

Background: We determined the role of presynaptic N-methyl-d-aspartate receptor (NMDAR) activity in spinal cords in opioid-induced hyperalgesia and tolerance.
Results: Chronic opioid increases presynaptic NMDAR activity at primary sensory nerve terminals through protein kinase C.
Conclusion: Increased presynaptic NMDAR activity potentiates nociceptive input and is responsible for opioid hyperalgesia and tolerance.
Significance: Understanding mechanisms of increased NMDAR activity is important for improving opioid therapies.
Keywords: G protein-coupled Receptors (GPCR); Glutamate; Glutamate Receptors Ionotropic (AMPA, NMDA); Opiate Opioid; Synaptic Plasticity
Abstract
Opioids are the most effective analgesics for the treatment of moderate to severe pain. However, chronic opioid treatment can cause both hyperalgesia and analgesic tolerance, which limit their clinical efficacy. In this study, we determined the role of pre- and postsynaptic NMDA receptors (NMDARs) in controlling increased glutamatergic input in the spinal cord induced by chronic systemic morphine administration. Whole-cell voltage clamp recordings of excitatory postsynaptic currents (EPSCs) were performed on dorsal horn neurons in rat spinal cord slices. Chronic morphine significantly increased the amplitude of monosynaptic EPSCs evoked from the dorsal root and the frequency of spontaneous EPSCs, and these changes were largely attenuated by blocking NMDARs and by inhibiting PKC, but not PKA. Also, blocking NR2A- or NR2B-containing NMDARs significantly reduced the frequency of spontaneous EPSCs and the amplitude of evoked EPSCs in morphine-treated rats. Strikingly, morphine treatment largely decreased the amplitude of evoked NMDAR-EPSCs and NMDAR currents of dorsal horn neurons elicited by puff NMDA application. The reduction in postsynaptic NMDAR currents caused by morphine was prevented by resiniferatoxin pretreatment to ablate TRPV1-expressing primary afferents. Furthermore, intrathecal injection of the NMDAR antagonist significantly attenuated the development of analgesic tolerance and the reduction in nociceptive thresholds induced by chronic morphine. Collectively, our findings indicate that chronic opioid treatment potentiates presynaptic, but impairs postsynaptic, NMDAR activity in the spinal cord. PKC-mediated increases in NMDAR activity at nociceptive primary afferent terminals in the spinal cord contribute critically to the development of opioid hyperalgesia and analgesic tolerance.
Introduction
Opioid analgesics are the mainstay of treatment for moderate to severe pain caused by cancer or by tissue and nerve injury. However, long-term administration of μ-opioid receptor agonists for chronic pain can lead to analgesic tolerance, which is a major obstacle to adequate pain relief. Opioid analgesic tolerance is a phenomenon in which repeated exposure to an opioid agonist results in the decreased therapeutic effect of the drug or the need for a higher dose to maintain the same analgesic effect. For all μ-opioid receptor agonists, analgesic tolerance occurs with repeated use. The cellular and signaling mechanisms responsible are poorly understood. The key question is where and how opioids induce neuroplasticity in the nervous system that leads to opioid tolerance. This problem has not been adequately studied in the nociceptive circuitry in which native opioid receptors are expressed. NMDA receptors (NMDARs),2 which play an important role in opioid-induced hyperalgesia and tolerance (1, 2), are located presynaptically on central terminals of primary afferent neurons as well as postsynaptically on dorsal horn neurons (3, 4). However, it is unclear what roles these receptors play in the development of opioid analgesic tolerance.
Many studies have shown that opioids paradoxically elicit abnormal pain or hyperalgesia (i.e. enhanced pain response to noxious stimuli). Opioid-induced hyperalgesia has been demonstrated in many animal studies (1, 5–8), and even brief exposure to fentanyl or morphine can induce long-lasting hyperalgesia (8, 9). Hyperalgesia also occurs in patients after administration of various opioids (10–12). The increased pain sensitivity requires the use of additional opioids to sustain the adequate analgesic effects and, consequently, could be interpreted as analgesic tolerance. The μ-opioid receptors at the spinal level are essential for the analgesic effect of opioids (13). We recently showed that in spinal lamina I and II neurons, brief opioid exposure can cause a long-lasting increase in glutamate release from nociceptive primary afferents (i.e. opioid-induced primary afferent hyperactivity) through activation of presynaptic NMDARs (14). It is not clear, however, how pre- and postsynaptic NMDARs help regulate glutamatergic synaptic input to spinal dorsal horn neurons after chronic opioid treatment. Identifying the origin of NMDAR activity associated with chronic opioid treatment is critical because it will influence whether research should focus on primary sensory neurons or spinal cord second-order neurons to improve the efficacy of opioid therapies.
In this study, we determined how chronic opioid treatment affects the function of pre- and postsynaptic NMDARs in the spinal cord. We provide new evidence showing that presynaptic NMDAR activity at primary afferent terminals is increased by chronic morphine and potentiates glutamatergic input to spinal dorsal horn neurons through protein kinase C (PKC). Strikingly, chronic morphine administration leads to a large reduction in postsynaptic NMDAR activity, which results from increased glutamate release from transient receptor potential vanilloid type 1 (TRPV1)-expressing primary afferents. This new information is important for understanding the mechanisms involved in NMDAR plasticity at the spinal level and their roles in opioid-induced hyperalgesia and analgesic tolerance.
EXPERIMENTAL PROCEDURES
Animals and Morphine Treatment
Male Sprague-Dawley rats (280–320 g; Harlan, Indianapolis, IN) were used in this study. Opioid hyperalgesia and analgesic tolerance were induced with daily intraperitoneal injections of morphine (10 mg/kg) for 8 consecutive days (15). Rats in the control group received daily intraperitoneal injections of the vehicle (saline). For behavioral experiments with live rats, intrathecal catheters were implanted during isoflurane-induced anesthesia. Briefly, each animal was placed prone on a stereotaxic frame, and a small incision was made at the back of the neck of the animal. A small puncture was made in the atlantooccipital membrane of the cisterna magna, and a catheter was then inserted such that the caudal tip reached the lumbar enlargement of the spinal cord (13, 15). We then exteriorized the rostral end of the catheter and closed the wound with sutures. The animals were allowed to recover for 5 days before intrathecal injections, and rats displaying signs of motor or neurological dysfunction were excluded from the study. Drugs were injected intrathecally in a volume of 10 μl followed by a 10-μl flush with normal saline. All surgical preparation and experimental protocols were approved by the Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center and conformed to National Institutes of Health guidelines on the ethical use of animals.
Behavioral Assessments of Mechanical and Thermal Nociception
The mechanical nociception of rats was assessed by the paw pressure test using an Ugo Basile analgesimeter (Varese, Italy). Noxious pressure was gradually applied to hind paws by pressing the device pedal to increase the force at a constant rate. The pedal was immediately released when the animal displayed pain by withdrawing the paw or vocalizing, and the nociceptive threshold of the animal was read on the scale. To avoid potential tissue injury, a maximum of 400 g was used as a cutoff (15, 16). Both hind paws were tested for each rat, and the mean value was used as the pressure nociceptive withdrawal threshold for that animal.
To quantitatively assess the thermal nociceptive threshold, rats were tested with a thermal testing apparatus (IITC Inc., Woodland Hills, CA). The rats were allowed to acclimate for 30 min on a glass surface with the temperature maintained at 30 °C. A mobile radiant heat source was focused onto the plantar surface of the hind paw. The paw withdrawal latency was recorded using a timer. A cutoff of 30 s was used to prevent potential tissue damage (15, 16). Both of the hind paws were tested in each animal, and the mean value was used as the thermal nociceptive withdrawal threshold.
Removal of TRPV1-expressing Dorsal Root Ganglion Neurons and Their Terminals in Spinal Cords
Originally isolated from the cactus-like plant Euphorbia resinifera, resiniferatoxin (RTX) is an ultrapotent TRPV1 agonist (17). Systemic injection of RTX selectively kills TRPV1-expressing primary afferent neurons and their central terminals in adult rats (14, 18, 19). Rats were anesthetized with isoflurane before receiving a single intraperitoneal injection of RTX (200 μg/kg, LC Laboratories, Woburn, MA) dissolved in a mixture of 10% ethanol and 10% Tween 80 in normal saline. Rats in the control group received a single intraperitoneal injection of the vehicle. Morphine treatment was conducted 7 days after the RTX and vehicle injections. The effect of RTX on TRPV1-expressing dorsal root ganglion neurons and central terminals was confirmed by using double labeling of TRPV1 and Griffonia simplicifolia isolectin B4 (IB4, a marker for unmyelinated afferent neurons and fibers) in the dorsal root ganglion and spinal dorsal horn in rats 7 days after RTX or vehicle injection, as described previously (13, 19).
