

Background: Chk1 regulates both nuclear and cytoplasmic checkpoints.
Results: The Chk1 polypeptide contains mechanisms mediating its cellular localization.
Conclusion: The nuclear (but not cytoplasmic) pool of Chk1 is essential for cell viability.
Significance: Dissecting mechanisms regulating the mobilization of Chk1 within different cellular compartments is critical for understanding checkpoint activation and its roles in cell biology.
Keywords: Cell Cycle, Checkpoint Control, DNA Damage, Nuclear Translocation, Signal Transduction, Checkpoints, Chk1, Protein Localization, Cell Viability, Somatic Gene Targeting
Abstract
Chk1 plays a key role in regulating the replication checkpoint and DNA damage response. Recent evidence suggests that mammalian Chk1 regulates both the nuclear and cytoplasmic checkpoint events. However, mechanisms regulating cellular mobilization of Chk1 were not well understood. Here, we report the identification of regions of human Chk1 that regulate its protein cellular localization and checkpoint function. We demonstrate that the two highly conserved motifs (CM1 and CM2) at the C terminus of Chk1 function as a nuclear export signal and nuclear localization signal, respectively. Mutating five highly conserved residues within these two motifs of Chk1 resulted in its accumulation mainly in the cytoplasm. These cytoplasmic Chk1 mutants were less stable and exhibited significantly reduced phosphorylation by DNA damage treatment, yet they retained, at least partially, checkpoint function. Using an adenovirus-mediated gene targeting technique, we attempted to create an HCT116 cell line in which endogenous Chk1 is mutated so that it is expressed exclusively in the cytoplasm. However, we failed to obtain homozygous mutant cell lines. We found that even the heterozygous mutant cell lines showed cell survival defects accompanied by spontaneous cell death. Together, these results reveal novel regulatory mechanisms that couple protein cellular localization with the checkpoint response and cell viability of Chk1.
Introduction
The genomes of eukaryotic cells are under constant assault by DNA-damaging agents presented both in the environment (such as UV light, ionization radiation, and chemical toxins) and within the cells (for instance, free radical species). If not properly handled, these DNA lesions could lead to the loss of genome integrity, fueling the selection of malignant clones that could develop into devastating cancers (1). Evolution has overlaid the core cell cycle machinery with elegant complex networks of pathways that respond to DNA damage. These genome surveillance pathways are collectively termed the DNA damage response, which, when activated, can slow or stall cell cycle progression and launch repair programs to fix the errors (1, 2). The DNA damage response contains three sequential steps: 1) the initial sensing of the damage, 2) the transduction of the damage signal, and 3) the execution of cell cycle progression inhibition and DNA damage repair (1, 3). Two large PI3K-related protein kinases, ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia mutated and Rad3-related), regulate the initial damage sensing. On the other hand, another two protein kinases, Chk1 and Chk2, are critical for relaying the DNA damage signal in step 2 via functioning as downstream targets of ATR and ATM, respectively (3).
Chk1 plays a crucial role in responding to a wide range of genotoxic stresses from S to G2/M phase of the cell division cycle (4). Consistent with this idea, loss of CHK1 leads to early embryonic lethality in mice (5, 6). Similarly, inhibition of Chk1 by either chemical inhibitors or RNA interference renders somatic cells vulnerable to genotoxic stress (7). Activation of Chk1 requires protein phosphorylation at two conserved sites, Ser-317 and Ser-345, by the upstream kinase ATR. This phosphorylation in turn activates Chk1, which then phosphorylates a number of downstream targets to control cell cycle transition and DNA damage repair.
Recent evidence suggests that Chk1 undergoes dynamic cellular mobilization both under normal growth conditions and upon DNA damage. Under normal growth conditions, Chk1 is expressed mainly in the nucleus, including the chromatin-enriched cellular compartment (8–10). In the absence of DNA damage, Chk1 phosphorylation by Cdk1 at sites distinct from Ser-317 and Ser-345 is required to trigger the nuclear export of Chk1 and to mediate the G2/M phase cell cycle transition (11). Upon DNA damage, Chk1 proteins on chromatin-enriched fractions are phosphorylated by ATR (8, 10, 12). This phosphorylation seems to trigger a rapid release of phosphorylated Chk1 from the chromatin-enriched compartment into the soluble nucleus and later to the cytoplasm (8, 10). Accumulating evidence suggests that phosphorylated Chk1 proteins are able to regulate downstream checkpoint events both in the nucleus and in the cytoplasm. For instance, they activate the Rad51-dependent damage repair in the nucleus (13). On the other hand, activated Chk1 may inhibit centrosomal cyclin B/Cdk1 activity and stabilize the mRNA in the cytoplasm (14, 15). At the later time point, phosphorylated Chk1 undergoes SCFFbx6 and/or Cul4A/DDB1 ubiquitin ligase-dependent degradation in the cytoplasm (16, 17). These results suggest that cellular localization of Chk1 is important for checkpoint function.
However, molecular mechanisms mediating Chk1 protein localization remained unclear. In this study, we identified a previously unknown function of Chk1 that controls the protein cellular localization, and we linked the cellular localization of Chk1 to checkpoint activation and maintenance of cell viability.
EXPERIMENTAL PROCEDURES
Cell Cultures, Transfection, Cell Proliferation, and Cell Death
HEK293T, HeLa, U2-OS, and A549 cells were cultured in DMEM with 10% FBS. HCT116 cells were grown in McCoy's 5A medium and 10% FBS. HEK293T cells were transfected with calcium phosphate, whereas other cell lines were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. To measure cell proliferation, WT Chk1 or F380D mutant knock-in heterozygous HCT116 cells were plated in 12-well plates at 1 × 104 cells/well and cultured in complete medium at 37 °C with 5% CO2. From day 3, three wells of cells from either WT or F380D Chk1 were trypsinized, and the number of total viable or dead cells was counted each day. Cell death was measured using trypan blue staining as described previously (16).
