

Background: Autophagy plays a role in cancer development.
Results: Oncogenic KRAS induces Vacuole Membrane Protein 1 (VMP1) through a novel AKT1-GLI3-p300 pathway and requires VMP1 to regulate autophagy in cancer cells.
Conclusion: Define a novel pathway initiated by the oncogene KRAS regulating autophagy.
Significance: These findings contribute to the understanding of the mechanism underlying oncogene-induced autophagy.
Keywords: Autophagy, Bioenergetics, Gene Expression, Oncogene, Signaling
Abstract
Autophagy is an evolutionarily conserved degradation process of cytoplasmic cellular constituents. It has been suggested that autophagy plays a role in tumor promotion and progression downstream oncogenic pathways; however, the molecular mechanisms underlying this phenomenon have not been elucidated. Here, we provide both in vitro and in vivo evidence of a novel signaling pathway whereby the oncogene KRAS induces the expression of VMP1, a molecule needed for the formation of the authophagosome and capable of inducing autophagy, even under nutrient-replete conditions. RNAi experiments demonstrated that KRAS requires VMP1 to induce autophagy. Analysis of the mechanisms identified GLI3, a transcription factor regulated by the Hedgehog pathway, as an effector of KRAS signaling. GLI3 regulates autophagy as well as the expression and promoter activity of VMP1 in a Hedgehog-independent manner. Chromatin immunoprecipitation assays demonstrated that GLI3 binds to the VMP1 promoter and complexes with the histone acetyltransferase p300 to regulate promoter activity. Knockdown of p300 impaired KRAS- and GLI3-induced activation of this promoter. Finally, we identified the PI3K-AKT1 pathway as the signaling pathway mediating the expression and promoter activity of VMP1 upstream of the GLI3-p300 complex. Together, these data provide evidence of a new regulatory mechanism involved in autophagy that integrates this cellular process into the molecular network of events regulating oncogene-induced autophagy.
Introduction
Autophagy is an important catabolic process mediating the degradation of cellular organelles and macromolecules and recycling bioenergetic components for cellular metabolic needs (1–3). Recent evidence suggests that autophagy can be associated with both cancer progression and tumor suppression (4–12). Yang et al. (10) have shown that autophagy is activated constitutively in oncogenic KRAS-driven tumors and that this cellular event is required for the development of these tumors. Pharmacological or genetic inactivation of autophagy impairs oncogenic KRAS-mediated tumorigenesis in vivo and can act in synergy with chemotherapy (10). Although the biological role of KRAS-induced autophagy has been studied intensively, the molecular mechanism underlying this phenomenon in cancer cells remains to be established.
Here, we explore the contribution of novel biochemical mechanisms to the regulation of autophagy induced by oncogenic KRAS in cancer cells. We indentify the transmembrane protein VMP1,6 a key mediator of autophagy (13–17), as a transcriptional target of oncogenic KRAS signaling in cancer cells. VMP1 has been identified as one of three essential autophagy genes conserved from worms to mammals. It regulates early steps of the autophagic pathway (13). The data we report here demonstrate that KRAS requires VMP1 to induce and maintain autophagy levels in cancer cells. By characterizing the signaling regulating VMP1 downstream of KRAS, we have been able to identify a novel AKT1-GLI3-p300 pathway mediating VMP1 promoter activity and expression in cancer cells. The transcription factor GLI3 binds to the VMP1 promoter, regulating its activity and expression. The histone acetyltransferase p300 complexes with GLI3 and cooperates with this transcription factor to modulate this promoter. Finally, we show that the activation of this new pathway leads to increased autophagy in cancer cells. Together, these findings define a novel signaling cascade controlling autophagy and help delineate the mechanism underlying oncogene-induced autophagy.
EXPERIMENTAL PROCEDURES
Cell Culture Conditions and Plasmids
PANC1, CHO, 293T, MCF10A, and HeLa cell lines were obtained from ATCC and were cultured as reported previously (18, 19). The L3.6 cells were kindly provided by Dr. Fidler (MD Anderson Cancer Center, Houston, TX) and HPDE6 cells by Dr. Tsao (University of Toronto, Toronto, Ontario, Canada). Experiments were performed using a luciferase reporter for the VMP1 promoter containing 3005 bp upstream of the transcriptional starting site. For the cloning of this regulatory sequence into the pGL3 vector (Promega, Madison, WI), we used the following primers: CGGGTCGACGTGGCGCCATCTCAGC and GGGAAGCTTCCCCAGATCCGCAATG. The expression vector FLAG-p300 was kindly provided by Dr. Donald Tindall (Mayo Clinic, Rochester, MN). The KRASG12D mutant was a gift from Dr. Daniel Billadeau (Mayo Clinic, Rochester, MN). The EGFP-LC3 and TK-pRL Renilla vector were provided generously by Dr. Scott Kaufmann (Mayo Clinic, Rochester, MN). The shRNA targeting KRAS, AKT1, AKT2, AKT3, GLI3, VMP1, and p300 and the non-targeting control (NT) were obtained from Sigma (St. Louis, MO). The pool of shRNA targeting p300 was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The GLI-luciferase reporter was provided kindly by Dr. Chi-chung Hui (University of Toronto, Toronto, Ontario, Canada). The constitutively active (ca) caPI3K, caAKT1 and dominant-negative AKT1 were kindly provided by Dr. Robert Scheaff (University of Tulsa, Tulsa, OK).
Transgenic Mice
krasG12D Cre transgenic mice and their corresponding wild-type littermates were kindly provided by Dr. Fergus Couch (Mayo Clinic, Rochester, MN), as described previously (21, 27). The recombined krasG12D allele was identified by PCR using the primer sets described previously (27). Briefly, DNA was harvested from mice tails using the DNeasy blood and tissue kit (Qiagen, Valencia, CA) and genotyped by PCR. Reaction conditions for Cre were 36 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min. Conditions for kras mutant were 40 cycles of 94 °C for 30 s, 63 °C for 30 s, and 72 °C for 1 min. Mice were housed under pathogen-free conditions in the Mayo Clinic facilities. All studies were conducted in compliance with the Mayo Clinic Institutional Animal Care and Use Committee guidelines.
Transfection
PANC1, HeLa, 293T, HPDE6, and CHO cells were transfected with Lipofectamine (Invitrogen) as described previously (18). 1.6 × 105 cells were plated into six-well plates with standard and transfected 24 h later. For each condition, 0.6 μg of VMP1 promoter reporter or 0.4 μg of GLI-luciferase reporter was used. In overexpression assays, 1.4 μg of KRASG12D, GLI3, caAKT1, caPI3K, or p300 vectors were used. For transfection of multiple constructs, 3 μg of DNA were used, consisting of 0.6 μg of VMP1 promoter reporter and equal amounts of the other vectors. In knockdown experiments, 0.6 μg of VMP1 promoter reporter was cotransfected with 2.4 μg of shRNA. In rescue experiments, 0.4 μg of VMP1 promoter reporter was used together with 1.2 μg of expression vector and 2.4 μg of shRNA. In each set of experiments, equal amounts of plasmid DNA were used by adding empty vectors to appropriate controls. L3.6 cells were transfected by electroporation at 320 V for 10 ms (BTX, Harvard Apparatus, Holliston, MA), and MCF10A cells were transfected at 375 V for 10 ms.