Spinal Cord Slice Preparation and Electrophysiological Recordings
The lumbar segment of the spinal cord was rapidly removed through laminectomy after the rats were anesthetized with 2% isoflurane. The lumbar spinal cord tissues were immediately placed in ice-cold sucrose artificial cerebrospinal fluid containing (in mm) 234 sucrose, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 25.0 NaHCO3, and 12.0 glucose. The spinal cord was placed in a gelatin block and glued onto the stage of a vibratome. Transverse spinal cord slices were cut (400 μm in thickness) in ice-cold sucrose artificial cerebrospinal fluid and then transferred in Krebs solution presaturated with 95% O2 and 5% CO2 at 34 °C for at least 1 h before being transferred to the recording chamber.
Neurons in the lamina II outer zone were visualized on a fixed stage microscope (BX50WI; Olympus, Tokyo, Japan) with differential interference contrast/infrared illumination. Lamina II of the spinal cord is the central site of termination of the majority of unmyelinated C-fibers carrying nociceptive information (20–22). Excitatory postsynaptic currents (EPSCs) were recorded using whole-cell voltage clamp techniques (14, 23). The impedance of the glass electrode was 4–7 MΩ when the pipette was filled with the internal solution containing (in mm) 135 potassium gluconate, 5 KCl, 2.0 MgCl2, 0.5 CaCl2, 5.0 HEPES, 5.0 EGTA, 5.0 ATP-Mg, 0.5 Na-GTP, and 10 QX314 (adjusted to pH 7.25 with 1.0 m CsOH, 280–300 mosm). The slice was continuously perfused with Krebs solution at 5.0 ml/min at 34 °C. Spontaneous EPSCs (sEPSCs) were recorded in the presence of 2 μm strychnine and 10 μm bicuculline at a holding potential of −60 mV. In addition, EPSCs were evoked from the dorsal root using a bipolar tungsten electrode connected to a stimulator (0.2 ms, 0.3–0.5 mA, 0.1 Hz; Grass Instruments, Quincy, MA). NMDAR-mediated EPSCs (NMDAR-EPSCs) were recorded in the presence of 2 μm strychnine, 10 μm bicuculline, and 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione at a holding potential of +40 mV (24). To determine the paired-pulse ratio, two EPSCs were evoked by a pair of stimuli given at 50-ms intervals. The paired-pulse ratio was expressed as the ratio of the amplitudes of the second and first synaptic responses (14). The input resistance was monitored, and the recording was abandoned if it changed by more than 15%. EPSCs were recorded using an amplifier (MultiClamp 700A, Axon Instruments, Foster City, CA), filtered at 1–2 kHz, and digitized at 10 kHz.
To directly determine postsynaptic NMDAR activity, currents were elicited by puff application of 100 μm NMDA to the recorded neuron in the extracellular solution containing a low concentration of Mg2+ (0.1 mm), 10 μm glycine, and 1 μm tetrodotoxin at a holding potential of −60 mV and using the pipette internal solution containing (in mm) 110.0 Cs2SO4, 2.0 MgCl2, 0.1 CaCl2, 1.1 EGTA, 10.0 HEPES, 2.0 MgATP, and 0.3 Na2GTP (pH was adjusted to 7.25 with 1.0 m CsOH (280–300 mosm)) (24). Similarly, to examine postsynaptic AMPAR function, currents were elicited similarly but with 100 μm AMPA (25). The puff electrode was placed about 150 μm away from the neuron being recorded. Puff application of NMDA or AMPA was performed using a Pressure System IIe (4 p.s.i., 15 ms; Toohey Co., Fairfield, NJ). All drugs were freshly prepared in artificial cerebrospinal fluid before the experiments and delivered via syringe pumps to reach their final concentrations. Chelerythrine, NMDA, and AMPA were purchased from Sigma-Aldrich. (Rp)-cAMPS was obtained from Tocris. Ro 25-6981, AP5, 6-cyano-7-nitroquinoxaline-2,3-dione, bicuculline, and tetrodotoxin were purchased from Ascent Scientific.
Data Analysis
All data are expressed as means ± S.E. To determine the amplitude of the evoked EPSCs, at least 10 consecutive EPSCs were averaged and analyzed off-line with Clampfit 10.0 software. sEPSCs were analyzed off-line using the Mini Analysis peak detection program (Synaptosoft, Leonia, NJ). The cumulative probability of the amplitude and the inter-event interval of the sEPSCs were compared using the Kolmogorov-Smirnov test, which estimates the probability that two distributions are similar. We used Student's t test to compare two groups and one-way analysis of variance followed by Dunnett's post hoc test to compare more than two groups. Two-way analysis of variance followed by Bonferroni's post hoc test was used to determine the difference in the development of hyperalgesia and opioid tolerance between the vehicle-treated and AP5-treated groups. A p value of < 0.05 was considered to be statistically significant.
RESULTS
Chronic Morphine Increases Glutamatergic Input to Dorsal Horn Neurons through Presynaptic NMDAR Activation
We previously showed that brief exposure to the μ-opioid receptor agonist (D-Ala2, N-Me-Phe4, Gly-ol5)-enkephalin increases glutamatergic input to dorsal horn neurons (14). To determine the effect of chronic morphine treatment on glutamatergic input to dorsal horn neurons, we recorded sEPSCs in the spinal cord slices from vehicle- and morphine-treated rats. The frequency of sEPSCs of lamina II neurons was significantly higher in the morphine-treated group (n = 11 neurons) than in the vehicle-treated group (n = 13 neurons), but the difference in the amplitude of sEPSCs was not significantly different (Fig. 1, A–C).
FIGURE 1.
