

Background: The hormone FGF21 is a potent regulator of carbohydrate and lipid metabolism and a promising drug for treating metabolic syndrome.
Results: Farnesoid X receptor (FXR) agonists and FGF19 induce hepatic FGF21 secretion via a transcriptional mechanism and posttranscriptional mechanism, respectively.
Conclusion: Activation of the FXR pathway stimulates FGF21 expression and secretion.
Significance: Activation of FXR is a potential approach to enhance endogenous FGF21 production and reverse metabolic syndrome.
Keywords: Bile Acid, Diabetes, Fatty Acid, Fibroblast Growth Factor (FGF), Hormone Receptors, Liver, Nuclear Receptors, Nutrition, Peroxisome Proliferator-activated Receptor (PPAR), Transcription Regulation
Abstract
Previous studies have shown that starvation or consumption of a high fat, low carbohydrate (HF-LC) ketogenic diet induces hepatic fibroblast growth factor 21 (FGF21) gene expression in part by activating the peroxisome proliferator-activated receptor-α (PPARα). Using primary hepatocyte cultures to screen for endogenous signals that mediate the nutritional regulation of FGF21 expression, we identified two sources of PPARα activators (i.e. nonesterified unsaturated fatty acids and chylomicron remnants) that induced FGF21 gene expression. In addition, we discovered that natural (i.e. bile acids) and synthetic (i.e. GW4064) activators of the farnesoid X receptor (FXR) increased FGF21 gene expression and secretion. The effects of bile acids were additive with the effects of nonesterified unsaturated fatty acids in regulating FGF21 expression. FXR activation of FGF21 gene transcription was mediated by an FXR/retinoid X receptor binding site in the 5′-flanking region of the FGF21 gene. FGF19, a gut hormone whose expression and secretion is induced by intestinal bile acids, also increased hepatic FGF21 secretion. Deletion of FXR in mice suppressed the ability of an HF-LC ketogenic diet to induce hepatic FGF21 gene expression. The results of this study identify FXR as a new signaling pathway activating FGF21 expression and provide evidence that FXR activators work in combination with PPARα activators to mediate the stimulatory effect of an HF-LC ketogenic diet on FGF21 expression. We propose that the enhanced enterohepatic flux of bile acids during HF-LC consumption leads to activation of hepatic FXR and FGF19 signaling activity and an increase in FGF21 gene expression and secretion.
Introduction
Fibroblast growth factor 21 (FGF21) is a member of the fibroblast growth factor family of signaling molecules that regulate cell growth, differentiation, and migration (1). Studies investigating the biological function of FGF21 have shown that FGF21 modulates carbohydrate and lipid metabolism in adult animals. For example, administration of FGF21 increases glucose uptake in adipocytes, an effect that is mediated by an elevation in the expression of the glucose transporter GLUT1 (2). FGF21 also increases rates of fatty acid oxidation and gluconeogenesis in liver (3–6). These effects are associated with an elevation in the expression of peroxisome proliferator-activated receptor-γ coactivator-1α, a transcriptional coactivator of genes comprising the fatty acid oxidation and gluconeogenic pathways. The stimulatory effect of FGF21 on gluconeogenic enzyme expression is dependent on the presence of peroxisome proliferator-activated receptor-γ coactivator-1α (5). In contrast to the effects of FGF21 on the expression of enzymes comprising the fatty acid oxidation and gluconeogenic pathways, FGF21 inhibits the expression of enzymes comprising the fatty acid synthesis pathway (3, 6). Alterations in hepatic fatty acid oxidation and fatty acid synthesis have been proposed to play a role in mediating the decrease in serum and hepatic triacylglycerols and increase in insulin sensitivity in obese/diabetic animals treated with exogenous FGF21 (5, 7, 8).
Studies analyzing the tissue/cell type distribution of FGF21 expression indicate that the liver is an important site of FGF21 synthesis in the body (1, 9). This observation plus the striking effects of FGF21 on carbohydrate and lipid metabolism has prompted studies investigating the regulation of hepatic FGF21 expression in intact animals. Feeding mice a high fat, low carbohydrate (HF-LC)2 ketogenic diet stimulates a 5–25-fold increase in hepatic FGF21 mRNA abundance (10, 11), an effect that is associated with a 1.7–9-fold elevation in FGF21 levels in the blood (10, 12, 13). This finding has led to the proposal that alterations in hepatic FGF21 expression play a role in mediating changes in carbohydrate and lipid metabolism caused by HF-LC ketogenic diet consumption. In this model, the HF-LC diet-induced increase in the synthesis and secretion of FGF21 activates hepatic fatty acid oxidation and gluconeogenesis. Alterations in these metabolic processes in turn facilitate the synthesis of ketone bodies from dietary fatty acids delivered to the liver. Support for this model is derived from studies demonstrating that knockdown of hepatic FGF21 expression attenuates the increase in hepatic fatty acid oxidation and ketogenesis caused by consumption of an HF-LC ketogenic diet (5, 14). The hepatic expression of FGF21 is also induced by starvation, another dietary condition in which rates of fatty acid oxidation, gluconeogenesis, and ketogenesis are elevated (4, 10, 15). Deletion of FGF21 abolishes the stimulatory effect of starvation on rates of fatty acid oxidation, gluconeogenesis, and ketogenesis (5). These findings provide further evidence that FGF21 is a key factor controlling carbohydrate and lipid metabolism during ketotic states.