Plasmid Construction and Mutagenesis
WT Chk1 or mutants were generated using PCR. WT Chk1 or mutants were cloned into either the pCMV-6×Myc or pEGFP-C1 vector. Point mutations were performed using the QuikChange mutagenesis kit (Stratagene) according to the manufacturer's protocol.2
Immunoblotting, Immunofluorescence, and Antibodies
Immunoblotting was carried out as described previously (16, 18). Anti-Chk1 (DCS-310 and G4) and anti-ATR (N-19) antibodies were from Santa Cruz Biotechnology. Anti-phospho-Ser-317 Chk1, anti-phospho-Ser-345 Chk1, anti-phospho-Ser-1981 ATM, and anti-phospho-Ser-216 Cdc25C antibodies were from Cell Signaling. Anti-MCM7 and anti-cyclin B antibodies were from Pharmingen. Anti-Cdc25A antibody was from NeoMarkers.
For immunofluorescence, U2-OS or HeLa cells grown on glass coverslips were transfected with Myc- or GFP-tagged WT Chk1 or mutants for 48 h. The cells were rinsed once with PBS, fixed with 3.75% formaldehyde and 0.2 m sucrose in PBS for 15 min at room temperature, washed three times with 0.1 m glycine in PBS, and stored in 0.1 m glycine and PBS at 4 °C. Fixed cells were permeabilized and blocked with 10% FBS and 0.2% Triton X-100 in PBS for 20 min at room temperature, washed three times with wash buffer (0.2% Triton X-100 and 0.1% BSA in PBS), and incubated or not with mouse monoclonal anti-Myc antibodies (1:500 dilution in 0.1% Triton X-100 in PBS) overnight at 4 °C. Cells were then washed three times with wash buffer, incubated in Texas Red-conjugated goat anti-mouse secondary antibodies (1:200 dilution in 0.1% Triton X-100 in PBS) for 1 h at room temperature, washed three times with wash buffer, mounted with antifade mounting solution (Invitrogen), stored overnight at −20 °C, and visualized under a fluorescence microscope.
DNA Replication by 5-Ethynyl-2′-deoxyuridine Incorporation
HeLa cells were plated on glass coverslips; transfected with doxycycline-controlled GFP, GFP-WT Chk1, or GFP-F380D Chk1 for 24 h; synchronized at G2/M phase with 100 ng/ml nocodazole for 20 h; washed with 1× PBS; and released into fresh medium. After 16 h, cells were labeled with the nucleotide analog 5-ethynyl-2′-deoxyuridine (10 μm) for 20 min; washed with ice-cold PBS; and then fixed with 3.7% formaldehyde at room temperature for 10 min, followed by use of the Click-iT kit (Invitrogen). 5-Ethynyl-2′-deoxyuridine incorporation into cells was analyzed by fluorescence microscopy.
Somatic Cell Gene Targeting
The approach for generating targeted cells with adeno-associated virus (AAV)3 was performed as described (19, 20). The targeting vectors were constructed by PCR with HCT116 genomic DNA as the template for the homologous arms. The targeting AAVs were packaged in 293AAV cells by transfecting equal amounts of the targeting vector, pHelper, and pRC plasmids. Viruses were harvested 72 h after transfection. HCT116 cells were infected with the targeting viruses and selected with Geneticin for 20 days. The Geneticin-resistant clones were then screened for homologous recombination by genomic PCR with primers derived from the neomycin resistance gene and the upstream region of the left homologous arm. After the first allele was targeted, the neomycin resistance gene was excised with Cre recombinase. We attempted to generate a homologous cell line through targeting two different heterozygous cell lines with the AAVs.
The primers used in the gene targeting were as follows: Chk1-F380D-LA-S, GGGAAAG/ideoxyU/catggtgaaaccccatctct; Chk1-F380D-LA-AS: GGAGACA/ideoxyU/tcctctctttcttccttgacca; Chk1-F380D-RA-S, GGTCCCA/ideoxyU/gggaggccttcatgcaaaat; Chk1-F380D-RA-AS, GGCATAG/ideoxyU/ttgaaaagccttaaagacagtttaga; Chk1-F380D-screening-S-1, agccaggtctgttgtcttca; Chk1-F380D-screening-AS-1, tttgctttgacacttgttctcc; Chk1-F380D-screening-S-2, tcctttagccaggtctgttg; Chk1-F380D-screening-AS-2, ttcacttcaactttgctttgaca; Chk1-F380D-Cre-S, gttacttggcaccccaggat; Chk1-F380D-Cre-AS, cattcttttgaccaaccgc; Neo-R, GTTGTGCCCAGTCATAGCCG; and Neo-F, TCTGGATTCATCGACTGTGG.
Clonogenic Assay
WT Chk1 or F380D mutant knock-in heterozygous HCT116 cell lines were treated with different doses of UV light, camptothecin (CPT), or hydroxyurea (HU) for 2 h. The cells were then replated into 6-well plates with 1 × 104 viable cells in each well with three wells per group and cultured for another 7 days or until the colonies grew to the appropriate size. The cells were washed once with prewarmed Hanks' balanced salt solution and stained with 2 ml of 0.2% crystal violet in 50% methanol for 30 min at room temperature. After washing five times with de-ionized water, pictures were taken under a microscope. Cells were then solubilized into 2 ml of 0.5% SDS in 50% methanol at 37 °C for 30 min. Samples were diluted, and the absorption at 595 nm was measured using a plate reader in 96-well plates.