Luciferase Reporter Assay
Cells were grown and transfected as indicated above. For luciferase reporter assays, cells were plated in triplicate into six-well plates in medium containing 10% FBS. Samples were harvested and prepared for luciferase assays in accordance with the manufacturer's protocol (Promega, Madison, WI): cells were harvested 36–48 h after transfection for overexpression studies and 72 h after transfection when performing shRNA knockdown assays. To control for intersample variations in transfection efficiency, the total protein of samples in each well was quantitated using the Bio-Rad protein assay (Bio-Rad), and luciferase readouts were normalized to protein content (Figs. 3–8) or Renilla readings (Promega) (supplemental Figs. 2–4). Relative luciferase activity represents luciferase readouts/protein concentrations or luciferase/Renilla readouts normalized to control cells within each experiment. Similar results were obtained using either normalization protocol.
FIGURE 3.
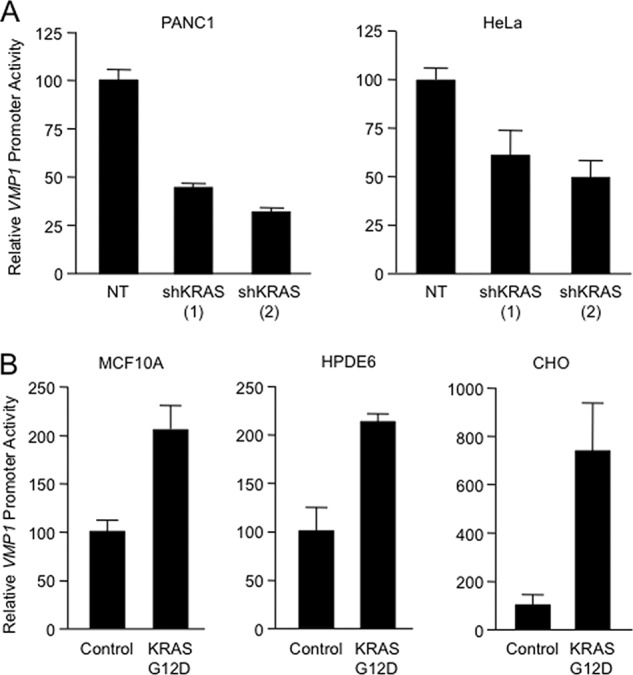
A, relative luciferase activity was determined in PANC1 and HeLa cells transfected with VMP1 promoter-luciferase reporter and two shKRAS constructs (shKRAS (1) and shKRAS (2)) or a non-targeting control vector (NT). B, relative changes in luciferase activity in MCF10A, HPDE6, and CHO cells transfected with the VMP1 promoter-luciferase reporter and either a control vector or a KRASG12D-expressing vector for 24 h. In all panels, bars represent the mean of triplicate experimental wells ± S.E.
FIGURE 4.
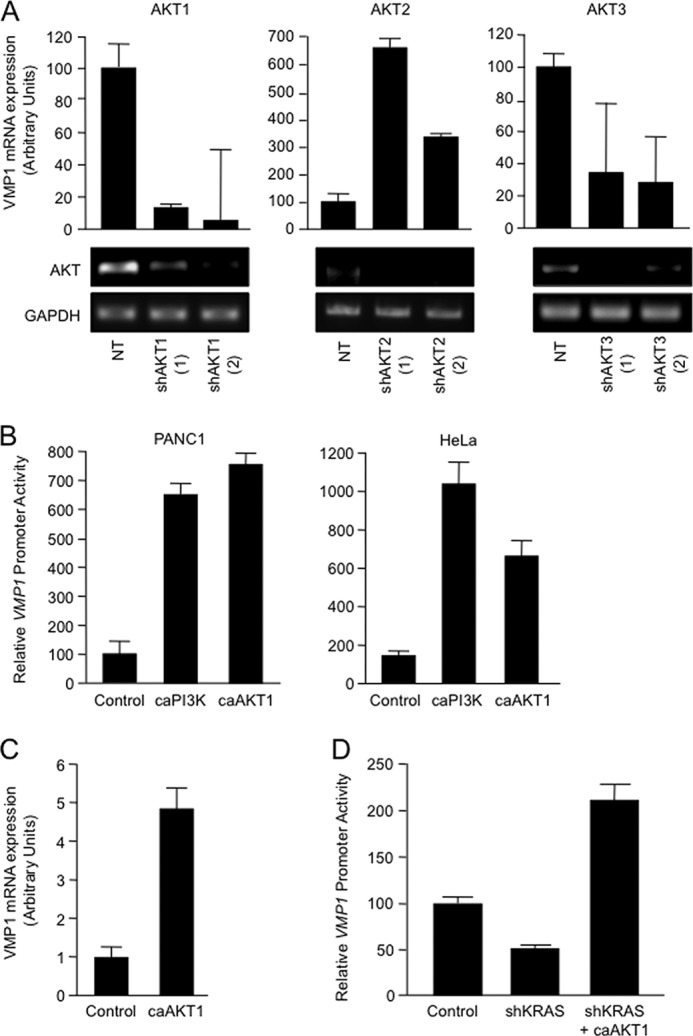
A, PANC1 cells were transfected with two shRNA vectors targeting AKT1 (shAKT1 (1) and shAKT1 (2)), AKT2 (shAKT2 (1) and shAKT2 (2)), AKT3 (shAKT3 (1) and shAKT3 (2)), and the non-targeting control vector (NT). VMP1 mRNA expression levels were determined after 72 h by real-time RT-PCR. The effect of knockdown on AKT1, AKT2, and AKT3 expression was examined by RT-PCR. B, PANC1 and HeLa cells were transfected with the VMP1-luciferase reporter construct and a constitutively active PI3K construct (caPI3K) or a constitutively active AKT1 construct (caAKT1) or the empty vector (control). After 24 h, VMP1 reporter activity was determined by luciferase assay. C, RNA was isolated from HeLa cells transfected with the construct caAKT1 or the control vector, and VMP1 mRNA expression was analyzed by real-time RT-PCR. D, PANC1 cells were transfected with the VMP1-luciferase reporter, a shKRAS construct (shKRAS) or NT control vector, and the constitutively active AKT1. After 72 h, reporter activity was determined by luciferase assay. In all panels, bars represent the mean of triplicate experimental wells ± S.E.
FIGURE 5.

