the phenotypic transition of medial VSMC. Indeed, this transition (initiated by macrophage secretion of proinflammatory cytokines and growth factors within the intima) forces these cells to switch from a contractile/quiescent to a secretory/proliferative state (referred to as the trans-differentiation process) allowing them to migrate and proliferate toward the intima and secrete, in turn, inflammatory molecules. Therefore, identifying the molecular mechanisms/entities, which control or participate to this phenotypic switch appears crucial to advance therapeutic strategies for atherosclerosis and/or to develop new molecular target(s) of stent-eluting molecules to improve the benefit of stent implantation.
Differentiated VSMC are associated with high expressions of several specific contractile proteins including SM22 and smooth muscle α-actin (27). Our results demonstrate that Notch inhibition via DAPT or Jagged1-Fc treatment in an inflammatory context modifies the VSMC phenotype by decreasing differentiation markers α-actin and SM22 (Fig. 4). This is in agreement with previous studies showing that Notch can directly or indirectly regulate expression of VSMC differentiation markers (24, 28–30). Similar results were observed in vivo in the rat carotid balloon injury model of restenosis, where SM22 expression drastically decreases postinjury concomitantly with Notch3 down-regulation and AC8 induction (Fig. 7A).
We (10, 14, 15) and others (5) have shown the involvement of both Notch and AC8 in the regulation of vascular smooth muscle cell transition toward a trans-differentiated phenotype. Nevertheless, none of these studies have established the link between Notch signaling and AC8 up-regulation. This study highlights, for the first time, the dependence of AC8 de novo expression on the regulation of the Notch signaling pathway. The most convincing arguments in support of this are the reported potentiating effect of the recombinant Jagged1-Fc on IL1β-induced AC8 expression (Fig. 3) and the observed AC8 down-regulation when overexpressing Notch3 or Notch 1 (Fig. 5) and HRT1 or HRT3 (Fig. 6). These findings also confirm this particular AC isoform as a marker of inflammatory VSMCs trans-differentiated status. The fact that neither AC3 nor AC6 expression (which are two AC isoforms well expressed in differentiated/contractile VSMCs) were affected in the same manner as AC8, by modulating Notch signaling, reinforces this conclusion (supplemental Fig. S1).
Taking our results into account and those obtained by others, we propose the Notch3 pathway as a key regulator of AC8 expression in VSMCs even though both intracellular domains, N3-ICD and N1-ICD, efficiently block its expression. Indeed, the mRNA transcript expression analysis of balloon-injured rat carotid artery tissues evidenced that only Notch3 inhibition parallels the emergence of AC8 (Fig. 7B and supplemental Fig. 3, upper right panel); earlier reports (10, 15, 31) and our results confirmed that Notch3 down-regulation precedes AC8 expression in response to IL1β treatment (Fig. 1) and in vivo in balloon-injured rat carotid arteries (Fig. 7). Notch1 expression showed not to be significantly different as compared with nontreated (control) cells; only a slight down-regulation of Notch2 expression was found (data not shown); Notch4 is not expressed neither in differentiated nor in trans-differentiated VSMCs (32).
Even though recombinant Jagged1-Fc has been shown to bind VSMCs (Fig. 3A, left panel), the Notch isoform(s) interacting with it remain(s) to be identified. Nevertheless, several arguments strongly suggest that Jagged1 could preferentially bind Notch3. For example, Liu et al. (33) published that Jagged1 is necessary for Notch3 expression in mural cells and that Notch3 could promote its own expression; Notch3 expression is strongly down-regulated (−70%, supplemental Fig. S2B) by Jagged1-Fc in VSMCs, whereas Notch1 expression is poorly affected (supplemental Fig. S2B); Shimizu et al. (34) reported that the purified extracellular region of Jagged1 binds Notch3 with a slightly higher affinity than Notch2 but significantly more than Notch1.
Regarding our results demonstrating that the Fc-bound soluble form of Notch ligand Jagged1 acts as an antagonist of the Notch pathway in VSMCs, it could be expected that the membrane-bound Jagged1 decreases IL1β-induced AC8 expression through Notch activation, maintaining their differentiated state. Consistent with this, Doi et al. (24) reported that Jagged1 signaling induces smooth muscle differentiation; results leading to similar conclusions were obtained by High et al. (35). The fact that this ligand is down-regulated in an inflammatory context (in presence of IL1β, supplemental Fig. S2A, right panel) is already in favor of this. Additional studies using the soluble versus full-length Jagged1 adenoviruses (36) should be performed to validate this hypothesis.