RESULTS
C Terminus of Chk1 Is Responsible for Its Nuclear-Cytoplasmic Shuttling
Previous studies suggest that Chk1 undergoes dynamic nuclear-cytoplasmic shuttling both under normal growth conditions and upon DNA damage (8, 10, 11). Importantly, this dynamic cellular mobilization seems to be critical for checkpoint function (8, 10). However, the detailed molecular mechanisms underlying this dynamic cellular mobilization of Chk1 remained unclear. Therefore, to understand how this nuclear-cytoplasmic shuttling is achieved, we attempted to define regions that regulate the cellular localization of human Chk1.
Chk1 contains a highly conserved N-terminal kinase domain and a C-terminal regulatory domain. The C terminus of Chk1 contains a Ser/Gln motif with the two ATR phosphorylation sites (Ser-317 and Ser-345) and two highly conserved motifs (CM1 and CM2) (Fig. 1A). To identify regions that regulate cellular localization of Chk1, we first generated truncated Chk1 mutants with deletions of the N-terminal kinase domain (mutant C), the C terminus (mutant N), the CM2 domain (mutant 1–421), or the CM1 and CM2 domains (mutant 1–368) (Fig. 1A). Full-length Chk1 or the truncated mutants were expressed as 6-Myc-tagged fusion proteins in U2-OS cells, and cellular localization was determined by immunofluorescence. The results showed that the full-length Chk1 protein was located mainly in the nucleus with diffuse cytoplasmic staining (Fig. 1B). The N-terminal kinase domain of Chk1 was located both in the nucleus and in the cytoplasm (Fig. 1B), similar to the GFP signal (Fig. 2A). This is typical for proteins that can freely pass the nuclear pore (21), suggesting that the N terminus of Chk1 does not actively mediate its cellular localization. In contrast, the Chk1 C terminus was localized mainly in the nucleus (Figs. 1B and 2A). Interestingly, deletion of the CM2 domain (mutant 1–421) significantly increased the cytoplasmic signal of Chk1 (Fig. 1B); however, further deletion of the CM1 domain (mutant 1–368) induced re-accumulation of Chk1 in the nucleus (Fig. 1B). These data indicate that the C terminus contains both a nuclear export signal (NES) and nuclear localization signal (NLS) that coordinately control the nuclear-cytoplasmic shuttling of Chk1.
FIGURE 1.

Cellular localization of Chk1. A, schematic diagram of human Chk1 and generation of deletion mutation strategy. SQ, Ser/Gln cluster; CM, conserved motif; FL, full-length (476 amino acids); N, N terminus (amino acids 1–264); C, C terminus (amino acids 265–476). B, U2-OS cells grown on glass coverslips were transfected with Myc-tagged vectors expressing full-length Chk1 or mutants for 48 h, fixed, stained with anti-Myc antibodies, and visualized using a fluorescence microscope (63× oil lens).
FIGURE 2.

Crm1-dependent nuclear export of Chk1. A, U2-OS cells grown on glass coverslips were transfected with GFP-tagged full-length (FL) Chk1 or mutants, fixed, and visualized under a fluorescence microscope (63× oil lens). B, U2-OS cells grown on glass coverslips were transfected with the GFP-Chk1 C7 fragment with the indicated mutations for 48 h, fixed, and visualized under a fluorescence microscope (63× oil lens). C, U2-OS cells grown on glass coverslips were transfected with the GFP-Chk1 C7 fragment for 48 h; treated with 6 ng/ml LMB for 0, 3, or 6 h; fixed; and visualized under a fluorescence microscope (63× oil lens).
CM1 and CM2 Domains Function as an NES and NLS, Respectively
To further define the regions of the C terminus of human Chk1 that function as an NES and NLS, we generated shorter Chk1 fragments targeting the C terminus of Chk1 (C1–C6) (Fig. 1A) as either Myc- or GFP-tagged fusion proteins and determined their localization in cells. The C1, C3, and C5 fragments were localized mainly in the nucleus, like the C-terminal domain (Fig. 1B). The C6 fragment (containing the CM2 domain of the distal C terminus of Chk1) was localized exclusively in the nucleus (Fig. 1B). However, the C4 fragment was located mainly in the cytoplasm (Figs. 1B and 2A). Previously, it was reported that sequences around the Ser-345 site of Chk1 might function as an NES (9, 11). However, the Chk1 C2 fragment, which covers this region of Chk1 (Fig. 1A), was strongly localized in the nucleus (Fig. 1B, Myc-Chk1 C2 panel). This suggests that residues flanking the Ser-345 site only marginally regulate the nuclear export of Chk1.
The C4 fragment comprises the C2 fragment, the CM1 domain, and a region with ∼20 residues immediately after the CM1 domain (Fig. 1A). Because the C2 fragment (containing the Ser-345 site) did not seem to actively regulate the nuclear export of Chk1, we hypothesized that the CM1 plus 20-amino acid domain is responsible for the nuclear export of Chk1. To directly test this idea, we generated a GFP-tagged C7 fragment, which covers this region (Fig. 1A). We showed that this GFP-Chk1 C7 fragment was localized exclusively in the cytoplasm (Fig. 2A). A summary of protein localization of these Chk1 mutants provided shown in supplemental Fig. S1A. Together, these results illustrate that regions flanking the CM1 and CM2 domains of Chk1 function as an NES and NLS, respectively (supplemental Fig. S1C).