A, expression of mouse gli3 and 18 S in mutant oncogenic kraSG12D-expressing pancreatic tissues was analyzed by RT-PCR as described under “Experimental Procedures.” B, LC3 and tubulin expression were analyzed by Western blot in HeLa, MCF10A, and 293T cells transfected with a GLI3-expressing construct or a parental control vector. C, MCF10A cells were transfected with EGFP-LC3 and either GLI3 or a parental control vector was analyzed by fluorescence microscopy to determine the distribution and levels of punctate LC3. D, LC3-II expression in KRASG12D mutant PANC1 cells transfected with two independent GLI3 shRNA constructs and a non-targeting control vector. The effect of knockdown on GLI3 expression was examined by RT-PCR. E, relative changes in luciferase activity in the indicated cells transfected with the VMP1 promoter-luciferase reporter and either a control vector or a GLI3-expressing construct. F, bioinformatics analysis of the VMP1 promoter identified candidate GLI (G) binding sites (upper panel). TSS, transcription start site. PANC1 cells were lysed, and a ChIP assay was performed using a GLI3 antibody (GLI3) or IgG control (IgG). PCR was performed using a specific set of primers as described under “Experimental Procedures” (lower panel). The positive amplicon for the ChIP assay is marked (−1059/−677). G, relative changes in luciferase activity in PANC1 and HeLa cells transfected with the VMP1 promoter-luciferase reporter and shRNA targeting GLI3 or the non-targeting control (NT). H, RNA PANC1 and HeLa cells transfected with GLI3 shRNA and a non-targeting control were used to examine VMP1 expression levels by real-time RT-PCR. All experiments were performed in triplicate wells with similar results. Bars represent average ± S.E.
FIGURE 6.
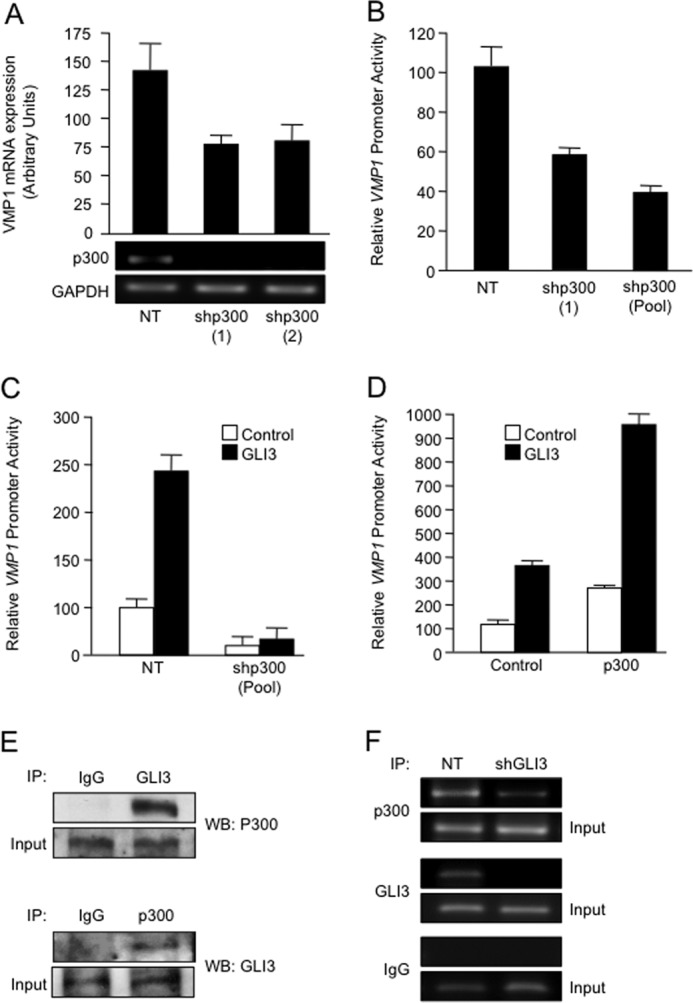
A, PANC1 cells were co-transfected with the VMP1 promoter-luciferase reporter and either a non-targeting control (NT) vector or one of two shRNAs targeting p300 (shp300 (1) and shp300 (2)). Seventy-two hours after transfection, RNA was isolated and subjected to RT-PCR to investigate mRNA levels of VMP1, p300, and GAPDH. B, lysates from PANC1 cells transfected with two different shRNA targeting p300 were isolated, and the relative changes in luciferase activity were analyzed. C, relative luciferase activity in PANC1 cells co-transfected with the VMP1 promoter-luciferase reporter and NT vector control or p300 shRNA (shp300 (Pool)), in the presence or absence of a GLI3-expressing vector. D, PANC1 cells were transfected with the VMP1 promoter-luciferase reporter construct and a p300-expressing construct or control vector, in the presence or absence of GLI3 expression. After 24 h, proteins were isolated and used to determine VMP1 reporter activity by luciferase assay. Bars represent average levels in each group ± S.E. E, PANC1 protein lysates were immunoprecipitated (IP) with the antibodies indicated, and Western blot (WB) was performed to demonstrate coimmunoprecipitation between GLI3 and p300. F, p300 and GLI3 ChIP assay in PANC1 cells transfected with non-targeting control (NT) and a GLI3 shRNA construct.
FIGURE 7.
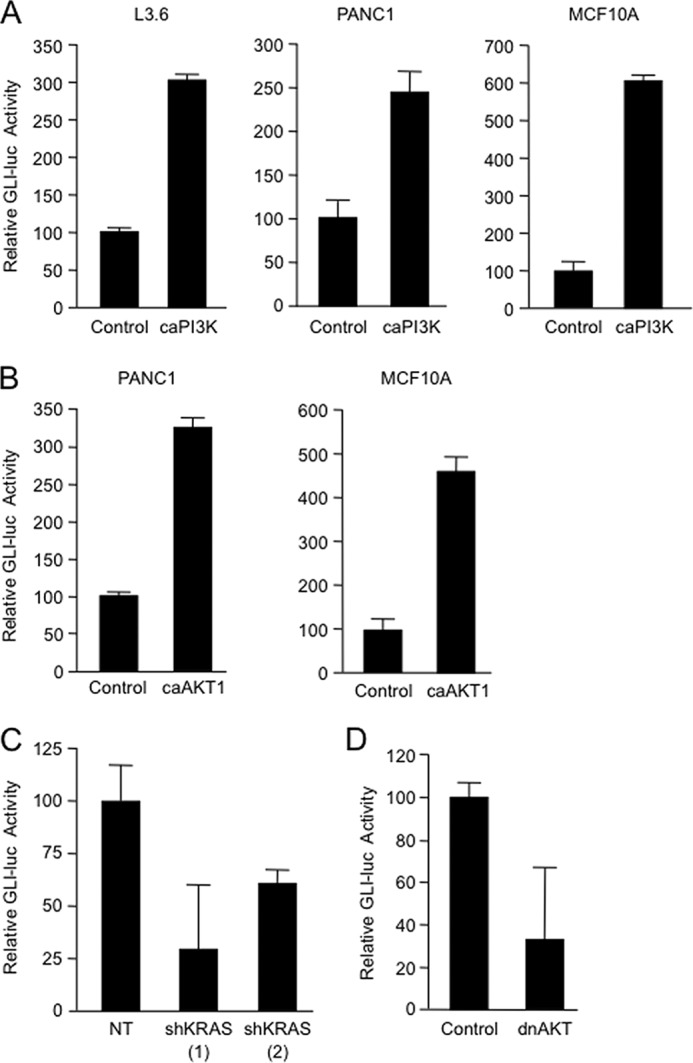
A, L3.6, PANC1 and MCF10A cells were co-transfected with the GLI-luciferase reporter and the caPI3K or a control vector; 24 h after transfection, cells were collected for luciferase assay analysis. B, relative changes in luciferase (luc) activity were determined in PANC1 and MCF10A cells transfected with the GLI-luciferase reporter and the caAKT1 or a control vector. Analysis was performed 24 h after transfection. C, relative luciferase activity was determined at 72 h in PANC1 cells transfected with the GLI-luciferase vector, two shKRAS constructs (shKRAS (1) and shKRAS (2)) or non-targeting vector (NT). D, PANC1 cells were co-transfected with the GLI-luciferase reporter and either the control vector or the dominant-negative AKT1 (dnAKT1); 48 h later, the cells were lysed and collected for luciferase assay. Bars represent average levels in each group ± S.E.