Notch preventing AC8 expression occurs via a RBP-Jκ-dependent, as opposed to independent, transcriptional pathway (25, 26, 37). Activation of the canonical RBP-Jκ-dependent Notch pathway leads to expression of HES and/or HRT transcriptional repressor factors. When treating VSMCs with Jagged1-Fc and/or IL1β (Fig. 3B, data not shown, and supplemental Fig. S2C), we showed that among all tested down-regulated target genes, HRT1 and HRT3 were the most sensitive for the Jagged1-Fc-induced Notch pathway inhibition. Although the Alexander et al. (31) study demonstrated HRT2 being the only Notch target gene down-regulated following IL1β treatment, HRT2 down-regulation does not seem to be involved in the AC8 increased expression. Indeed, overexpression of N3-ICD or N1-ICD blocks AC8 expression along with an increase of HRT1 and -3 (Fig. 5). Finally, overexpressing HRT1 and HRT3 significantly decreased IL1β-induced AC8 expression (Fig. 6, left panel) and the only Notch target genes to be down-regulated with comparable kinetics to Notch3 are HRT1 and HRT3 (supplemental Fig. S2C). Altogether, this demonstrates that HRT1 and/or HRT3 are involved in Jagged1-Notch-dependent AC8 regulation. Because HRT1 and HRT3 mRNA levels were significantly down-regulated in post-balloon-injured rat carotids (Refs. 7 and 8, our data, and supplemental Fig. S3), the AC8 gene silencing in VSMC can rely on the in vivo HRT3 and/or HRT1 repressing effect. By causing decreased expression of HRT1 and HRT3 (supplemental Fig. S2C), IL1β treatment could liberate E-boxes of the AC8 promoter to allow the NF-κB-dependent transcription of this gene, because IL1β/NF-κB pathways are known to be involved in the inflammatory response of VSMC (10, 11, 37). Preventing NF-κB activation by overexpressing a mutated nonphosphorylatable/nondegradable form of IκBα(32–36), reverses the AC8 up-regulation mediated by IL1β (supplemental Fig. S5). (Of note: Notch target protein families Hes and HRT are known to bind E-box DNA sequences (CACNAG and CANNTG (4).) Because our unpublished data are in favor of the existence of two distinct tissue-specific promoter regions (one driving AC8 expression in the brain and another driving its expression in trans-differentiated VSMCs), demonstrating HRTs and NF-κB direct binding to E-boxes and NF-κB responsive elements would require the cloning and characterization of the VSMC-specific AC8 promoter.4
Compiling our in vitro and in vivo studies and according to study by Doi et al. (24) stating that Jagged-1 is the main ligand for VSMC related signaling, it is most likely that the repression of AC8 expression in differentiated VSMCs occurs through Jagged1 activation of the Notch3-HRT3 and/or HRT1 pathway. Therefore, the most relevant experiment to be performed in vivo would be to follow AC8 expression and neointimal formation in mice overexpressing HRT3 and/or HRT1 in SMC. Because the very recent paper by Wu et al. (38) shows a cause to effect relationship between the Jagged1-Notch pathway inhibition and the VSMC de/trans-differentiation leading to the increase of neointimal formation using jagged1 null mice (38), our hypothesis linking neointimal formation and AC8 expression is even more likely.
Acknowledgments
We extend our thanks to Nathalie Mougenot and Adeline Jacquet for their technical contribution to the balloon injury procedures, Urban Lendahl for kindly sending us the N1-ICD and N3-ICD viruses, and Deepak Srivastava for kindly sending us the HRT1–3 plasmids.
This work was supported by the Pierre and Marie Curie University and l'Agence Nationale de la Recherche Grant 11 BSV1 034 01.

This article contains supplemental Figs. S1–S5.