Nuclear Export of Chk1 is Crm1-dependent
The region flanking the CM2 domain has been shown to regulate nuclear localization of Xenopus Chk1 (22). It contains multiple putative NLSs, e.g. clusters of basic residues (KK and (R/K)X(K/R)). Protein nuclear export is often regulated by Crm1, which binds to a leucine-rich (LXXXLXXLXL) NES in the substrate (21). Even though the CM1 region of Chk1 does not contain canonical NESs, it does have an NES-like sequence (LXXXMXXFF) located at residues 371–381 in human Chk1, in which Leu-373 is absolutely conserved (supplemental Fig. S1C). In addition, the region immediately following the CM1 domain contains three Leu residues (Leu-384, Leu-393, and Leu-400 in human Chk1) that are conserved in mammals (supplemental Fig. S1C), which may form a functional NES when assembled into a three-dimensional structure (21). To confirm that this is the region that regulates Chk1 nuclear export, we mutated several highly conserved hydrophobic residues in this region of the GFP-Chk1 C7 fragment. The results showed that mutating the absolutely conserved Leu-373 residue to Arg or Ala led to significant accumulation of the GFP-Chk1 C7 fragment in the nucleus (Fig. 2B and data not shown). Mutating Val-374 to Asn (the V374N mutant) also induced nuclear accumulation of the GFP-Chk1 C7 fragment (Fig. 2B). However, mutating those three less well conserved Leu residues (Leu-384, Leu-393, and Leu-400) had little or no effect (Fig. 2B). These data indicate that the CM1 domain functions as a non-canonical NES for Chk1 nuclear export.
One possibility for the cytoplasmic localization of the GFP-Chk1 C7 fragment is the lack of an NLS in this fragment. If this were the case, then we would expect that blocking Crm1-dependent nuclear export should not induce nuclear accumulation of this Chk1 mutant. However, the results showed that treatment with leptomycin B (LMB), a Crm1 inhibitor (21), induced accumulation of the GFP-Chk1 C7 fragment in the nucleus in a time-dependent fashion (Fig. 2C). As a control, LMB treatment induced further accumulation of the C6 mutant in the nucleus (supplemental Fig. S1B). These data suggest that the cytoplasmic localization of the GFP-Chk1 C7 fragment is driven by an NES but is not due to the lack of NLSs.
Coupling Cellular Localization and Protein Phosphorylation of Chk1
To further understand the mechanisms regulating Chk1 cellular localization, we intended to identify critical residues within the CM1 and CM2 domains that control Chk1 cellular localization. To this end, we mutated essentially every residue within these two domains and examined protein localization of these point mutants. The results showed that although the majority of point mutants retained the similar cellular localization as WT Chk1, mutating one of five residues (Phe-380, Phe-441, Gly-446, Phe-451, or Phe-455) caused cytoplasmic accumulation of Chk1 (supplemental Fig. S2), suggesting that these residues may play important roles in controlling Chk1 cellular localization. To understand how these mutants accumulated in the cytoplasm, we treated cells expressing these mutants with LMB and monitored the protein localization. The results showed that LMB treatment caused nuclear accumulation of all five Chk1 mutants (Fig. 3A and data not shown). These data indicate that cytoplasmic accumulation of these mutants is due to Crm1-dependent nuclear export as well.
FIGURE 3.

Protein cellular localization and phosphorylation of Chk1. A, U2-OS cells grown on glass coverslips were transfected with GFP-tagged Chk1 mutants for 48 h, treated or not with 6 ng/ml LMB for 6 h, fixed, and visualized under a fluorescence microscope (63× oil lens). B, HEK293T cells were transfected with GFP-tagged full-length (FL) Chk1 or mutants for 48 h, treated or not with 500 nm CPT for 2 h, and immunoblotted with the indicated antibodies. Exogenous (Exo.) and endogenous (Endo.) Chk1 proteins are shown by arrows.
Under normal conditions, Chk1 is localized and phosphorylated by ATR mainly in the nucleus (8, 10). This led us to test the protein phosphorylation of these five Chk1 mutants upon DNA damage. We expressed GFP-WT Chk1 or mutants in HEK293T cells, treated the cells with a DNA-damaging agent (the topoisomerase I inhibitor CPT), and examined the protein phosphorylation of both exogenous GFP-Chk1 and endogenous Chk1 proteins. The results showed that although GFP-WT Chk1 was highly phosphorylated by CPT treatment, phosphorylation of the five cytoplasmic mutants of Chk1 was rarely detected (Fig. 3B, lanes 5–12). The failure to detect Ser-345 phosphorylation of these cytoplasmic Chk1 mutants was not due to the lack of DNA damage response, as endogenous Chk1 was equally phosphorylated in cells expressing each mutant (Fig. 3B). A summary of protein localization and phosphorylation for these point mutants of Chk1 is shown in supplemental Fig. S3. These data provide experimental evidence to illustrate a correlation between the cytoplasmic localization and protein phosphorylation defects of Chk1, an idea suggested previously (8, 10).
Cytoplasmic Chk1 Partially Retains Checkpoint Function
During our analyses, we noticed that the cytoplasmic Chk1 mutants were less stable than WT Chk1 (Fig. 3B and supplemental Fig. S4A), consistent with the idea that Chk1 undergoes proteasome-dependent degradation in the cytoplasm (8, 16, 17). Therefore, the lack of phosphorylation of these mutants might be due to reduced protein expression. To address this issue, we investigated whether blocking protein degradation by inhibiting the proteasome could enable us to detect phosphorylation of the cytoplasmic Chk1 mutants. We observed that treatment with the proteasome inhibitor MG132 alone caused accumulation of Ser-345-phosphorylated proteins for GFP-WT Chk1, GFP-F380D, and GFP-G446N (Fig. 4A, lanes 1 and 3, and supplemental Fig. S4B). Addition of CPT increased the phosphorylation level of both GFP-WT Chk1 and mutant proteins (Fig. 4A, lanes 1–4). These data demonstrate that cytoplasmic Chk1 mutant proteins can still be phosphorylated at ATR sites in cell cultures. However, we do realize that the CPT-induced phosphorylation of the cytoplasmic mutants of Chk1 was much less than that of WT or endogenous Chk1 (Fig. 4A). This is probably because those mutants spent much less time in the nucleus, thereby showing less phosphorylation by ATR than WT or endogenous Chk1.