FIGURE 8.
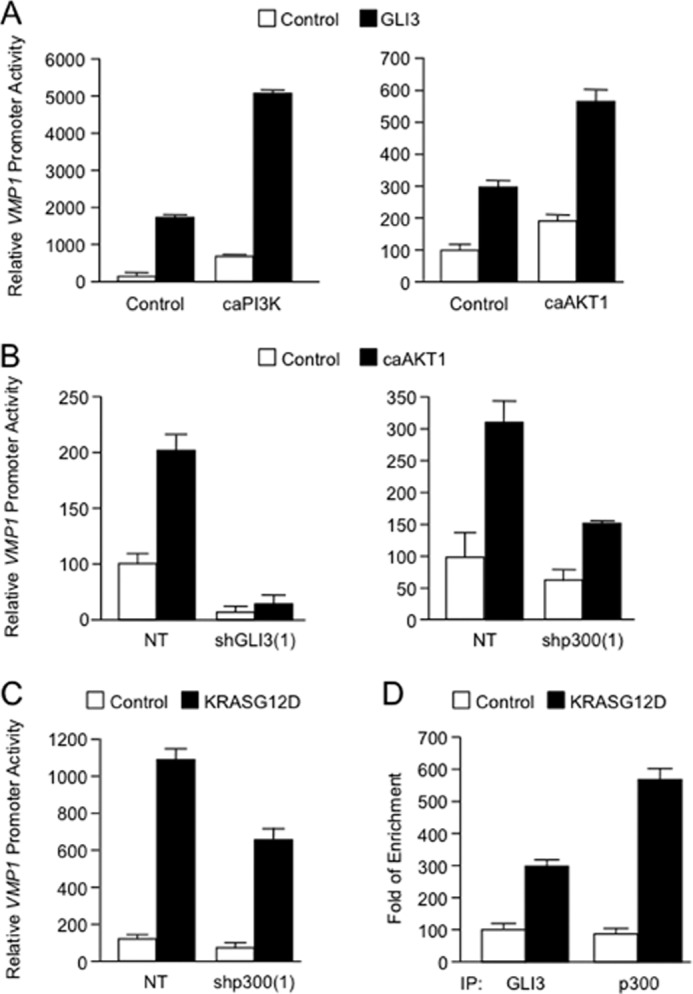
A, PANC1 cells were transfected with the VMP1 promoter-luciferase reporter with caPI3K (left), caAKT1 (right), or control vector in the presence or absence of GLI3-expressing vector. Twenty-four hours after transfection, the relative luciferase activity was determined. B, PANC1 cells were transfected with the VMP1 promoter-luciferase reporter construct, NT vector, or shRNA targeted for GLI3 or p300, along with a control vector or a constitutively active AKT1 construct. Seventy-two hours after transfection, samples were collected for luciferase assay. C, relative luciferase activity in PANC1 cells transfected with the VMP1 promoter-luciferase reporter construct, an shRNA targeting p300, or a non-targeting vector in the presence or absence of oncogenic KRASG12D. Bars represent average levels in each group ± S.E. D, ChIP assay in HeLa cells transfected with control vector or oncogenic mutant KRASG12D. Lysed cells were immunoprecipitated with GLI3 antibody (GLI3), p300 antibody (p300), or IgG control (IgG). Real-time PCR was performed using a specific set of primers as described under “Experimental Procedures.”
Reverse Transcriptase PCR (RT-PCR)
Total RNA was extracted from cultured cells or pancreatic tissue using TRIzol reagent (Invitrogen). Moloney murine leukemia virus reverse transcriptase was used to reverse transcribe 5 μg of RNA (Invitrogen). A portion of the total cDNA was amplified by PCR using 94 °C denaturation, 58 °C annealing, and 72 °C extension temperatures for 40 cycles with Ex TaqTM DNA polymerase (Takara Bio, Inc., Otsu, Shiga, Japan). For AKT2 amplification, an annealing temperature of 60 °C was used. Sense and antisense primers used for amplification of each mRNA species were as follows: human GLI3, CCAACGGAAATCAATAGGAGTTGAA and GAGGGTGGTTTGAGTGTAACAATAC; mouse gli3, AACCTCACTCTGCAACAAGGACAG and GTGTTTGTGGTCCTCCTTGCCTAC; KRAS, GAACCCAGCAGTTACCTTAAAGCTG and ACACTGTAACCCAGTTAGCTCTGTG; p300, TGCCAGTCTACAGGCCTATCAGCA and AGTCTGAGTTATCGGTGCTGAGTC; AKT1, GGCACCAGATGCAACCTCACTATG and ACAGCGGAAAGGTTAAGCGTCGAA; AKT2, TTTGAACAGTGACTGGGCACAGTG and AGACACAGTCTCATTGTCCCAGGCT; AKT3, TAGGAAATGGGCAGTGAAGGCTAA and GTCACTTGCTCTTTCATACAATGCA; human GAPDH, GACCTGACCTGCCGTCTAGAAAAA and ACCACCCTGTTGCTGTAGCCAAAT; mouse 18 S, AACCCGTTGAACCCCATTCGTGAT and CAGGTTCACCTACGGAAACCTTGT. Amplified products were visualized under UV illumination after electrophoresis on ethidium bromide-stained 2% agarose gels. Amplification of the appropriate gene fragments was confirmed by comparison with molecular weight markers run on the same gel.
Real-time expression analysis of VMP1 and 18 S transcript was performed using TaqMan® fluorescence methodology and ABI 7900 (Applied Biosystems, Foster City, CA). A predesigned primer/probe set for mouse and human VMP1 and 18 S expression was purchased from Applied Biosystems. RNA (5 μg) was extracted from cultured cells or pancreatic tissue and was reverse-transcribed using a high capacity cDNA synthesis kit (Applied Biosystems). From each sample, 2 μl of the cDNA synthesis reaction were used for quantitative PCR analysis. The amount of VMP1 transcript was calculated and expressed as the difference relative to the control gene 18 S (2ΔCt, where ΔCt represents the difference in threshold cycles between the target and control gene).