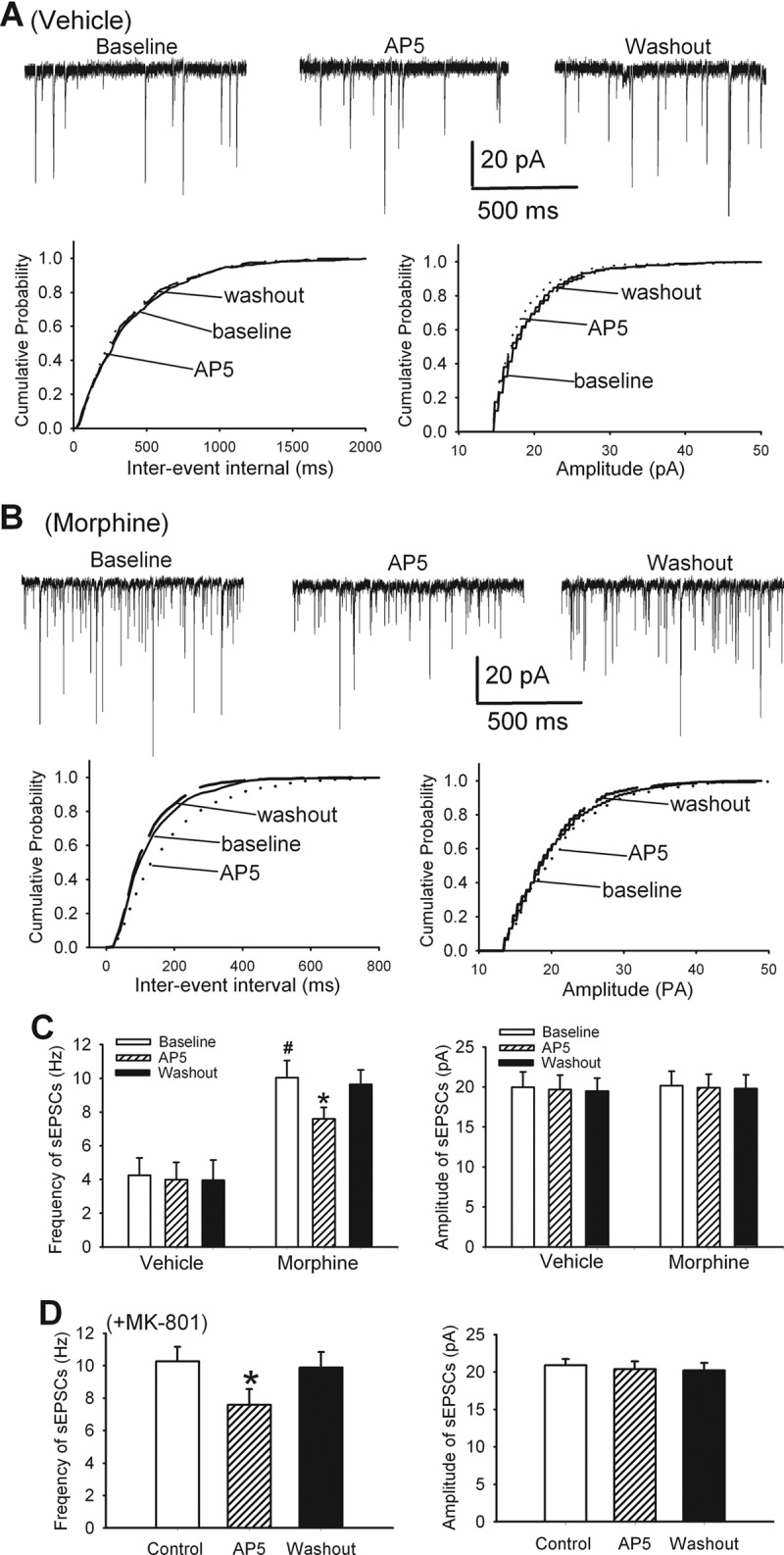
Chronic morphine increases glutamatergic input and presynaptic NMDAR activity in the spinal cord. A, original recording traces and cumulative plots show the effect of bath application of 50 μm AP5 on the frequency and amplitude of sEPSCs of a lamina II neuron from a vehicle-treated rat. B, original recordings and cumulative plots show the effect of 50 μm AP5 on sEPSCs of a lamina II neuron from a morphine-treated rat. C, summary data of the effect of 50 μm AP5 on the mean frequency and amplitude of sEPSCs in vehicle-treated (n = 13 neurons) and morphine-treated (n = 11 neurons) rats. D, group data of the effects of 50 μm AP5 on the mean frequency and amplitude of sEPSCs, recorded with 1 mm MK-801 included in the pipette solution, in morphine-treated rats (n = 11 neurons).*, p < 0.05 when compared with respective base-line controls. #, p < 0.05 when compared with base-line controls in vehicle-treated group.
To determine the role of NMDARs in the control of glutamatergic input to dorsal horn neurons, we tested the effect of AP5, a selective NMDAR antagonist (26), on sEPSCs. We have shown that 50 μm AP5 completely blocks NMDAR-mediated EPSCs (24). Bath application of 50 μm AP5 significantly decreased the frequency, but not the amplitude, of sEPSCs of lamina II neurons in the same morphine-treated rats (n = 11 neurons, Fig. 1, B and C). However, AP5 application had no significant effects on the frequency and amplitude of sEPSCs of lamina II neurons in vehicle-treated rats (n = 13 neurons, Fig. 1, A and C).
To determine the role of presynaptic NMDARs in glutamatergic input increased by morphine treatment, we recorded sEPSCs in separate lamina II neurons by including the NMDAR channel blocker MK-801 in the pipette solution to block the postsynaptic NMDARs. We have shown that intracellular dialysis of 1 mm MK-801 completely blocked the postsynaptic NMDARs within 15 min (14). In all 11 neurons recorded with MK-801, AP5 still significantly reduced the frequency of sEPSCs in morphine-treated rats (p < 0.05, Fig. 1D). These results suggest that chronic morphine increases glutamatergic input to spinal dorsal horn neurons by enhancing presynaptic NMDAR activity.
Chronic Morphine Increases NMDAR Activity at Primary Afferents in the Spinal Cord
To determine whether NMDARs at primary afferent terminals play a role in increased glutamate release to spinal dorsal horn neurons, monosynaptic EPSCs were evoked from the dorsal root in vehicle- and morphine-treated rats. These EPSCs were identified on the basis of the constant latency and absence of conduction failure of evoked EPSCs in response to a 20-Hz electrical stimulation, as we described previously (14, 27). In this protocol, we used a fixed stimulation intensity to evoke AMPAR-EPSCs in vehicle-treated and morphine-treated rats. The amplitude of AMPAR-EPSCs of lamina II neurons was significantly higher in morphine-treated rats than in vehicle-treated rats (n = 9 neurons, Fig. 2, A and B). Bath application of 50 μm AP5 significantly reduced the amplitude of monosynaptic EPSCs of lamina II neurons in morphine-treated rats (n = 11 neurons, Fig. 2, A and B). Also, the effect of AP5 on evoked monosynaptic EPSCs of morphine-treated rats was associated with a decrease in the paired-pulse ratio (Fig. 2C). However, AP5 had no significant effect on the amplitude of monosynaptic EPSCs of lamina II neurons recorded from vehicle-treated rats (n = 14 neurons, Fig. 2, A and B).
FIGURE 2.
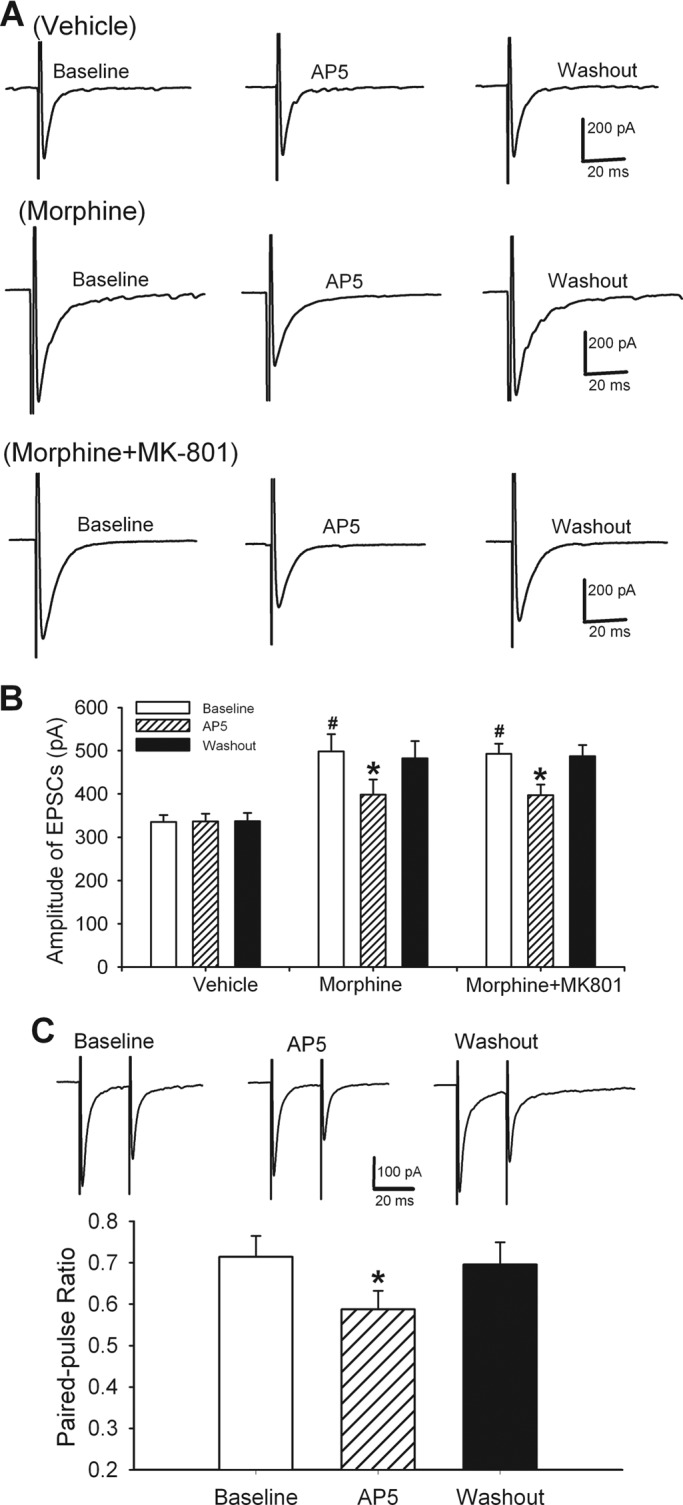
Potentiation of glutamatergic input to spinal dorsal horn neurons through increased activity of NMDARs at primary afferent terminals by chronic morphine. A, original recordings show the effect of 50 μm AP5 on monosynaptic AMPAR-EPSCs of the lamina II neuron evoked from the dorsal root in a vehicle-treated and a morphine-treated rat (recorded with and without 1 mm MK-801 in the pipette solution). B, group data show the effect of 50 μm AP5 on the mean amplitude of evoked AMPAR-EPSCs of lamina II neurons in vehicle-treated (n = 9 neurons) as well as morphine-treated rats recorded without MK-801 (n = 11 neurons) or with MK-801 (n = 9 neurons). C, AP5-produced inhibition of the EPSC amplitude in morphine-treated rats was associated with a decreased paired-pulse ratio (n = 11 neurons). *, p < 0.05 when compared with respective base-line controls. #, p < 0.05 when compared with base-line controls in vehicle-treated group.