FIGURE 4.
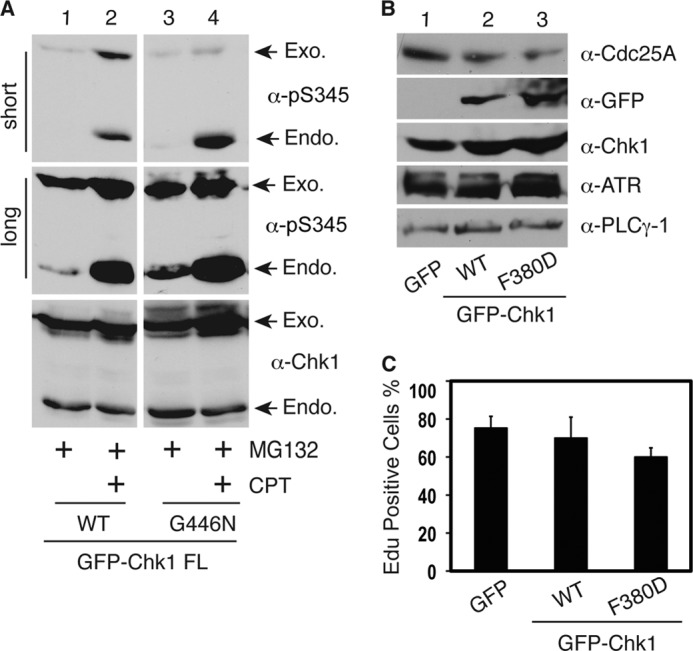
Cytoplasmic Chk1 mutants retain checkpoint function. A, HEK293T cells were transfected with GFP-tagged vectors expressing WT Chk1 or the G446N mutant for 48 h, treated or not with the proteasome inhibitor MG132 (2 μm) for 12 h, treated or not with 500 nm CPT for an additional 2 h, and immunoblotted with the indicated antibodies. Short and long exposure results for the anti-phospho-Ser-345 antibodies are shown. Exogenous (Exo.) and endogenous (Endo.) Chk1 proteins are indicated by arrows. B, HEK293T cells were transfected with vectors expressing GFP, GFP-WT Chk1, or GFP-F380D for 48 h and immunoblotted with the indicated antibodies. α-PLCγ-1, anti-phospholipase Cγ1 antibody. C, HeLa cells plated on glass coverslips were transfected with GFP, GFP-WT Chk1, or GFP-F380D for 24 h; synchronized at G2/M phase by 100 ng/ml nocodazole treatment for 20 h; washed; and released into fresh medium. After 16 h, cells were labeled with 10 μm 5-ethynyl-2′-deoxyuridine (EdU; a nucleotide analog) for 20 min and fixed, and 5-ethynyl-2′-deoxyuridine-positive cells were counted under a fluorescence microscope. Data represent the mean ± S.D. from three independent experiments.
Because these cytoplasmic Chk1 mutants can still be phosphorylated by ATR, we investigated whether they are functional. To address this issue, we first expressed GFP-tagged WT Chk1 or mutants in HEK293T cells, immunoprecipitated GFP-Chk1 proteins by anti-GFP antibodies, and used these Chk1 proteins to perform in vitro kinase assays. A GST-Cdc25C fragment (residues 200–256) was used as the substrate. This Cdc25C fragment contains a known Chk1 phosphorylation site (Ser-216); thus, it is a surrogate for assessing the catalytic activity of Chk1 (16). Our data showed that all mutants retained the basal catalytic activity toward Cdc25C, which is comparable with WT Chk1 (supplemental Fig. S4C and data not shown), suggesting that mutation of these sites does not alter the basic catalytic activity of Chk1. We then transfected HEK293T cells with vectors expressing GFP, GFP-WT Chk1, or GFP-F380D and examined the expression level of Cdc25A. Cdc25A is a key Chk1 downstream target that plays a crucial role in regulating both the entry into and progression of S phase (23). Activation of Chk1 leads to proteasome-dependent degradation of Cdc25A, followed by S phase progression inhibition (23). Our results showed that when expression of the F380D mutant protein was equivalent to that of WT Chk1 in cells, it caused a similar reduction in the level of Cdc25A compared with WT Chk1 (Fig. 4B, lanes 2 and 3).
In consistence with the reduction in Cdc25A, we observed that cells expressing the F380D mutant and, to a lesser extent, WT Chk1 demonstrated a delayed S phase cell cycle entry as indicated by the reduced incorporation of 5-ethynyl-2′-deoxyuridine (a nucleotide analog) into DNA compared with the GFP control (Fig. 4C). Together, these data indicate that the cytoplasmic mutants of Chk1 can still localize to chromatin to be phosphorylated by ATR, followed by release from the chromatin into the nucleus and cytoplasm, like WT Chk1. However, this dynamic cellular mobilization process appears to be significantly accelerated for the mutants probably due to the enhanced NES (the F380D mutant) or disrupted NLS (Phe-441, Gly-446, Phe-451, and Phe-455 mutants). As a result, their phosphorylation at ATR sites was barely detected in the absence of proteasome inhibition because the phosphorylated proteins were quickly degraded in the cytoplasm. Nevertheless, these findings suggest that cytoplasmic Chk1 retains, at least partially, checkpoint function (see model in Fig. 8).