Chromatin Immunoprecipitation (ChIP) Assay
Chromatin immunoprecipitation was conducted following the Magna ChIP kit protocol (EMD Millipore, Bedford, MA). Briefly, PANC1 cells (7 × 106) were cross-linked with 1% formaldehyde directly into the media for 10 min at room temperature. The cells were then washed and scraped with phosphate-buffered saline and collected by centrifugation at 800 × g for 5 min at 4 °C, resuspended in cell lysis buffer and incubated on ice for 15 min. The pellet was then resuspended in nuclear lysis buffer and sheared to fragment DNA to ∼700 bp. Samples were then immunoprecipitated using a GLI3 antibody (Santa Cruz Biotechnology), p300 antibody (Santa Cruz Biotechnology), normal goat IgG (Santa Cruz Biotechnology), or normal rabbit IgG (Santa Cruz Biotechnology) overnight at 4 °C on a rotating wheel. Following immunoprecipitation, samples were washed and eluted using the chromatin immunoprecipitation kit in accordance with the manufacturer's instructions. Cross-links were removed at 62 °C for 2 h, followed by 10 min at 95 °C, and immunoprecipitated DNA was purified (EMD Millipore, Bedford, MA) and subsequently amplified by PCR. PCR was performed using seven primer sets for the seven areas containing potential GLI binding sites in the VMP1 promoter sequence (Fig. 5E): 1) CACGTACATGGGATTACAGGCACC (sense) and CAGTTCCTTCTCCAGATAGTACCGG (antisense); 2) CCGGTACTATCTGGAGAAGGAACT (sense) and GGCAACATAGCAAGACCAAGTCCA (antisense); 3) GCCTAGTGCCTAAGACTAAGGTA (sense) and AGTGGCTTAAGGGTCAGAGAAGA (antisense); 4) GTTCAAGTCATTCTCCTGCCTCAG (sense) and GTATCGATGCGCTCTCTCCGAAAT (antisense); 5) CTGAGACGAAGTCTCACTCTGTCG (sense) and CATACCCTCAGCTGGGCACTTATG (antisense); 6) CATAAGTGCCCAGCTGAGGGTATG (sense) CCTCGTTGCATTACCCACCTTCTTC (antisense); and 7) AGCGAACACGTTTCTATCGCGTGA (sense) and CTGAGTTAGTTTACCTACCCGCTC (antisense). PCR products were visualized by 2% agarose gel.
Immunoblotting
Western blot was performed as described previously (14, 18). 5 × 105 PANC1, MCF10A, 293T, and HeLa cells were plated in standard medium with 10% FBS and used 24 h later for transfection experiments. Cells were lysed 48 h after transfection for overexpression experiments and 72 h for the RNAi studies. LC3-II, AKT, and phospho-AKT antibodies were obtained from Cell Signaling (Danvers, MA). Anti-tubulin and β-actin antibody was obtained from Sigma-Aldrich, and anti-VMP1 was purchased from Abcam (Cambridge, MA). The p62 antibody was purchased from Santa Cruz Biotechnology. Peroxidase-conjugated secondary antibodies were used, and immunoreactive proteins were detected by chemiluminescence (GE Healthcare).
Fluorescence Microscopy
MCF10A, PANC1, and HeLa cells were transfected with EGFP-LC3 to identify the autophagosome. Punctate fluorescence was imaged by fluorescence microscopy using an LSM510 microscope (Zeiss, Heidelberg, Germany).
Immunoprecipitation
PANC1 cells were washed twice with cold phosphate-buffered saline and lysed in lysis buffer (150 mm NaCl, 0.5% Nonidet P-40, 50 mm Tris-HCl, pH 7.5, 20 mm MgCl2) supplemented with Complete protease inhibitor tablets (Roche Applied Science, Penzberg, Germany) for 1 h on ice. After the lysates were cleared at 15,000 × g for 20 min, supernatants were collected and subjected to immunoprecipitation following the Dynabeads Protein G immunoprecipitation kit protocol (Invitrogen). The following antibodies were cross-linked to Dynabeads Protein G for 1 h at room temperature: anti-p300, anti-GLI3, and normal rabbit IgG from Santa Cruz Biotechnology. Then, Dynabeads-antibody complexes and lysates were incubated overnight at 4 °C with rotation. Proteins were eluted by addition of SDS sample buffer and incubation at 95 °C for 5 min. These eluates were subjected to Western blot analysis as described above, using anti-p300 and anti-GLI3 antibodies (Santa Cruz Biotechnology).
RESULTS
KRAS Requires VMP1 to Induce Autophagy
The GTPase KRAS is a central transducer of multiple signaling pathways involved in cell growth and survival. Mutations such as the G→A transition in codon 12 are highly prevalent in human tumors. This mutation results in a glycine to aspartic acid substitution (G12D) in the expressed protein, compromising its GTPase activity and resulting in constitutive downstream signaling and subsequent cellular transformation (19–21). Recent reports have shown that oncogenes, including KRAS, can promote autophagy in cancer cells (20–24). Using three well established markers of this cellular process, we demonstrated that KRASG12D increases autophagy. This was shown by the increase in the punctate distribution of microtubule-associated protein light chain 3 tagged with EGFP (EGFP-LC3) in MCF10A cells transfected with oncogenic KRAS mutant (Fig. 1A). Similarly, overexpression of KRASG12D led to an increase in LC3-II, the processed form of LC3, in MCF10A, 293T, and HeLa cells (Fig. 1B). To complement these results, we determined the expression of the polyubiquitin-binding protein p62/SQSTM1, a molecule that is degraded during the progression of autophagy. As demonstrated by supplemental Fig. 1A, KRASG12D induces the degradation of p62 in HeLa cells. Conversely, knockdown of KRAS in cultured pancreatic cancer cells (PANC1) carrying the G12D oncogenic mutant of this GTPase led to a decrease in processing of LC3 (Fig. 1C) and EGFP-LC3 punctae (Fig. 1D). Expression of KRAS by RT-PCR was used as a control for the efficiency of shRNA knockdown (Fig. 1C, lower panel). In addition, we determined KRAS signaling activity by studying AKT activation using antibodies against the phosphorylated form of AKT (in cells overexpressing a mutant KRASG12D (supplemental Fig. 1B) or transfected with shRNA targeting KRAS (supplemental Fig. 1C). As expected, KRASG12D overexpression increased the phosphorylation of AKT, and the shRNA targeting this GTPase diminished the phosphorylation of this kinase. Together, these findings establish a role for KRAS in the induction and maintenance of autophagy in cancer cells.
FIGURE 1.

A, MCF10A cells transfected with EGFP-LC3 and either KRASG12D mutant or parental control vector were analyzed by fluorescence microscopy to determine the distribution of punctate EGFP-LC3 fluorescence. The right panel shows the quantification of EGFP-LC3 positive over the total number of cells between the experimental groups. B, the indicated cells were transfected with control vector or KRASG12D. At 48 h post-transfection, LC3 and tubulin were determined by Western blot. C, PANC1 cells carrying an oncogenic mutant KRASG12D were transfected with two independent KRAS shRNA (shKRAS (1) and shKRAS (2)) constructs and NT control. The levels of LC3 lipidation and TUBULIN were determined by Western blot. KRAS and GAPDH mRNA expression levels by RT-PCR were included as a control for the shRNA knockdown. D, punctate EGFP-LC3 distribution of PANC1 cells transfected with KRAS shRNA constructs and NT control.