In another nine lamina II neurons from morphine-treated rats, 1 mm MK-801 was included in the internal pipette solution and dialyzed for 15 min to block postsynaptic NMDARs before EPSCs were recorded. In all nine neurons, bath application of 50 μm AP5 still significantly reduced the amplitude of monosynaptic AMPAR-EPSCs evoked from the dorsal root (Fig. 2, A and B). Taken together, these results indicate that chronic morphine increases presynaptic NMDAR activity at primary afferent terminals in the spinal cord.
PKC, but Not PKA, Mediates the Increase in Presynaptic NMDAR Activity in the Spinal Cord by Chronic Morphine
Chronic opioid treatment results in significant increases in adenylyl cyclase activity and in basal levels of cAMP in the central nervous system (28, 29). To determine whether cAMP-dependent PKA is involved in increased presynaptic NMDAR activity by chronic morphine, we used (Rp)-cAMPS, a highly selective and membrane-permeable PKA inhibitor. (Rp)-cAMPS competes with cAMP for the binding sites on the regulatory subunit of PKA and, when bound, prevents release of the free catalytic subunit (30). It has been shown that 100 μm (Rp)-cAMPS inhibits PKA activity in brain slices (31). Incubation with 100 μm (Rp)-cAMPS in the spinal cord slices from morphine-treated rats for 1–2 h had no significant effect on the base-line frequency of sEPSCs (n = 11 neurons) or the amplitude of monosynaptic EPSCs (n = 12 neurons) of lamina II neurons evoked from the dorsal root (Fig. 3, A and C). Furthermore, bath application of 50 μm AP5 still significantly inhibited the frequency of sEPSCs and the amplitude of evoked EPSCs in all lamina II neurons tested in the spinal cord slices treated with (Rp)-cAMPS (Fig. 3, B and D).
FIGURE 3.

Lack of a role of PKA in glutamatergic input to spinal dorsal horn neurons increased by chronic morphine. A, original recordings and cumulative plots show the effect of 50 μm AP5 on the frequency and amplitude of sEPSCs of a lamina II neuron from a spinal cord slice pretreated with 100 μm (Rp)-cAMPS in one morphine-treated rat. B, original traces show the effect of 50 μm AP5 on the amplitude of monosynaptic EPSCs of the lamina II neuron from a spinal cord slice pretreated with (Rp)-cAMPS in a morphine-treated rat. C and D, summary data of the effect of 50 μm AP5 on the mean frequency and amplitude of sEPSCs (n = 11 neurons) and the amplitude of evoked EPSCs (n = 12 neurons) in spinal cord slices pretreated with (Rp)-cAMPS in morphine-treated rats. *, p < 0.05 when compared with respective base-line controls.
Intrathecal administration of PKC inhibitors can reverse or attenuate opioid tolerance induced by chronic morphine administration (32). Increased PKC activity can promote NMDAR surface expression and regulate the channel gating (33, 34). We thus determined whether PKC plays a role in increased presynaptic NMDAR activity in the spinal cord by chronic morphine treatment. Chelerythrine is a membrane-permeant inhibitor of the catalytic site of PKC (35). It has been shown that 10 μm chelerythrine inhibits PKC activity in brain slices (36). Incubation for 1–2 h with 10 μm chelerythrine, a specific PKC inhibitor, in the spinal cord slices from morphine-treated rats largely reduced the base-line frequency of sEPSCs (n = 10 neurons) and the amplitude of monosynaptic EPSCs (n = 12 neurons) of lamina II neurons evoked from the dorsal root (Fig. 4, A–D). In those neurons, additional treatment with 50 μm AP5 failed to significantly affect the frequency of sEPSCs and the amplitude of evoked EPSCs (Fig. 4, A–D). Thus, our data indicate that PKC plays a critical role in increased presynaptic NMDAR activity in the spinal cord caused by chronic opioid treatment.
FIGURE 4.

PKC contributes to augmented glutamatergic input to spinal dorsal horn neurons induced by chronic morphine. A, representative recordings and cumulative plots show the lack of effect of 50 μm AP5 on the frequency and amplitude of sEPSCs of a lamina II neuron from a spinal cord slice pretreated with 10 μm chelerythrine in one morphine-treated rat. B, original traces show the effect of 50 μm AP5 on the amplitude of monosynaptic EPSCs of the lamina II neuron from a spinal cord slice pretreated with chelerythrine in a morphine-treated rat. C and D, summary data of the effect of 50 μm AP5 on the mean frequency and amplitude of sEPSCs (n = 10 neurons) and the amplitude of evoked EPSCs (n = 12 neurons) in spinal cord slices pretreated with chelerythrine in morphine-treated rats. Note that the base-line frequency of sEPSCs by chelerythrine treatment in morphine-treated rats was normalized to that of vehicle-treated rats.
Presynaptic NR2A and NR2B Subunits Contribute to Increased Synaptic Glutamatergic Input Induced by Chronic Morphine
NMDAR is a heteromeric complex composed of the NR1 subunit and at least one of the NR2 subunits (37, 38). To determine the role of NR2A and NR2B subunits in the increased glutamate release to dorsal horn neurons induced by chronic morphine injections, we used 0.6 μm Ro 25-6981, a highly specific NR2B-containing NMDAR antagonist (39), as well as a low concentration of AP5 (5 μm), which preferentially blocks NR2A-containing NMDARs at this level (40). At 0.1 μm, Ro 25-6981 blocks ∼90% of NR2B-mediated NMDA currents (39). Previous studies have demonstrated that bath application of 0.3–0.6 μm Ro 25-6981 maximally blocks NR2B-containing NMDAR-EPSCs in brain slices (41, 42). Highly specific blockers for NR2A-containing NMDARs are not available at the present time. Ro 25-6981 significantly decreased the frequency of sEPSCs of lamina II neurons in morphine-treated rats (n = 14 neurons, Fig. 5, A and C). In these neurons, treatment with 5 μm AP5 again significantly reduced the frequency of sEPSCs. Neither Ro 25-6981 nor 5 μm AP5 had a significant effect on the amplitude of sEPSCs of lamina II neurons (Fig. 5, A and C).
FIGURE 5.

Presynaptic NR2A- and NR2B-containing NMDARs contribute to increased glutamatergic input to spinal dorsal horn neurons by chronic morphine. A, representative current traces and cumulative plots show the effect of bath application of 0.6 μm Ro 25-6981 (Ro) and 5 μm AP5 (Ro+AP5) on the frequency and amplitude of sEPSCs of a lamina II neuron in one morphine-treated rat. B, original traces show the effect of Ro 25-6981 (Ro) and 5 μm AP5 (Ro+AP5) on the amplitude of evoked monosynaptic EPSCs of a lamina II neuron in one morphine-treated rat. C and D, group data of the effects of Ro 25-6981 (Ro) and 5 μm AP5 (Ro+AP5) on the frequency and amplitude of sEPSCs (n = 14 neurons) and the mean amplitude of evoked EPSCs (n = 11 neurons) in morphine-treated rats. *, p < 0.05 when compared with respective base-line controls. #, p < 0.05 when compared with the effect of Ro 25-6981.