FIGURE 8.
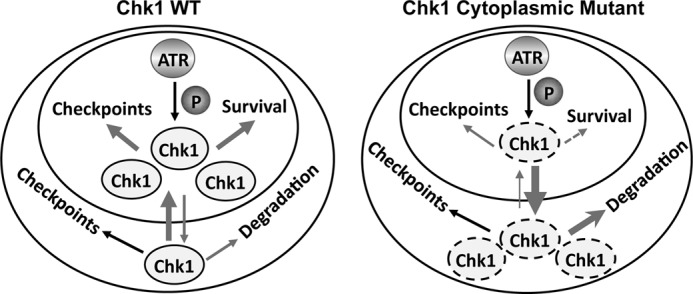
Model of correlation between cellular localization and function of Chk1. The inner and outer circles represent the nucleus and cytoplasm, respectively. Cytoplasmic Chk1 mutants (dashed ovals) exhibit a significantly accelerated pace of nuclear export, leaving them little time to accumulate in the nucleus to be phosphorylated by ATR or to perform functions essential for cell viability and, to a lesser extent, checkpoints. In the meantime, the mutant form of Chk1 in the cytoplasm is unstable, causing the reduction of the total level of the Chk1 mutant in the cells. The arrow thickness roughly indicates the intensity of events. The dashed arrow indicates functional defects. See “Discussion” for more detail.
Cytoplasmic Chk1 Does Not Support Cell Viability
Recently, increasing evidence suggests that Chk1 regulates both nuclear and cytoplasmic checkpoint events (15, 24). CHK1 is an essential gene (5). Thus, an important question is which pool, nuclear or cytoplasmic, contributes to the cell essential function of Chk1? To address this question, we utilized the AAV-mediated gene targeting technique to create an HCT116 colorectal cancer cell line (19, 20) in which CHK1 loci are mutated to express Chk1 mutant proteins that will exclusively accumulate in the cytoplasm. Because all five cytoplasmic Chk1 mutants behaved equally in terms of protein location, catalytic activity, and ability to be phosphorylated by ATR, we reasoned that generating one mutant cell line is adequate to address this question. We chose to mutate Phe-380 to Asp (see experimental design in Fig. 5A) because the genomic DNA around this residue appears to be suitable for gene targeting. We successfully generated heterozygous cell lines with one allele of CHK1 mutated as confirmed by genomic DNA sequencing (Fig. 5B). However, after screening >300 clones, we were unable to identify a single homozygous clone with both alleles mutated. Resistant clones appeared to be either retargeting the same allele or drug-resistant (data not shown).
FIGURE 5.

Somatic mutation of CHK1. A, schematic diagram of the generation of a somatic cell line with the Chk1 F380D mutation. The endogenous (Endo) locus is targeted to mutate Phe-380 to Asp (shown by red arrows). B, sequencing results of genomic DNA from heterozygous cell lines. Note the mixed WT and F380D mutant signal. C, asynchronously growing WT and heterozygous HCT116 cell lines were lysed, and equal amounts of total proteins were loaded for detection of the indicated proteins. α-PCNA, anti-proliferating cell nuclear antigen antibody. D, WT and heterozygous HCT116 cell lines were synchronized at G2/M phase by 100 ng/ml nocodazole treatment for 20 h, released into different cell cycle stages, and immunoblotted with the indicated antibodies. Chk1 protein expression from WT and one heterozygous clone (B13) is shown. α-PLCγ-1, anti-phospholipase Cγ1 antibody.
Subsequently, we analyzed the heterozygous cell lines in more detail. First, we examined Chk1 protein expression in both heterozygous and WT cell lines. We found that the heterozygous cell line expressed slightly less Chk1 proteins than the WT line (Fig. 5C and data not shown). This is consistent with the observation that cytoplasmic Chk1 mutant proteins were less stable than WT Chk1. Chk1 protein expression peaks in S-to-G2 phase (25). We showed that the cell cycle-dependent Chk1 expression pattern was also reserved in the heterozygous cell line, despite a slight reduction in the level of total Chk1 (Fig. 5D). Second, we examined cell cycle progression in both heterozygous and WT cell lines. We synchronized cells at the G2/M boundary with nocodazole, released them at different time points, and measured the cell cycle profile by flow cytometry. The results showed that the heterozygous cell line entered S phase at nearly the same pace as the WT cell line except that the heterozygous cells stayed in S phase longer than the WT cells (data not shown). However, when we analyzed cell proliferation, we noticed a significant growth defect in the heterozygous cell line compared with the WT cell line (Fig. 6A). This growth defect was accompanied by high levels of spontaneous cell death of the heterozygous cells (Fig. 6B). These data suggest that mutating one allele of CHK1 reduces cell proliferation ability.
FIGURE 6.

Spontaneous cell death of HCT116 heterozygous cell lines. 1 × 104 cells from WT or heterozygous HCT116 cell lines were plated in 12-well plates at day 0, and the cell number was counted at days 1 and 3–8 using the trypan blue assay. The viable cells and the rate of cell death are shown in A and B, respectively. Data represent the mean ± S.D. from three experiments.