Expression studies of different components of the machinery essential for the formation of the autophagosome demonstrated that KRASG12D induces the expression of VMP1. HeLa cells transfected with KRASG12D showed a 5-fold increase in the mRNA (Fig. 2A, upper panel) and protein (Fig. 2A, lower panel) expression of VMP1 as compared with the control transfected cells. In agreement with the in vitro data, mutant krasG12D-expressing tissues (27) showed autophagy (10) and a nearly 10-fold increase in VMP1 expression compared with the tissues of their wild-type littermates (Fig. 2B), supporting the role of this oncogene in the regulation of VMP1. Furthermore, RNAi knockdown of KRAS using two independent shRNA constructs led to a >50% decrease in the VMP1 expression in cells carrying a KRAS mutant allele (PANC1) or wild-type allele (HeLa) (Fig. 2C). To define the role of VMP1 in oncogenic KRAS-induced autophagy, we used two independent shRNA constructs targeting VMP1. As shown in Fig. 2, D and E, knockdown of VMP1 diminishes the processing of LC3 in mutant KRAS PANC1 cells (Fig. 2D) and decreases LC3 punctae (Fig. 2E). Similarly, targeting of VMP1 impairs mutant KRAS-induced LC3-II lipidation and punctate distribution of EGFP-LC3 in MCF10A cells (Fig. 2F). Further characterization of this phenomenon showed that this GTPase modulates the promoter activity of VMP1. RNAi knockdown of KRAS diminished VMP1 promoter activity in cells carrying a KRAS mutant allele (PANC1) or wild-type allele (HeLa) (Fig. 3A). Conversely, overexpression of oncogenic mutant KRASG12D in non-transformed cells (MCF10A, HPDE6, and CHO) carrying wild-type copies of this GTPase augmented VMP1 promoter activity (Fig. 3B). Together, these findings define a novel mechanism controlling autophagy in cancer downstream of the oncogene KRAS and identify VMP1 as a target for this oncogenic signaling cascade.
FIGURE 2.

A, HeLa cells were transfected with KRASG12D for 48 h, and RNA was subsequently extracted. VMP1 expression levels were determined by real-time RT-PCR (upper panel) and Western blot (lower panel). B, RNA was isolated from wild-type and krasG12D murine pancreatic tissue at 9 months and used to determine expression of vmp1 relative to 18 S by real-time RT-PCR. C, VMP1 mRNA expression levels were determined by real-time RT-PCR in PANC1 and HeLa cells transfected with shKRAS constructs or NT control vector. D, LC3-II expression by Western blot in PANC1 cells carrying an oncogenic mutant KRASG12D transfected with two independent VMP1 shRNA constructs or a non-targeting control vector. VMP1 mRNA expression levels by RT-PCR were included as a control of knockdown efficiency. E, EGFP-LC3 distribution in PANC1 cells co-transfected with VMP1 shRNA or non-targeting control. F, Western blot for LC3-II in MCF10A cells transfected with VMP1 shRNA and either a control vector or KRASG12D (lower left panel). MCF10A cells were transfected with EGFP-LC3 and with KRASG12D or control vector in the presence or absence of VMP1 shRNA. Left, the percentage of autophagic cells with punctate EGFP-LC3 fluorescence was calculated relative to all EGFP-positive cells. Right, a representative punctate EGFP-LC3 fluorescence by confocal microscopy.
AKT1 Mediates KRAS Induction of VMP1
Analysis of signaling downstream of KRAS demonstrated that an active PI3K-AKT1 pathway is required to maintain the promoter activity and expression of VMP1. Knockdown of AKT kinases (AKT1, AKT2, and AKT3) in PANC1 cells suggested that the regulatory function of this signaling pathway is mediated mainly by AKT1, as RNAi inactivation of this kinase results in a robust reduction of VMP1 expression (Fig. 4A). Overexpression of caPI3K or caAKT1 increased the promoter activity (Fig. 4B) and expression (Fig. 4C) of VMP1, further supporting the role of this pathway in the regulation of VMP1 expression. Finally, we demonstrated that caAKT1 was able to rescue the effect of shRNA targeting the oncogenic KRASG12D in PANC1 cells (Fig. 4D). Indeed, overexpression of caAKT1 restores the activity of the VMP1 promoter in cells transfected with shRNA targeting KRAS. Taken together, these findings suggest the PI3K-AKT1 axis as a mediator of KRAS-induced VMP1 expression.
KRAS-GLI3 Pathway Couples Membrane Signals to Transcriptional Activation of VMP1
Initial bioinformatics analysis and promoter deletion studies have identified GLI proteins (GLI1, GLI2, and GLI3) as candidate mediators of KRAS-PI3K-AKT1 activation of the VMP1 promoter in cancer cells (data not shown). These transcription factors are known effectors of the Hedgehog pathway and essential for regulation of the cellular functions mediated by this signaling cascade (28, 29). GLI1 and GLI2 function predominantly as activators, whereas GLI3 is thought to act as a transcriptional repressor (28, 29). Thus, we used a combination of transcriptional and chromatin assays to determine a possible involvement of GLI proteins in VMP1 activation. Luciferase and expression studies showed that GLI3 can regulate the expression and promoter activity of this molecule in cancer cells. GLI3 is highly expressed in cells carrying a mutant KRASG12D allele (30) and is induced in vivo by oncogenic KRAS. As shown in Fig. 5A, transgenic animals expressing mutant krasG12D showed higher levels of gli3 compared with their wild-type littermates. In MCF10A, 293T, and HeLa cells, overexpression of GLI3 induced autophagy, as shown by the increase of LC3-II (Fig. 5B) and EGFP-LC3 punctae (Fig. 5C). Knockdown of GLI3 led to a decrease in LC3-II in KRAS mutant PANC1 cells (Fig. 5D). Reporter studies demonstrate that overexpression of GLI3 led to an increase in the promoter activity of VMP1 in both KRASG12D-positive PANC1 cells and wild-type HeLa cells (Fig. 5E). This regulatory sequence contains 10 candidate binding sites for GLI transcription factors within the −3000 bp upstream of the transcriptional starting site (Fig. 5F, upper panel). Using a ChIP assay, we confirmed binding of endogenous GLI3 to a region located −1059 bp to −677 bp upstream of the first intron. Fig. 5F (lower panel) shows GLI3 occupancy of this regulatory sequence in PANC1 cells. RNAi targeting GLI3 resulted in a reduction in promoter activity (Fig. 5G) and expression (Fig. 5H) in PANC1 and HeLa cells. These results demonstrate that VMP1 is a novel direct target of the GLI3 transcription factor and identify a previously unknown activator function for GLI3.
KRAS Activates VMP1 by Inducing GLI3-dependent Recruitment of Histone Acetyltranferases
Transcription factors affect gene expression not only through their inherent activation/repression properties but also through functional interactions with co-regulator molecules. Here, we tested whether activation by GLI3 involved histone acetyltransferases, molecules that have been shown to cooperate with GLI proteins during development (31, 32). RNAi knockdown using two independent shRNAs or a pooled of shRNAs targeting the acetyltransferase p300 led to a decrease in VMP1 expression (Fig. 6A) and promoter activity (Fig. 6B). Moreover, p300 shRNA constructs impaired GLI3-mediated activation of the VMP1 promoter in cancer cells (Fig. 6C). Overexpression of GLI3 and p300 led to a synergistic activation of the VMP1 promoter (Fig. 6D). Endogenous immunoprecipitation studies showed that GLI3 and p300 can complex in cancer cells (Fig. 6E). Moreover, ChIP assays showed that p300 is present in the VMP1 promoter and that RNAi knockdown of GLI3 diminished the presence of this acetyltransferase on the VMP1 promoter (Fig. 6F), thus suggesting that p300 requires GLI3 for its recruitment to the VMP1 promoter. Combined, these data show that the GLI3-p300 activator/co-activator complex is part of the novel signaling pathway controlling the expression and promoter activity of this autophagy-related gene in cancer cells.