J. H. F. de Baaij, R. Blaise, and I. Limon, unpublished data.
- VSMC
- vascular smooth muscle cell
- AC
- adenylyl cyclase
- ADAM
- a disintegrin and metalloproteinase
- CD31
- cluster of differentiation 31 also known as platelet endothelial cell adhesion molecule
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- HES
- hairy and enhancer of split
- HRT
- hairy-related transcription factors
- NF-κB
- nuclear factor-κB
- NICD
- Notch intracellular domain
- RBP-Jκ
- recombinant signal-binding protein 1 for Jκ
- RBP-Jκ-DN
- RBP-Jκ dominant-negative
- SM22
- smooth muscle 22α
- ICD
- intracellular domain
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Artavanis-Tsakonas S., Muskavitch M. A., Yedvobnick B. (1983) Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 80, 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999) Notch signaling. Cell fate control and signal integration in development. Science 284, 770–776 [DOI] [PubMed] [Google Scholar]
- 3. Tamura K., Taniguchi Y., Minoguchi S., Sakai T., Tun T., Furukawa T., Honjo T. (1995) Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H). Curr. Biol. 5, 1416–1423 [DOI] [PubMed] [Google Scholar]
- 4. Fischer A., Gessler M. (2007) Delta-Notch–and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 35, 4583–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morrow D., Guha S., Sweeney C., Birney Y., Walshe T., O'Brien C., Walls D., Redmond E. M., Cahill P. A. (2008) Notch and vascular smooth muscle cell phenotype. Circ. Res. 103, 1370–1382 [DOI] [PubMed] [Google Scholar]
- 6. Sweeney C., Morrow D., Birney Y. A., Coyle S., Hennessy C., Scheller A., Cummins P. M., Walls D., Redmond E. M., Cahill P. A. (2004) Notch1 and -3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jκ dependent pathway. FASEB J. 18, 1421–1423 [DOI] [PubMed] [Google Scholar]
- 7. Wang W., Campos A. H., Prince C. Z., Mou Y., Pollman M. J. (2002) Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. J. Biol. Chem. 277, 23165–23171 [DOI] [PubMed] [Google Scholar]
- 8. Campos A. H., Wang W., Pollman M. J., Gibbons G. H. (2002) Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells. Implications in cell-cycle regulation. Circ. Res. 91, 999–1006 [DOI] [PubMed] [Google Scholar]
- 9. Proweller A., Pear W. S., Parmacek M. S. (2005) Notch signaling represses myocardin-induced smooth muscle cell differentiation. J. Biol. Chem. 280, 8994–9004 [DOI] [PubMed] [Google Scholar]
- 10. Clément N., Gueguen M., Glorian M., Blaise R., Andréani M., Brou C., Bausero P., Limon I. (2007) Notch3 and IL-1β exert opposing effects on a vascular smooth muscle cell inflammatory pathway in which NF-κB drives cross-talk. J. Cell Sci. 120, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 11. Delbosc S., Glorian M., Le Port A. S., Bereziat G., Andreani M., Limon I. (2008) The benefit of docosahexanoic acid on the migration of vascular smooth muscle cells is partially dependent on Notch regulation of MMP-2/-9. Am. J. Pathol. 172, 1430–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Espinosa L., Inglés-Esteve J., Robert-Moreno A., Bigas A. (2003) IκBα and p65 regulate the cytoplasmic shuttling of nuclear corepressors. Cross-talk between Notch and NFκB pathways. Mol. Biol. Cell 14, 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Selzman C. H., Shames B. D., McIntyre R. C., Jr., Banerjee A., Harken A. H. (1999) The NF-κB inhibitory peptide, IκBα, prevents human vascular smooth muscle proliferation. Ann. Thorac. Surg. 67, 1227–1231; discussion 1231–1222 [DOI] [PubMed] [Google Scholar]
- 14. Clément N., Glorian M., Raymondjean M., Andréani M., Limon I. (2006) PGE2 amplifies the effects of IL-1β on vascular smooth muscle cell de-differentiation. A consequence of the versatility of PGE2 receptors 3 due to the emerging expression of adenylyl cyclase 8. J. Cell Physiol. 208, 495–505 [DOI] [PubMed] [Google Scholar]
- 15. Gueguen M., Keuylian Z., Mateo V., Mougenot N., Lompré A. M., Michel J. B., Meilhac O., Lipskaia L., Limon I. (2010) Implication of adenylyl cyclase 8 in pathological smooth muscle cell migration occurring in rat and human vascular remodelling. J. Pathol. 221, 331–342 [DOI] [PubMed] [Google Scholar]
- 16. Mallat Z., Steg P. G., Benessiano J., Tanguy M. L., Fox K. A., Collet J. P., Dabbous O. H., Henry P., Carruthers K. F., Dauphin A., Arguelles C. S., Masliah J., Hugel B., Montalescot G., Freyssinet J. M., Asselain B., Tedgui A. (2005) Circulating secretory phospholipase A2 activity predicts recurrent events in patients with severe acute coronary syndromes. J. Am. Coll. Cardiol. 46, 1249–1257 [DOI] [PubMed] [Google Scholar]
- 17. Willoughby D., Baillie G. S., Lynch M. J., Ciruela A., Houslay M. D., Cooper D. M. (2007) Dynamic regulation, desensitization, and cross-talk in discrete subcellular microdomains during β2-adrenoceptor and prostanoid receptor cAMP signaling. J. Biol. Chem. 282, 34235–34249 [DOI] [PubMed] [Google Scholar]