We then tested the response of the heterozygous cell lines to DNA damage. First, we detected Chk1 protein phosphorylation by DNA damage. The results showed that the heterozygous cell lines exhibited reduced Chk1 phosphorylation compared with the WT cell line when treated with a replicative stress, HU (Fig. 7A, lanes 3, 6, and 9). These results are consistent with the overexpression data showing that phosphorylation of cytoplasmic Chk1 mutants was significantly reduced (Fig. 3B). Second, we consistently detected elevated levels of phosphorylated Chk1 and ATM (at Ser-1981) proteins in the absence of DNA damage in the heterozygous cell lines compared with the WT cells (Fig. 7A, lanes 1, 4, and 7, and data not shown), indicating that the heterozygous cells have spontaneous DNA damage. Third, we measured long-term cell survival after DNA damage by clonogenic assay. Consistent with the short-term cell proliferation data (Fig. 6A), the heterozygous cell lines exhibited significantly reduced ability to form colonies compared with the WT cell line in the absence of DNA damage (supplemental Fig. S5, A–C, at the 0 dose). However, the heterozygous cell lines did not appear to further enhance the growth suppression effect imposed by DNA-damaging agents compared with the WT cell line (Fig. 7, B–D), probably because Chk1 proteins from the remaining WT allele in the heterozygous cell lines are regulating the DNA damage response.
FIGURE 7.

Cytoplasmic mutant of Chk1 does not support the cell essential function of Chk1. A, asynchronously growing WT or heterozygous HCT116 cell lines were treated with the proteasome inhibitor MG132 (2 μm) for 12 h or with 2 mm HU for 2 h and immunoblotted with the indicated antibodies. WT or heterozygous HCT116 cell lines were treated or not with indicated doses of UV light, HU, or CPT for 2 h; reseeded at a density of 1000 viable cells/6-well plate; and cultured for 2 weeks. The cells were fixed and stained with crystal violet and destained, and the absorbance was measured at a wavelength of 594 nm. The results for UV light, HU, and CPT are shown in B–D, respectively. Data represent the mean ± S.D. from three wells. The non-treated groups (0 dose) in the WT and heterozygous cell lines were set to 1.
DISCUSSION
The nuclear-cytoplasmic shuttling of Chk1 appears to occur both under normal growth conditions (11, 26) and upon DNA damage (8, 10). In the former, Chk1 phosphorylation by Cdk1 at sites distinct from ATR is required to trigger the Crm1-dependent nuclear export of Chk1 and to mediate the G2/M phase cell cycle transition (11). Upon DNA damage, Chk1 phosphorylation at ATR sites triggers a chromatin → soluble nucleus → cytoplasm mobilization. However, the precise molecular mechanisms regulating this nuclear-cytoplasmic shuttling were not well understood. Previously, it was reported that the motif flanking the Ser-345 site of Chk1 has NES activity (9, 11). However, our results show that this region has marginal NES activity at best. In this study, we defined regions of the Chk1 polypeptide that regulate its own cellular localization. We confirmed that the distal C terminus of human Chk1, which contains the highly conserved CM2 domain, functions as an NLS, similar to its Xenopus counterpart (22). Furthermore, we identified a previously unknown non-canonical NES that overlaps with the CM1 domain. Therefore, our studies demonstrate that the highly conserved CM1 and CM2 domains at the C terminus of human Chk1 function as an NES and NLS, respectively.
The CM1 domain is a non-canonical NES, as it does not contain the canonical Leu-rich NES, yet it contains similar hydrophobic residues as a canonical NES. We also showed that this non-canonical NES regulates Chk1 nuclear export through the Crm1-dependent pathway. Interestingly, the CM1 domain is also involved in Chk1 degradation in the cytoplasm through interaction with the F box protein Fbx6 (16). In addition, this domain was also reported to be able to interact with phosphatase, as well as with proliferating cell nuclear antigen (PCNA), through a PCNA-interacting protein (PIP) box within this CM1 domain (27, 28). Consistent with our results, mutation of the PIP box within the CM1 domain led to Chk1 phosphorylation defects (28). These studies suggest that evolution has overlaid multiple layers of function on the same CM1 domain of Chk1. However, we speculate that the cellular localization function of this domain might play a dominative role in determining Chk1 function, as Chk1 mutants that accumulate in the cytoplasm are defective in nuclear functions, including phosphorylation and cell viability maintenance. Nevertheless, further studies are needed to address this issue.
CHK1 is an essential gene for cell viability (5). Under normal conditions, Chk1 is expressed mainly in the nucleus. Upon DNA damage, Chk1 regulates both nuclear and cytoplasmic checkpoints (15, 24). This phosphorylation-mediated protein translocation of Chk1 from the nucleus to the cytoplasm after DNA damage seems to be important for regulating the entire cellular checkpoint response. This raises an important question of which cellular compartment of Chk1, nuclear or cytoplasmic, is essential for the maintenance of cell viability. Using the AAV-mediated gene targeting technique, we attempted to generate cell lines in which endogenous CHK1 is mutated so that the Chk1 product is supposed to be expressed exclusively in the cytoplasm. Unfortunately, we were not able to identify a single clone with two CHK1 alleles mutated. Nevertheless, the heterozygous cell lines showed severe cell proliferation defects accompanied by significantly elevated spontaneous cell death, a phenotype very similar to CHK1 heterozygous mice (29). These data indicate that it is the nuclear pool of Chk1 that supports the essential function of Chk1. Our results also demonstrate that even the heterozygous cell line with one allele of CHK1 mutated has a growth defect. The growth inhibition in the heterozygous cell line is accompanied by high rates of spontaneous cell death in a way similar to the CHK1+/− mouse cell line (29). Together, these data indicate a tightly regulated pattern of Chk1 function in cell cycle checkpoints and cell survival (see model in Fig. 8).
Supplementary Material
Acknowledgments
We thank Tony Hunter for discussion and suggestions and Paul MacDonald for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Howard Temin Career Development Award R00CA126173 and Grant R01 CA163214 from NCI (to Y. W. Z.). This work was also supported by American Cancer Society Pilot Grant IRG-91-022-15 (to Y. W. Z.).