GLI3 Acts as Downstream Mediator of KRAS-PI3K-AKT1 Pathway
GLI transcription factors are regulated by Hedgehog and mediate many of its cellular functions during development as well as disease (28). Interestingly, the cancer cell lines used in our studies do not have an active Hedgehog pathway and do not respond to activator ligand in culture (30, 33). These results prompted us to examine whether the KRAS-PI3K-AKT1 pathway can regulate GLI activity in cancer cells. L3.6, PANC1, and MCF10A cells overexpressing a caPI3K (Fig. 7A) or AKT1 (Fig. 7B) showed increased GLI activity, as demonstrated by the induction of a GLI luciferase reporter containing eight consecutive GLI binding sites upstream of the luciferase gene (GLI-luciferase). Conversely, inactivation of this pathway using shRNA targeting oncogenic KRASG12D (Fig. 7C) or a dominant-negative AKT1 (Fig. 7D) demonstrated lower GLI-luciferase activity compared with control transfected cells. Thus, these results provide biochemical evidence supporting a role for the KRAS-PI3K-AKT1 pathway in the modulation of GLI activity in cancer cells.
Next, we sought to determine whether GLI3 was required for the activation of VMP1 by the KRAS-PI3K-AKT1 pathway. PANC1 cells were co-transfected with GLI3 and the constitutively active variants of PI3K and AKT1. The results included in Fig. 8A show that GLI3 is able to synergize with these mutants to activate the VMP1 promoter. Conversely, we found that PANC1 cells co-transfected with a constitutively active AKT1 expression vector as well as shRNA targeting GLI3 or p300, and VMP1 reporter luciferase constructs showed impaired VMP1 promoter activation (Fig. 8B). Similar results were observed in cells co-transfected with p300 shRNA and the KRASG12D oncogenic mutant (Fig. 8C). Finally, overexpression of mutant KRAS increased in the presence of GLI3 and p300 in the VMP1 promoter in HeLa cells (Fig. 8D). Taken together, these results define the PI3K-AKT1-GLI3-p300 pathway as the signaling pathway mediating KRAS induction of the autophagy regulator, VMP1.
DISCUSSION
Autophagy plays a major role in sustaining cellular metabolism and nutrient homeostasis in tumor cells (1–3, 5–7). For instance, autophagy provides cancer cells with a mechanism for escaping unfavorable conditions and surviving nutrient-limiting conditions (4, 5). Numerous signaling pathways have been associated with the initiation and regulation of autophagy in cancer cells (1–10, 13). Many of these cascades play a critical role as oncogene or tumor suppressors during tumorigenesis (7). However, the molecular mechanism underlying this phenomenon is still poorly understood. Here, we show that KRAS, an oncogene that induces autophagy, triggers the activation of VMP1 in vitro and in vivo. VMP1, a transmembrane protein essential for the formation of the autophagosome, plays a role in autophagy during stress responses (13–16). Genetic screens in Caenorhabditis elegans identified VMP1 as one of three essential components of the metazoan-specific autophagic pathway (34).
Our study expands on these findings and provides new mechanistic insights into the role of the autophagic process in cancer cells. We define a novel signaling pathway initiated by the oncogene KRAS using the transcription factor GLI3 as an effector to regulate VMP1 expression and autophagy in cancer cells (Fig. 9). Recent studies have demonstrated that KRAS is capable of inducing autophagy in cancer cells through an increase in reactive oxygen species (10, 21–23). The increase in intracellular reactive oxygen species induced by oncogenic KRAS activates JNK and promotes autophagy and the expression of ATG5 and ATG7, which are components of the autophagosome (21). Interestingly, KRASG12D-induced increases in intracellular reactive oxygen species were attenuated by p38 MAPK inhibition, which also suppressed autophagy and subsequent cellular transformation (21). In the current study, we present data supporting a novel mechanism downstream of KRAS that regulates components of autophagic machinery in cancer cells. This mechanism involves the canonical PI3K-AKT cascade, a novel transcriptional complex (GLI3-p300) and a new target, VMP1, which is an essential molecule during early stages of the formation of the autophagosome and one that could play a functional role in KRASG12D-induced transformation.
FIGURE 9.

Schematic representation of a novel KRAS-initiated pathway leading to the activation of VMP1 in cancer cells.
Recent reports by several groups, including ours (18, 30, 32), have demonstrated regulation of the GLI proteins by Hedgehog and non-Hedgehog signaling in multiple cancer types. Here, we provide evidence that the transcription factor GLI3 is a novel effector of the KRAS-PI3K-AKT1 pathway in cancer cells. As mentioned above, GLI3 is a downstream effector of the Hedgehog signaling pathway, a cascade that plays a major role during development (28) as well as carcinogenesis (28, 29). Activation of these transcription factors occurs when the Hedgehog ligand binds to the receptor PATCHED. The binding blocks an inhibitory effect that PATCHED has over SMOOTHENED, a signaling component of the Hedgehog receptor complex, freeing SMOOTHENED to activate GLI transcription factors. These proteins in turn regulate Hedgehog target genes (28).
In this study, we have demonstrated that GLI3 activity is regulated in a SMOOTHENED-independent manner by KRAS-PI3K-AKT1. We have defined GLI3 as a downstream mediator of this signaling cascade and have identified a novel transcriptional activation function for this transcription factor over the autophagy-inducing gene VMP1 in cancer cells. On the basis of our data, we propose a novel pathway that uses GLI3 as the effector for its cellular function and expands the repertoire of signaling cascades that, in addition to Hedgehog, regulate GLI transcriptional activity in tumors.
In summary, our results identify a novel pathway supporting the role of autophagy during carcinogenesis and demonstrate an alternative cascade used by KRAS to induce autophagy in cancer cells. The molecular mechanisms that link oncogenic KRAS to the induction of autophagy and subsequent malignant transformation may provide new insights into the relationship between autophagy and cancer. Lastly, because pharmacological inhibitors for KRAS, PI3K, AKT1, and autophagy (e.g. chloroquine) are being tested in clinical trials for cancer, the biochemical evidence reported here extends the conceptual framework for the better design and interpretation of therapeutic approaches to these dismal diseases.
Supplementary Material
Acknowledgments
We thank Emily Porcher and Kimberly McGhee for assistance in preparing the submitted manuscript and Drs. Lucas Nacusi, Raul Urrutia, and Gwen Lomberk for critical reading of the manuscript.