We also determined the role of NR2A and NR2B subunits at primary afferent terminals in controlling increased glutamatergic input to dorsal horn neurons induced by chronic morphine treatment. Monosynaptic AMPAR-EPSCs were evoked from the dorsal root in morphine-treated rats. Bath application of 0.6 μm Ro 25-6981 significantly inhibited the amplitude of monosynaptic EPSCs of 11 lamina II neurons in the morphine-treated rats. Further blocking NR2A with 5 μm AP5 also significantly decreased the amplitude of EPSCs of these neurons (Fig. 5, B and D). These results suggest that chronic morphine increases glutamate release via presynaptic NR2A and NR2B subunit-containing NMDARs in the spinal cord.
Chronic Morphine Decreases Postsynaptic NMDAR Currents of Dorsal Horn Neurons
Next, we determined whether chronic morphine alters the postsynaptic NMDAR function in the spinal dorsal horn. We first recorded NMDAR-mediated EPSCs of lamina II neurons monosynaptically evoked from the dorsal root stimulation in vehicle-treated and morphine-treated rats. In this protocol, we adjusted the stimulation intensity so that the amplitude of AMPAR-EPSCs was the same in both sets of rats. We unexpectedly observed that the amplitude of NMDAR-EPSCs and the ratio of NMDAR-EPSCs to AMPAR-EPSCs of lamina II neurons were significantly reduced in the morphine-treated rats (n = 14 neurons) when compared with those of vehicle-treated rats (n = 16 neurons, Fig. 6, A and B).
FIGURE 6.

Chronic morphine impairs postsynaptic NMDAR activity of dorsal horn neurons. A, representative traces show the difference in the amplitude of AMPAR-EPSCs and NMDAR-EPSCs of the same lamina II neurons in a vehicle-treated and a morphine-treated rat. AMPAR-EPSCs and NMDAR-EPSCs were evoked from the same lamina II neuron at holding potentials of −60 mV and +40 mV, respectively. B, group data show the difference in the mean amplitude of NMDAR currents and the mean ratio of NMDAR-EPSCs to AMPAR-EPSCs of lamina II neurons in vehicle-treated (n = 16 neurons) and morphine-treated (n = 14 neurons) rats. C, original recordings show the inward NMDAR and AMPAR currents of the lamina II neuron elicited by puff application of NMDA and AMPA in a vehicle-treated and a morphine-treated rat. D, summary data show the difference in the mean amplitude of NMDAR (vehicle, n = 14; morphine, n = 11) and AMPAR (vehicle, n = 20; morphine, n = 22) currents of lamina II neurons in vehicle-treated and morphine-treated rats. *, p < 0.05 when compared with the value in vehicle controls.
We then examined the postsynaptic NMDAR currents elicited by direct puff application of 100 μm NMDA to lamina II neurons. The amplitude of the inward NMDAR currents of all lamina II neurons was significantly smaller in morphine-treated (n = 11 neurons) than in vehicle-treated rats (n = 14 neurons, p < 0.05; Fig. 6, C and D). In contrast, the amplitude of AMPAR currents elicited by puff application of 100 μm AMPA did not differ significantly between vehicle-treated (n = 20 neurons) and morphine-treated rats (n = 22 neurons, Fig. 6, C and D). These data indicate that chronic opioid treatment diminishes the postsynaptic NMDAR function of spinal dorsal horn neurons.
TRPV1-expressing Primary Afferents Are Critical for the Chronic Morphine-induced Reduction of Postsynaptic NMDAR Function in the Spinal Cord
Because of our unexpected finding that chronic morphine causes a large reduction in postsynaptic NMDAR activity in the spinal dorsal horn, we explored the mechanism for this phenomenon. We previously showed that brief opioid application followed by washout increases glutamate release from TRPV1-expressing primary afferents (14). Thus, we assessed whether persistent release of glutamate from these afferents induced by repeated morphine injections contributes to the reduction in postsynaptic NMDAR currents in the spinal cord. Immunocytochemical analysis confirmed that systemic RTX treatment of rats eliminated TRPV1-expressing dorsal root ganglion neurons and their central terminals in the spinal dorsal horn (supplemental Fig. 1), similar to the findings we reported previously (14, 15).
In the vehicle-treated rats, chronic morphine treatment significantly decreased the amplitude of NMDAR-EPSCs and the ratio of NMDAR-EPSCs to AMPAR-EPSCs of lamina II neurons (n = 14, Fig. 7, A and B). In contrast, these changes were largely prevented in RTX-treated rats (n = 21 neurons, Fig. 7, A and B).
FIGURE 7.

Ablation of TRPV1-expressing primary afferents blocks the reduction in postsynaptic NMDAR activity in the spinal dorsal horn caused by chronic morphine. A, original traces show the effect of morphine treatment on monosynaptic NMDAR- and AMPAR-EPSCs of a lamina II neuron evoked by dorsal root stimulation in a RTX-treated and a vehicle-treated rat. B, group data show the effect of chronic morphine on the mean amplitude of evoked NMDAR- and AMPAR-EPSCs and the ratio of NMDAR-EPSCs to AMPAR-EPSCs in vehicle-treated (n = 14 neurons) and RTX-treated (n = 21 neurons) rats. Data from control rats not treated with morphine (n = 16 neurons) were included for comparison. C, original traces show the effect of chronic morphine on NMDAR currents elicited by puff application of 100 μm NMDA to a lamina II neuron in a vehicle-treated rat and an RTX-treated rat. D, group data show the effect of morphine on the mean amplitude of puff-induced NMDAR currents in vehicle-treated (n = 11 neurons) and RTX-treated (n = 16 neurons) rats. Note that data from control rats not treated with morphine (n = 14 neurons) were shown for comparison. *, p < 0.05 when compared with the base-line value in controls not treated with morphine.
To further determine the role of TRPV1-expressing primary afferents in the reduction in postsynaptic NMDAR function induced by morphine, NMDAR currents were elicited by puff 100 μm NMDA directly to the dorsal horn neurons. The morphine-induced reduction in the amplitude of NMDAR currents of lamina II neurons in vehicle-treated rats (n = 11 neurons) was abolished in RTX-treated rats (n = 16 neurons, Fig. 7, C and D). Taken together, these results suggest that increased glutamate release from TRPV1-expressing primary afferents by chronic morphine is responsible for the diminished postsynaptic NMDAR activity of dorsal horn neurons.
Blocking Spinal NMDARs Attenuates the Development of Opioid Tolerance and Hyperalgesia
It has been shown that systemic administration of NMDAR antagonists attenuates opioid analgesic tolerance (2, 8). The results from our current study indicate that increased presynaptic NMDAR activity by chronic morphine increases glutamate release to spinal dorsal horn neurons, a mechanism that plays a major role in both hyperalgesia and analgesia tolerance induced by opioids. We therefore assessed the role of NMDARs at the spinal level in the development of both hyperalgesia and tolerance produced by systemic morphine administration. Morphine tolerance was induced by daily intraperitoneal injection of morphine (10 mg/kg) in rats for 8 consecutive days. AP5 (20 μg) or vehicle was also injected daily for 8 consecutive days through the intrathecal catheter in morphine-treated rats. To test the analgesic effect of morphine, we first injected 5 mg/kg of morphine intraperitoneally and measured the time course of its effect on the paw withdrawal threshold in response to application of the noxious pressure and thermal stimulus. To assess the role of NMDARs at the spinal level in the development of morphine analgesic tolerance, we injected 10 μg of AP5 or vehicle 5 min before morphine administration. Nociceptive testing was performed before and 30 min after morphine injection. The remaining 10 μg of AP5 or vehicle was then injected intrathecally along with another dose of morphine (5 mg/kg, intraperitoneal) 2 h after the behavioral test each day.
For the group of rats injected with vehicle only, daily intraperitoneal injection of morphine gradually attenuated the antinociceptive effect of morphine on the paw withdrawal threshold in response to application of noxious pressure or thermal stimuli. Within 5 days of morphine treatment, acute injection of morphine (5 mg/kg, intraperitoneal) failed to produce a significant effect on the paw withdrawal threshold in vehicle-treated rats (n = 9 rats, Fig. 8, A and B). In contrast, the antinociceptive effect of morphine on the withdrawal threshold was gradually reduced but largely sustained in AP5-treated rats during the 8-day period of morphine treatment (n = 9 rats, Fig. 8, A and B). Even on the last day of treatment, morphine (10 mg/kg, intraperitoneal) still significantly increased the paw withdrawal threshold in the AP5 group.