- 18. Ostrom R. S., Liu X., Head B. P., Gregorian C., Seasholtz T. M., Insel P. A. (2002) Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells. Expression in caveolin-rich and noncaveolin domains. Mol. Pharmacol. 62, 983–992 [DOI] [PubMed] [Google Scholar]
- 19. del Monte F., Harding S. E., Schmidt U., Matsui T., Kang Z. B., Dec G. W., Gwathmey J. K., Rosenzweig A., Hajjar R. J. (1999) Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 100, 2308–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin S., Hansson E. M., Tikka S., Lanner F., Sahlgren C., Farnebo F., Baumann M., Kalimo H., Lendahl U. (2008) Notch signaling regulates platelet-derived growth factor receptor-β expression in vascular smooth muscle cells. Circ. Res. 102, 1483–1491 [DOI] [PubMed] [Google Scholar]
- 21. Parks A. L., Curtis D. (2007) Presenilin diversifies its portfolio. Trends Genet. 23, 140–150 [DOI] [PubMed] [Google Scholar]
- 22. Boyer-Di Ponio J., Wright-Crosnier C., Groyer-Picard M. T., Driancourt C., Beau I., Hadchouel M., Meunier-Rotival M. (2007) Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome. Inhibitory effect on Notch signaling. Hum. Mol. Genet. 16, 2683–2692 [DOI] [PubMed] [Google Scholar]
- 23. Trifonova R., Small D., Kacer D., Kovalenko D., Kolev V., Mandinova A., Soldi R., Liaw L., Prudovsky I., Maciag T. (2004) The non-transmembrane form of Delta1, but not of Jagged1, induces normal migratory behavior accompanied by fibroblast growth factor receptor 1-dependent transformation. J. Biol. Chem. 279, 13285–13288 [DOI] [PubMed] [Google Scholar]
- 24. Doi H., Iso T., Sato H., Yamazaki M., Matsui H., Tanaka T., Manabe I., Arai M., Nagai R., Kurabayashi M. (2006) Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jκ-dependent pathway. J. Biol. Chem. 281, 28555–28564 [DOI] [PubMed] [Google Scholar]
- 25. Iso T., Chung G., Hamamori Y., Kedes L. (2002) HERP1 is a cell type-specific primary target of Notch. J. Biol. Chem. 277, 6598–6607 [DOI] [PubMed] [Google Scholar]
- 26. Waltzer L., Bourillot P. Y., Sergeant A., Manet E. (1995) RBP-Jκ repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transcription factor EBNA2. Nucleic Acids Res. 23, 4939–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 28. Noseda M., Fu Y., Niessen K., Wong F., Chang L., McLean G., Karsan A. (2006) Smooth Muscle α-actin is a direct target of Notch/CSL. Circ. Res. 98, 1468–1470 [DOI] [PubMed] [Google Scholar]
- 29. Tang Y., Urs S., Boucher J., Bernaiche T., Venkatesh D., Spicer D. B., Vary C. P., Liaw L. (2010) Notch and transforming growth factor-β (TGFβ) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J. Biol. Chem. 285, 17556–17563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang Y., Urs S., Liaw L. (2008) Hairy-related transcription factors inhibit Notch-induced smooth muscle α-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ. Res. 102, 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexander M. R., Murgai M., Moehle C. W., Owens G. K. (2012) Interleukin-1β modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-κB-dependent mechanisms. Physiol. Genomics 44, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uyttendaele H., Marazzi G., Wu G., Yan Q., Sassoon D., Kitajewski J. (1996) Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development 122, 2251–2259 [DOI] [PubMed] [Google Scholar]
- 33. Liu H., Kennard S., Lilly B. (2009) NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 104, 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shimizu K., Chiba S., Kumano K., Hosoya N., Takahashi T., Kanda Y., Hamada Y., Yazaki Y., Hirai H. (1999) Mouse jagged1 physically interacts with Notch2 and other Notch receptors. Assessment by quantitative methods. J. Biol. Chem. 274, 32961–32969 [DOI] [PubMed] [Google Scholar]
- 35. High F. A., Lu M. M., Pear W. S., Loomes K. M., Kaestner K. H., Epstein J. A. (2008) Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. U.S.A. 105, 1955–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caolo V., Schulten H. M., Zhuang Z. W., Murakami M., Wagenaar A., Verbruggen S., Molin D. G., Post M. J. (2011) Soluble Jagged-1 inhibits neointima formation by attenuating Notch-Herp2 signaling. Arterioscler. Thromb. Vasc. Biol. 31, 1059–1065 [DOI] [PubMed] [Google Scholar]
- 37. Shen J., Yang M., Ju D., Jiang H., Zheng J. P., Xu Z., Li L. (2010) Disruption of SM22 promotes inflammation after artery injury via nuclear factor κB activation. Circ. Res. 106, 1351–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu X., Zou Y., Zhou Q., Huang L., Gong H., Sun A., Tateno K., Katsube K., Radtke F., Ge J., Minamino T., Komuro I. (2011) Role of Jagged1 in arterial lesions after vascular injury. Arterioscler. Thromb. Vasc. Biol. 31, 2000–2006 [DOI] [PubMed] [Google Scholar]