This article contains supplemental Figs. S1–S5.
Primer information is available upon request.
- AAV
- adeno-associated virus
- CPT
- camptothecin
- HU
- hydroxyurea
- NES
- nuclear export signal
- NLS
- nuclear localization signal
- LMB
- leptomycin B.
REFERENCES
- 1. Jackson S. P., Bartek J. (2009) The DNA damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang Y. W., Hunter T., Abraham R. T. (2006) Turning the replication checkpoint on and off. Cell Cycle 5, 125–128 [DOI] [PubMed] [Google Scholar]
- 3. Abraham R. T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196 [DOI] [PubMed] [Google Scholar]
- 4. Stracker T. H., Usui T., Petrini J. H. (2009) Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 8, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., Donehower L. A., Elledge S. J. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 14, 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 6. Takai H., Tominaga K., Motoyama N., Minamishima Y. A., Nagahama H., Tsukiyama T., Ikeda K., Nakayama K., Nakanishi M. (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1−/− mice. Genes Dev. 14, 1439–1447 [PMC free article] [PubMed] [Google Scholar]
- 7. Ma C. X., Janetka J. W., Piwnica-Worms H. (2010) Death by releasing the breaks: Chk1 inhibitors as cancer therapeutics. Trends Mol. Med. 17, 88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y. W., Otterness D. M., Chiang G. G., Xie W., Liu Y. C., Mercurio F., Abraham R. T. (2005) Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 19, 607–618 [DOI] [PubMed] [Google Scholar]
- 9. Jiang K., Pereira E., Maxfield M., Russell B., Goudelock D. M., Sanchez Y. (2003) Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J. Biol. Chem. 278, 25207–25217 [DOI] [PubMed] [Google Scholar]
- 10. Smits V. A., Reaper P. M., Jackson S. P. (2006) Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA damage checkpoint response. Curr. Biol. 16, 150–159 [DOI] [PubMed] [Google Scholar]
- 11. Enomoto M., Goto H., Tomono Y., Kasahara K., Tsujimura K., Kiyono T., Inagaki M. (2009) Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J. Biol. Chem. 284, 34223–34230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao H., Piwnica-Worms H. (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21, 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sørensen C. S., Hansen L. T., Dziegielewski J., Syljuåsen R. G., Lundin C., Bartek J., Helleday T. (2005) The cell cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201 [DOI] [PubMed] [Google Scholar]
- 14. Krämer A., Mailand N., Lukas C., Syljuåsen R. G., Wilkinson C. J., Nigg E. A., Bartek J., Lukas J. (2004) Centrosome-associated Chk1 prevents premature activation of cyclin B-Cdk1 kinase. Nat. Cell Biol. 6, 884–891 [DOI] [PubMed] [Google Scholar]
- 15. Reinhardt H. C., Hasskamp P., Schmedding I., Morandell S., van Vugt M. A., Wang X., Linding R., Ong S. E., Weaver D., Carr S. A., Yaffe M. B. (2010) DNA damage activates a spatially distinct late cytoplasmic cell cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 40, 34–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y. W., Brognard J., Coughlin C., You Z., Dolled-Filhart M., Aslanian A., Manning G., Abraham R. T., Hunter T. (2009) The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol. Cell 35, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leung-Pineda V., Huh J., Piwnica-Worms H. (2009) DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 69, 2630–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J., Engle S., Zhang Y. (2011) A new in vitro system for activating the cell cycle checkpoint. Cell Cycle 10, 500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rago C., Vogelstein B., Bunz F. (2007) Genetic knock-outs and knock-ins in human somatic cells. Nat. Protoc. 2, 2734–2746 [DOI] [PubMed] [Google Scholar]
- 20. Zhang X., Guo C., Chen Y., Shulha H. P., Schnetz M. P., LaFramboise T., Bartels C. F., Markowitz S., Weng Z., Scacheri P. C., Wang Z. (2008) Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat. Methods 5, 163–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. la Cour T., Kiemer L., Mølgaard A., Gupta R., Skriver K., Brunak S. (2004) Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17, 527–536 [DOI] [PubMed] [Google Scholar]
- 22. Katsuragi Y., Sagata N. (2004) Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol. Biol. Cell 15, 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartek J., Lukas J. (2007) DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 19, 238–245 [DOI] [PubMed] [Google Scholar]
- 24. Reinhardt H. C., Yaffe M. B. (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneko Y. S., Watanabe N., Morisaki H., Akita H., Fujimoto A., Tominaga K., Terasawa M., Tachibana A., Ikeda K., Nakanishi M. (1999) Cell cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 18, 3673–3681 [DOI] [PubMed] [Google Scholar]
- 26. Xu N., Libertini S., Black E. J., Lao Y., Hegarat N., Walker M., Gillespie D. A. (2011) Cdk-mediated phosphorylation of Chk1 is required for efficient activation and full checkpoint proficiency in response to DNA damage. Oncogene 31, 1086–1094 [DOI] [PubMed] [Google Scholar]
- 27. Ohta M., Guo Y., Halfter U., Zhu J. K. (2003) A novel domain in the protein kinase SOS2 mediates interaction with the protein phosphatase 2C ABI2. Proc. Natl. Acad. Sci. U.S.A. 100, 11771–11776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scorah J., Dong M. Q., Yates J. R., 3rd, Scott M., Gillespie D., McGowan C. H. (2008) A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 283, 17250–17259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lam M. H., Liu Q., Elledge S. J., Rosen J. M. (2004) Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6, 45–59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.