FIGURE 8.

Blocking NMDARs at the spinal level attenuates the development of hyperalgesia and analgesic tolerance caused by chronic morphine. A, time course of changes in the base-line nociceptive withdrawal thresholds, tested by applying a noxious pressure stimulus to the hind paw, and the analgesic effect of morphine in rats treated with intrathecal injections of vehicle (saline) or AP5 (n = 9 rats in each group). B, time course of changes in the base-line nociceptive withdrawal thresholds, tested by applying a noxious heat stimulus to the hind paw, and the analgesic effect of morphine in rats treated with intrathecal injections of saline or AP5 (n = 9 rats in each group). The base-line withdrawal threshold was measured before morphine injection each day, and the analgesic effect of morphine was tested 30 min after each morphine injection (5 mg/kg, intraperitoneal). *, p < 0.05 when compared with the corresponding morphine effect in the vehicle control. #, p < 0.05 when compared with the corresponding base-line value in the vehicle control.
The base-line paw withdrawal thresholds of vehicle-treated rats in response to the noxious pressure and thermal stimuli, measured 30 min before morphine injection each day, were significantly reduced within 4 days of morphine treatment (n = 9 rats, Fig. 8). However, the base-line paw withdrawal thresholds of AP5-treated rats were not significantly altered during the 8 days of morphine administration (n = 9 rats, Fig. 8, A and B). Even at day 8, the base-line withdrawal threshold in the AP5 group was similar to that measured before the start of morphine treatment. Therefore, these results suggest that increased NMDAR activity at the spinal level plays a critical role in the development of both hyperalgesia and analgesic tolerance induced by systemic opioid administration.
DISCUSSION
Our knowledge of the mechanisms underlying opioid-induced hyperalgesia and tolerance is still fragmentary. Opioid-induced hyperalgesia and tolerance may occur via a similar mechanism. For example, repeated morphine injections into rats progressively reduce the nociceptive threshold of the animals, which occurs in parallel with the development of tolerance (15). Also, many agents that can reduce opioid-induced hyperalgesia, such as PKC inhibitors (1, 32) and NMDAR antagonists (1, 7, 43), attenuate opioid analgesic tolerance as well. Some evidence suggests a link between opioid-induced hyperalgesia and tolerance (44, 45), but the unifying cellular and molecular mechanisms for these two important phenomena remain unknown. Interestingly, brief application of (D-Ala2, N-Me-Phe4, Gly-ol5)-enkephalin, a potent and short-acting μ-opioid receptor agonist, induces a prolonged increase in glutamatergic input to lamina I and II neurons, which is mediated by presynaptic NMDARs (14). We used spinal cord slices for this study because they preserve the intrinsic connection between primary afferents and second-order sensory neurons in the spinal cord and express native μ-opioid receptors. Whole-cell voltage clamp recording in spinal cord slices is a powerful approach because it allows measurement of the synaptic release of glutamate at the cellular level in real time through recordings of the glutamatergic EPSCs in dorsal horn neurons. We found that chronic morphine treatment significantly increased the frequency of sEPSCs of dorsal horn neurons as well as the amplitude of monosynaptic EPSCs of dorsal horn neurons evoked from the dorsal root. Our findings suggest that a persistent increase in glutamate release from nociceptive primary afferents induced by repeated morphine treatment is likely a common mechanism shared by opioid-induced hyperalgesia and analgesic tolerance. Therefore, the concurrent development of hyperalgesia could counteract the opioid analgesic effect, producing an effect that mimics tolerance.
A salient finding of our study is that chronic opioid treatment increases presynaptic NMDAR activity in the spinal dorsal horn. We found that blocking NMDARs with the selective NMDAR antagonist AP5 significantly reduced the frequency, but not the amplitude, of sEPSCs of dorsal horn neurons in morphine-treated rats. AP5 also significantly inhibited the amplitude of monosynaptic EPSCs evoked from the dorsal root in morphine-treated rats; however, it had no significant effect on the frequency of sEPSCs or the amplitude of evoked EPSCs in vehicle-treated rats. Additional evidence for the involvement of presynaptic NMDARs is provided by our observations that AP5 reduced the paired-pulse ratio of evoked EPSCs and that blocking postsynaptic NMDAR channels with the receptor channel blocker MK-801 did not alter the inhibitory effects of AP5 on sEPSCs and evoked EPSCs of lamina II neurons in morphine-treated rats. Increased presynaptic NMDAR activity has been shown in the spinal cord of neonatal rats after chronic morphine exposure (43). Our findings collectively indicate that increased NMDAR activity at the primary afferent terminals plays a critical role in augmented glutamatergic input to spinal dorsal horn neurons by chronic morphine.
The precise signaling mechanisms responsible for increased NMDAR activity at primary afferents after chronic stimulation of the μ-opioid receptors are not fully known. In this study, we found that inhibition of PKC completely normalized the increase in the frequency of sEPSCs in morphine-treated rats, suggesting that this protein kinase plays an essential role in morphine-induced increases in presynaptic NMDAR activity in the spinal cord. In contrast, inhibiting PKA had no significant effect on morphine-induced increases in the frequency of sEPSCs of dorsal horn neurons. Opioid treatment can cause translocation of PKC to the plasma membrane (46, 47) and PKC activation (48). Increased PKC activity could potentiate presynaptic NMDAR function by reducing the Mg2+ block of NMDARs and promoting NMDAR trafficking to the plasma membrane (33, 34).
NMDARs are mainly composed of NR1, NR2A, and NR2B subunits in the spinal cord (49, 50). We found that blocking NR2B-containing NMDARs with Ro 25-6981 and NR2A-containing NMDARs with a low concentration of AP5 significantly reduced the frequency of sEPSCs in morphine-treated rats. Similarly, both Ro 25-6981 and a low concentration of AP5 significantly reduced the amplitude of monosynaptic EPSCs of lamina II neurons evoked from the dorsal root in morphine-treated rats. Triheteromeric NMDARs containing NR1/NR2A/NR2B subunits exist in neurons (51). Thus, it is possible that at least some presynaptic NMDARs in the spinal cord may contain both NR2A and NR2B subunits. Our findings indicate that increased synaptic glutamate release in the spinal cord by chronic morphine is mediated by NR2A- and NR2B-containing NMDARs on primary afferent terminals.
We were surprised to find that the postsynaptic NMDAR activity in the spinal cord is profoundly reduced by chronic morphine treatment. In this regard, the amplitudes of NMDAR-EPSCs and postsynaptic NMDAR currents of dorsal horn neurons elicited by puff NMDA application were significantly decreased in chronic morphine-treated rats. Another interesting finding of our study is that removal of TRPV1-expressing primary afferents by RTX prevented these decreases. These results suggest that the reduction in the postsynaptic NMDAR function induced by morphine is an adaptive response secondary to the increase in presynaptic glutamate release from TRPV1-expressing primary afferents. Ablation of TRPV1-expressing primary afferents by RTX also completely abolished the increase in glutamatergic input to lamina I and II neurons after brief opioid exposure (14). Hence, the decreased postsynaptic NMDAR function by morphine results from persistent glutamate release from TRPV1-expressing primary afferent terminals in the spinal cord. Our findings provide further evidence that TRPV1-expressing primary afferents counteract the analgesic effect of opioids and that opioid-induced hyperalgesia originates from the hyperactivity of nociceptive primary afferents. It is not yet clear how increased glutamate release from these primary afferents leads to the reduction in postsynaptic NMDAR activity in the spinal dorsal horn. It has been shown that NMDARs are subject to agonist-induced internalization through clathrin-mediated endocytosis (52, 53). Also, persistent glutamate release may activate the calcium-dependent protease calpain, which can lead to the cleavage of postsynaptic NMDARs (54, 55). Furthermore, glutamate release augmented by chronic morphine treatment may increase calcineurin activity to down-regulate postsynaptic NMDARs (56).