This work was supported by the National Institutes of Health Grant CA136526, Mayo Clinic Pancreatic SPORE Grant P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology Grant P30 DK84567 (to M. E. F.-Z.); Agencia Nacional de Promoción Científica y Tecnológica Grants ANPCyT-PICT01627 and ANPCyT-PICT0411 (to A. R.); Consejo Nacional de Investigaciones Científicas y Técnicas Grant CONICET-PIP2527; and University of Buenos Aires Grant UBA-UBACyTM072 (to M. I. V.).

This article contains supplemental Figs. 1–4.
- VMP1
- vacuole membrane protein 1
- NT
- non-targeting control
- ca
- constitutively active
- EGFP
- enhanced GFP
- LC3
- light chain 3.
REFERENCES
- 1. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lum J.J., DeBerardinis R. J., Thompson C.B. (2005) Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 6, 439–448 [DOI] [PubMed] [Google Scholar]
- 3. Kundu M., Thompson C. B. (2008) Autophagy: Basic principles and relevance to disease. Annu. Rev. Pathol. 3, 427–455 [DOI] [PubMed] [Google Scholar]
- 4. Chen N., Debnath J. (2010) Autophagy and tumorigenesis. FEBS Lett. 584, 1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim J. H., Kim H. Y., Lee Y. K., Yoon Y. S., Xu W. G., Yoon J. K., Choi S. E., Ko Y. G., Kim M. J., Lee S. J., Wang H. J., Yoon G. (2011) Involvement of mitophagy in oncogenic K-Ras-induced transformation: Overcoming a cellular energy deficit from glucose deficiency. Autophagy 7, 1187–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. White E., Karp C., Strohecker A. M., Guo Y., Mathew R. (2010) Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 22, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amaravadi R. K., Yu D., Lum J. J., Bui T., Christophorou M. A., Evan G. I., Thomas-Tikhonenko A., Thompson C. B. (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 117, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenfeldt M. T., Ryan K. M. (2011) The multiple roles of autophagy in cancer. Carcinogenesis 32, 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Notte A., Leclere L., Michiels C. (2011) Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 82, 427–434 [DOI] [PubMed] [Google Scholar]
- 10. Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J. M., Dell'antonio G., Mautner J., Tonon G., Haigis M., Shirihai O. S., Doglioni C., Bardeesy N., Kimmelman A. C. (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 12.Mathew R., Kongara S., Beaudoin B., Karp C. M., Bray K., Degenhardt K., Chen G., Jin S., White E. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vaccaro M. I., Ropolo A., Grasso D., Iovanna J. L. (2008) A novel mammalian trans-membrane protein reveals an alternative initiation pathway for autophagy. Autophagy 4, 388–390 [DOI] [PubMed] [Google Scholar]
- 14. Ropolo A., Grasso D., Pardo R., Sacchetti M. L., Archange C., Lo Re A., Seux M., Nowak J., Gonzalez C. D., Iovanna J. L., Vaccaro M. I. (2007) The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 282, 37124–37133 [DOI] [PubMed] [Google Scholar]
- 15. Grasso D., Ropolo A., Lo Ré A., Boggio V., Molejón M. I., Iovanna J. L., Gonzalez C. D., Urrutia R., Vaccaro M. I. (2011) Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J. Biol. Chem. 286, 8308–8324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grasso D., Sacchetti M. L., Bruno L., Lo Ré A., Iovanna J. L., Gonzalez C. D., Vaccaro M. I. (2009) Autophagy and VMP1 expression are early cellular events in experimental diabetes. Pancreatology 9, 81–88 [DOI] [PubMed] [Google Scholar]
- 17. Grasso D., Garcia M. N., Iovanna J. L. (2012) Autophagy in pancreatic cancer. Int. J. Cell Biol. 2012, 760498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elsawa S. F., Almada L. L., Ziesmer S. C., Novak A. J., Witzig T. E., Ansell S. M., Fernandez-Zapico M. E. (2011) GLI2 transcription factor mediates cytokine cross-talk in the tumor microenvironment. J. Biol. Chem. 286, 21524–21534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim M. J., Woo S. J., Yoon C. H., Lee J. S., An S., Choi Y. H., Hwang S. G., Yoon G., Lee S. J. (2011) Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286, 12924–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pardo R., Lo Ré A., Archange C., Ropolo A., Papademetrio D. L., Gonzalez C. D., Alvarez E. M., Iovanna J. L., Vaccaro M. I. (2010) Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 10, 19–26 [DOI] [PubMed] [Google Scholar]
- 21. Tuveson D. A., Hingorani S. R. (2005) Ductal pancreatic cancer in humans and mice. Cold Spring Harb. Symp. Quant. Biol. 70, 65–72 [DOI] [PubMed] [Google Scholar]
- 22. Pérez-Mancera P. A., Tuveson D. A. (2006) Physiological analysis of oncogenic K-ras. Methods Enzymol. 407, 676–690 [DOI] [PubMed] [Google Scholar]
- 23. Mariño G., Martins I., Kroemer G. (2011) Autophagy in Ras-induced malignant transformation: Fatal or vital? Mol. Cell 42, 1–3 [DOI] [PubMed] [Google Scholar]
- 24. Guo J. Y., Chen H. Y., Mathew R., Fan J., Strohecker A. M., Karsli-Uzunbas G., Kamphorst J. J., Chen G., Lemons J. M., Karantza V., Coller H. A., Dipaola R.S., Gelinas C., Rabinowitz J. D., White E. (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lock R., Roy S., Kenific C. M., Su J. S., Salas E., Ronen S. M., Debnath J. (2011) Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lock R., Debnath J. (2011) Ras, autophagy and glycolysis. Cell Cycle 10, 1516–1517 [DOI] [PubMed] [Google Scholar]
- 27. Hingorani S. R., Petricoin E. F., Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V., Hruban R. H., Lowy A. M., Tuveson D. A. (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 [DOI] [PubMed] [Google Scholar]
- 28. Hui C. C., Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 [DOI] [PubMed] [Google Scholar]
- 29. Stecca B., Ruiz I., Altaba A. (2010) Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell. Biol. 2, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nolan-Stevaux O., Lau J., Truitt M. L., Chu G. C., Hebrok M., Fernández-Zapico M. E., Hanahan D. (2009) GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akimaru H., Chen Y., Dai P., Hou D. X., Nonaka M., Smolik S. M., Armstrong S., Goodman R. H., Ishii S. (1997) Drosophila CBP is a co-activator of cubitus interruptus in hedgehog signaling. Nature 386, 735–738 [DOI] [PubMed] [Google Scholar]
- 32. Lin W., Zhang Z., Srajer G., Chen Y. C., Huang M., Phan H. M., Dent S. Y. (2008) Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev. Dyn. 237, 928–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yauch R. L., Gould S. E., Scales S. J., Tang T., Tian H., Ahn C. P., Marshall D., Fu L., Januario T., Kallop D., Nannini-Pepe M., Kotkow K., Marsters J. C., Rubin L. L., de Sauvage F. J. (2008) A paracrine requirement for hedgehog signaling in cancer. Nature 455, 406–410 [DOI] [PubMed] [Google Scholar]
- 34. Tian Y., Li Z., Hu W., Ren H., Tian E., Zhao Y., Lu Q., Huang X., Yang P., Li X., Wang X., Kovács A. L., Yu L., Zhang H. (2010) C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141, 1042–1055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.