In addition, we demonstrated in this study that NMDARs at the spinal level contribute to the development of both hyperalgesia and analgesic tolerance induced by chronic morphine treatment. We found that the analgesic effect of this treatment was largely preserved in rats treated with intrathecal injections of AP5. Also, intrathecal AP5 treatment prevented the reduction in the base-line paw withdrawal thresholds caused by repeated morphine injections. Our data suggest that increased presynaptic NMDAR activity and enhanced glutamatergic input in the spinal cord, induced by chronic opioid treatment, are important for both opioid-induced hyperalgesia and opioid-induced tolerance. Consistent with the critical role of presynaptic NMDARs on primary afferents in the development of opioid tolerance, we have previously shown that the duration of the opioid analgesic effect is profoundly increased and that the development of opioid analgesic tolerance is attenuated in RTX-treated rats (15, 18). It is notable that blocking of the NMDARs at the spinal level completely blocked hyperalgesia but only partially attenuated the analgesic tolerance produced by systemic administration of morphine. It is possible that other mechanisms such as reduced function of μ-opioid receptors on primary afferent terminals (15) also contribute to the development of opioid tolerance.
In summary, our study provides important new evidence that chronic opioid treatment increases presynaptic NMDAR activity, but suppresses postsynaptic NMDAR function, in the spinal cord. PKC-mediated increases in presynaptic NMDAR activity contribute to increased glutamatergic input from primary afferents and to opioid-induced hyperalgesia and analgesic tolerance (Fig. 9). Opioid-induced NMDAR hyperactivity at primary afferents is critically involved in this hyperalgesia and tolerance. The pivotal role of presynaptic NMDARs on TRPV1-expressing primary afferents in opioid-induced hyperalgesia and analgesic tolerance is not well recognized; this subpopulation of primary sensory neurons could be targeted to reduce or prevent hyperalgesia and tolerance caused by chronic opioid treatment. This new information is important to our understanding of the underlying mechanisms of opioid-induced hyperalgesia and analgesic tolerance and suggests new strategies to improve the efficacy of opioids in treating various painful conditions.
FIGURE 9.

Diagram depicting the mechanisms involved in changes in pre- and postsynaptic NMDARs in the spinal cord and hyperalgesia and tolerance induced by chronic opioid treatment. Chronic stimulation of μ-opioid receptors leads to increased PKC activity, which augments the activity of NR2A- and NR2B-containing NMDARs present on TRPV1-expressing primary afferent terminals in the spinal cord. This increased activity of presynaptic NMDARs (primary afferent hyperactivity) potentiates synaptic glutamate release and nociceptive input to spinal dorsal horn neurons, and consequently, causes hyperalgesia and opioid tolerance. In contrast, the postsynaptic NMDARs in the spinal dorsal horn are down-regulated in response to increased synaptic glutamate release from TRPV1-expressing primary afferents after chronic opioid treatment.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM064830 and NS073935 (to H. L. P.). This work was also supported by the N. G. and Helen T. Hawkins Endowment (to H. L. P.).

This article contains supplemental Fig. 1.
- NMDAR
- N-methyl-d-aspartic acid receptor
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- AP5
- 2-amino-5-phosphonopentanoic acid
- EPSC
- excitatory postsynaptic current
- sEPSC
- spontaneous excitatory postsynaptic current
- NMDAR-EPSCs
- NMDAR-mediated EPSCs
- AMPAR-EPSCs
- AMPAR-mediated EPSCs
- RTX
- resiniferatoxin
- TRPV1
- transient receptor potential vanilloid type 1
- (Rp)-cAMPS
- adenosine-3′,5′-cyclic monophosphorothioate, Rp isomer.
REFERENCES
- 1. Mao J., Price D. D., Mayer D. J. (1994) Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J. Neurosci. 14, 2301–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trujillo K. A., Akil H. (1991) Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251, 85–87 [DOI] [PubMed] [Google Scholar]
- 3. Antal M., Fukazawa Y., Eördögh M., Muszil D., Molnár E., Itakura M., Takahashi M., Shigemoto R. (2008) Numbers, densities, and colocalization of AMPA- and NMDA-type glutamate receptors at individual synapses in the superficial spinal dorsal horn of rats. J. Neurosci. 28, 9692–9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bardoni R., Torsney C., Tong C. K., Prandini M., MacDermott A. B. (2004) Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J. Neurosci. 24, 2774–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yaksh T. L., Harty G. J., Onofrio B. M. (1986) High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: clinical and theoretic implications. Anesthesiology 64, 590–597 [DOI] [PubMed] [Google Scholar]
- 6. Yaksh T. L., Harty G. J. (1988) Pharmacology of the allodynia in rats evoked by high dose intrathecal morphine. J. Pharmacol. Exp. Ther. 244, 501–507 [PubMed] [Google Scholar]
- 7. Larcher A., Laulin J. P., Celerier E., Le Moal M., Simonnet G. (1998) Acute tolerance associated with a single opiate administration: involvement of N-methyl-d-aspartate-dependent pain facilitatory systems. Neuroscience 84, 583–589 [DOI] [PubMed] [Google Scholar]
- 8. Célèrier E., Rivat C., Jun Y., Laulin J. P., Larcher A., Reynier P., Simonnet G. (2000) Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiology 92, 465–472 [DOI] [PubMed] [Google Scholar]
- 9. Van Elstraete A. C., Sitbon P., Trabold F., Mazoit J. X., Benhamou D. (2005) A single dose of intrathecal morphine in rats induces long-lasting hyperalgesia: the protective effect of prior administration of ketamine. Anesth. Analg. 101, 1750–1756 [DOI] [PubMed] [Google Scholar]
- 10. Chia Y. Y., Liu K., Wang J. J., Kuo M. C., Ho S. T. (1999) Intraoperative high dose fentanyl induces postoperative fentanyl tolerance. Can. J. Anaesth. 46, 872–877 [DOI] [PubMed] [Google Scholar]
- 11. Devulder J. (1997) Hyperalgesia induced by high-dose intrathecal sufentanil in neuropathic pain. J. Neurosurg. Anesthesiol. 9, 146–148 [DOI] [PubMed] [Google Scholar]
- 12. Stubhaug A., Breivik H., Eide P. K., Kreunen M., Foss A. (1997) Mapping of punctate hyperalgesia around a surgical incision demonstrates that ketamine is a powerful suppressor of central sensitization to pain following surgery. Acta Anaesthesiol. Scand. 41, 1124–1132 [DOI] [PubMed] [Google Scholar]
- 13. Chen S. R., Pan H. L. (2006) Blocking mu opioid receptors in the spinal cord prevents the analgesic action by subsequent systemic opioids. Brain Res. 1081, 119–125 [DOI] [PubMed] [Google Scholar]
- 14. Zhou H. Y., Chen S. R., Chen H., Pan H. L. (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J. Neurosci. 30, 4460–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S. R., Prunean A., Pan H. M., Welker K. L., Pan H. L. (2007) Resistance to morphine analgesic tolerance in rats with deleted transient receptor potential vanilloid type 1-expressing sensory neurons. Neuroscience 145, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen S. R., Pan H. L. (2003) Antinociceptive effect of morphine, but not μ-opioid receptor number, is attenuated in the spinal cord of diabetic rats. Anesthesiology 99, 1409–1414 [DOI] [PubMed] [Google Scholar]
- 17. Szallasi A., Blumberg P. M., Annicelli L. L., Krause J. E., Cortright D. N. (1999) The cloned rat vanilloid receptor VR1 mediates both R-type binding and C-type calcium response in dorsal root ganglion neurons. Mol. Pharmacol. 56, 581–587 [DOI] [PubMed] [Google Scholar]
- 18. Chen S. R., Pan H. L. (2006) Loss of TRPV1-expressing sensory neurons reduces spinal μ-opioid receptors but paradoxically potentiates opioid analgesia. J. Neurophysiol. 95, 3086–3096 [DOI] [PubMed] [Google Scholar]
- 19. Zhou H. Y., Chen S. R., Chen H., Pan H. L. (2009) The glutamatergic nature of TRPV1-expressing neurons in the spinal dorsal horn. J. Neurochem. 108, 305–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan H. L., Khan G. M., Alloway K. D., Chen S. R. (2003) Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J. Neurosci. 23, 2911–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pan Y. Z., Pan H. L. (2004) Primary afferent stimulation differentially potentiates excitatory and inhibitory inputs to spinal lamina II outer and inner neurons. J. Neurophysiol. 91, 2413–2421 [DOI] [PubMed] [Google Scholar]
- 22. Woodbury C. J., Ritter A. M., Koerber H. R. (2000) On the problem of lamination in the superficial dorsal horn of mammals: a reappraisal of the substantia gelatinosa in postnatal life. J. Comp. Neurol. 417, 88–102 [DOI] [PubMed] [Google Scholar]
- 23. Chen S. R., Chen H., Yuan W. X., Pan H. L. (2011) Increased presynaptic and postsynaptic α2-adrenoceptor activity in the spinal dorsal horn in painful diabetic neuropathy. J. Pharmacol. Exp. Ther. 337, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li D. P., Yang Q., Pan H. M., Pan H. L. (2008) Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J. Physiol. 586, 1637–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li D. P., Byan H. S., Pan H. L. (2012) Switch to glutamate receptor 2-lacking AMPA receptors increases neuronal excitability in hypothalamus and sympathetic drive in hypertension. J. Neurosci. 32, 372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olverman H. J., Jones A. W., Watkins J. C. (1984) l-Glutamate has higher affinity than other amino acids for [3H]-d-AP5 binding sites in rat brain membranes. Nature 307, 460–462 [DOI] [PubMed] [Google Scholar]
- 27. Zhou H. Y., Chen S. R., Chen H., Pan H. L. (2008) Sustained inhibition of neurotransmitter release from nontransient receptor potential vanilloid type 1-expressing primary afferents by μ-opioid receptor activation-enkephalin in the spinal cord. J. Pharmacol. Exp. Ther. 327, 375–382 [DOI] [PubMed] [Google Scholar]
- 28. Avidor-Reiss T., Nevo I., Saya D., Bayewitch M., Vogel Z. (1997) Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J. Biol. Chem. 272, 5040–5047 [DOI] [PubMed] [Google Scholar]
- 29. Jolas T., Nestler E. J., Aghajanian G. K. (2000) Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience 95, 433–443 [DOI] [PubMed] [Google Scholar]
- 30. Connelly P. A., Botelho L. H., Sisk R. B., Garrison J. C. (1987) A study of the mechanism of glucagon-induced protein phosphorylation in isolated rat hepatocytes using (Sp)-cAMPS and (Rp)-cAMPS, the stimulatory and inhibitory diastereomers of adenosine cyclic 3′,5′-phosphorothioate. J. Biol. Chem. 262, 4324–4332 [PubMed] [Google Scholar]
- 31. Xie C. W., Lewis D. V. (1997) Involvement of cAMP-dependent protein kinase in μ-opioid modulation of NMDA-mediated synaptic currents. J. Neurophysiol. 78, 759–766 [DOI] [PubMed] [Google Scholar]
- 32. Granados-Soto V., Kalcheva I., Hua X., Newton A., Yaksh T. L. (2000) Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain 85, 395–404 [DOI] [PubMed] [Google Scholar]
- 33. Lan J. Y., Skeberdis V. A., Jover T., Grooms S. Y., Lin Y., Araneda R. C., Zheng X., Bennett M. V., Zukin R. S. (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat. Neurosci. 4, 382–390 [DOI] [PubMed] [Google Scholar]
- 34. Chen L., Huang L. Y. (1992) Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356, 521–523 [DOI] [PubMed] [Google Scholar]
- 35. Herbert J. M., Augereau J. M., Gleye J., Maffrand J. P. (1990) Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 172, 993–999 [DOI] [PubMed] [Google Scholar]
- 36. Hansel C., Linden D. J. (2000) Long-term depression of the cerebellar climbing fiber-Purkinje neuron synapse. Neuron 26, 473–482 [DOI] [PubMed] [Google Scholar]
- 37. Monyer H., Sprengel R., Schoepfer R., Herb A., Higuchi M., Lomeli H., Burnashev N., Sakmann B., Seeburg P. H. (1992) Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256, 1217–1221 [DOI] [PubMed] [Google Scholar]
- 38. Vicini S., Wang J. F., Li J. H., Zhu W. J., Wang Y. H., Luo J. H., Wolfe B. B., Grayson D. R. (1998) Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J. Neurophysiol. 79, 555–566 [DOI] [PubMed] [Google Scholar]
- 39. Fischer G., Mutel V., Trube G., Malherbe P., Kew J. N., Mohacsi E., Heitz M. P., Kemp J. A. (1997) Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit: characterization in vitro. J. Pharmacol. Exp. Ther. 283, 1285–1292 [PubMed] [Google Scholar]
- 40. Kimura R., Matsuki N. (2008) Protein kinase CK2 modulates synaptic plasticity by modification of synaptic NMDA receptors in the hippocampus. J. Physiol. 586, 3195–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu L. J., Toyoda H., Zhao M. G., Lee Y. S., Tang J., Ko S. W., Jia Y. H., Shum F. W., Zerbinatti C. V., Bu G., Wei F., Xu T. L., Muglia L. J., Chen Z. F., Auberson Y. P., Kaang B. K., Zhuo M. (2005) Up-regulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 25, 11107–11116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ye Z. Y., Li L., Li D. P., Pan H. L. (2012) Casein kinase 2-mediated synaptic GluN2A up-regulation increases N-methyl-d-aspartate receptor activity and excitability of hypothalamic neurons in hypertension. J. Biol. Chem. 287, 17438–17446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeng J., Thomson L. M., Aicher S. A., Terman G. W. (2006) Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J. Neurosci. 26, 12033–12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laulin J. P., Célèrier E., Larcher A., Le Moal M., Simonnet G. (1999) Opiate tolerance to daily heroin administration: an apparent phenomenon associated with enhanced pain sensitivity. Neuroscience 89, 631–636 [DOI] [PubMed] [Google Scholar]
- 45. Colpaert F. C. (1996) System theory of pain and of opiate analgesia: no tolerance to opiates. Pharmacol. Rev. 48, 355–402 [PubMed] [Google Scholar]
- 46. Kramer H. K., Simon E. J. (1999) Role of protein kinase C (PKC) in agonist-induced μ-opioid receptor down-regulation: I. PKC translocation to the membrane of SH-SY5Y neuroblastoma cells is induced by μ-opioid agonists. J. Neurochem. 72, 585–593 [DOI] [PubMed] [Google Scholar]
- 47. Mayer D. J., Mao J., Price D. D. (1995) The development of morphine tolerance and dependence is associated with translocation of protein kinase C. Pain 61, 365–374 [DOI] [PubMed] [Google Scholar]
- 48. Chen L., Huang L. Y. (1991) Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a μ-opioid. Neuron 7, 319–326 [DOI] [PubMed] [Google Scholar]
- 49. Momiyama A. (2000) Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurons of the adult rat spinal cord. J. Physiol. 523, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagy G. G., Watanabe M., Fukaya M., Todd A. J. (2004) Synaptic distribution of the NR1, NR2A and NR2B subunits of the N-methyl-d-aspartate receptor in the rat lumbar spinal cord revealed with an antigen-unmasking technique. Eur. J. Neurosci. 20, 3301–3312 [DOI] [PubMed] [Google Scholar]
- 51. Rauner C., Köhr G. (2011) Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem. 286, 7558–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nong Y., Huang Y. Q., Ju W., Kalia L. V., Ahmadian G., Wang Y. T., Salter M. W. (2003) Glycine binding primes NMDA receptor internalization. Nature 422, 302–307 [DOI] [PubMed] [Google Scholar]
- 53. Vissel B., Krupp J. J., Heinemann S. F., Westbrook G. L. (2001) A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 4, 587–596 [DOI] [PubMed] [Google Scholar]
- 54. Guttmann R. P., Sokol S., Baker D. L., Simpkins K. L., Dong Y., Lynch D. R. (2002) Proteolysis of the N-methyl-d-aspartate receptor by calpain in situ. J. Pharmacol. Exp. Ther. 302, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 55. Wu H. Y., Yuen E. Y., Lu Y. F., Matsushita M., Matsui H., Yan Z., Tomizawa K. (2005) Regulation of N-methyl-d-aspartate receptors by calpain in cortical neurons. J. Biol. Chem. 280, 21588–21593 [DOI] [PubMed] [Google Scholar]
- 56. Tong G., Shepherd D., Jahr C. E. (1995) Synaptic desensitization of NMDA receptors by calcineurin. Science 267, 1510–1512 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.