FGF21 has also come into focus as a potential pharmacological agent for treating metabolic syndrome. Administration of recombinant FGF21 or transgenic expression of the FGF21 gene in mouse and non-human primate models of obesity and type 2 diabetes causes an increase in energy expenditure and a decrease in adiposity and triacylglycerol accumulation in the blood and liver (2, 3, 6, 8, 16). Treatment of obese/diabetic animals with FGF21 also reduces blood glucose levels and increases glucose tolerance and insulin sensitivity. Another attractive feature of FGF21 is that it is not mitogenic and increases resistance to agents inducing tumorigenesis (2, 7, 17). The striking ability of FGF21 to reverse obesity and diabetes without deleterious side effects has generated a strong interest in the physiological and molecular mechanisms controlling FGF21 expression.
The mechanisms mediating the nutritional regulation of FGF21 expression have not been completely defined. The peroxisome proliferator-activated receptor-α (PPARα) is a nuclear receptor that mediates the stimulatory effect of starvation and high fat consumption on the expression of genes involved in fatty acid oxidation (18). Previous studies have shown that deletion of the PPARα gene suppresses the ability of HF-LC consumption and starvation to induce hepatic FGF21 mRNA abundance (4, 10, 11). This observation provides support for a role of PPARα in mediating diet-induced changes in FGF21 gene expression. As deletion of PPARα does not abolish the effects of HF-LC consumption and starvation on FGF21 mRNA abundance, a PPARα-independent pathway(s) also contributes to the nutritional regulation of FGF21 gene expression. The objective of the present study was to identify endogenous signaling factors that mediate the nutritional regulation of hepatic FGF21 expression. We have identified two sources of PPARα activators (i.e. nonesterified unsaturated fatty acids and chylomicron remnants) that induce of FGF21 expression. In addition, we have discovered that endogenous activators of the farnesoid X receptor (FXR) (i.e. bile acids) and an endocrine component of the FXR signaling pathway (i.e. FGF19) cause a robust increase in FGF21 expression and/or FGF21 secretion. We also show that FXR plays a role in mediating the stimulatory effect of an HF-LC ketogenic diet on FGF21 expression.
EXPERIMENTAL PROCEDURES
Animals
Male Sprague-Dawley rats (125–150 g), C57BL/6J mice, and Fxr−/− mice on a C57BL/6J background (19) were maintained at 24 °C on a 12:12 h light-dark cycle. All animals were allowed ad libitum access to food and water. Animals were fed a chow diet comprising 6.2% fat, 18.6% protein, and 44.2% digestible carbohydrate (all w/w) (Harlan Teklad, 18% protein rodent diet); a standard purified diet comprising 7.2% fat, 17.7% protein, and 60.1% carbohydrate (all w/w) (Harlan Teklad, AIN-93G); or an HF-LC ketogenic diet comprising 78.9% fat, 9.5% protein, and 0.76% carbohydrate (all w/w) (Bio-Serv, F3666). All studies were approved by the Institutional Animal Care and Use Committees of West Virginia University and Northeast Ohio Medical University.
Cell Culture
Hepatocytes were isolated from 24-h starved male Sprague-Dawley rats (∼200 g) as described by Stabile et al. (20). Cells (3 × 106) were plated on 60-mm collagen-coated dishes containing Waymouth's medium MD752/1 supplemented with 20 mm HEPES, pH 7.4, 0.5 mm serine, 0.5 mm alanine, 100 μg/ml penicillin, 100 μg/ml streptomycin, 50 mg/ml gentamicin, and 5% newborn calf serum. At 4 h of incubation, the medium was replaced with one of the same composition lacking newborn calf serum. A Matrigel overlay (0.3 mg/ml) and insulin (50 nm) were added at this time. At 24 h of incubation, the cells were washed in serum-free Medium 199, and the incubation was continued in serum-free Medium 199. At 48 h of incubation, the medium was replaced with one of the same composition containing the treatments indicated in the figure legends. Hepatocyte cultures were maintained in a humidified chamber at 37 °C in 5% CO2 and 95% air.
Human HepG2 hepatoma cells were plated on 60-mm dishes containing Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum. After the cells reached 80% confluence, the medium was changed to one of the same composition lacking fetal bovine serum. After 24 h of incubation, the medium was replaced with serum-DMEM containing the treatments indicated in the figure legends.
Fatty acids (Nu Chek Prep) were prepared as 1 mm stock solutions in complex with bovine serum albumin (BSA) at a 1:4 molar ratio (21). Intestinal lymph was obtained from lymph fistula rats intubated intraduodenally with a lipid emulsion containing safflower oil (0.36 g/animal) (22, 23). The procedure for preparing whole chylomicrons and chylomicron remnants from the intestinal lymph has been described (24). The triacylglycerol content of chylomicrons and chylomicron remnants was determined using a Triglyceride and Free Glycerol kit supplied by Sigma-Aldrich. The fatty acid composition of chylomicrons and chylomicron remnants was determined by gas chromatography. Chenodeoxycholic acid, taurochenodeoxycholic acid, taurocholic acid, taurodeoxycholic acid, cholic acid, glucagon-like peptide-1, GW4064, 3,5,3′-triiodothyronine (T3), corticosterone, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) were obtained from Sigma-Aldrich. GW7647 and GW0742 were purchased from Cayman Chemical. Bovine insulin and glucagon were gifts from Lilly. Recombinant human FGF19 (R&D Systems), rat leptin (Alexis Biochemicals), and tauro-β-muricholic acid (Steraloids) were obtained from the indicated sources.
Isolation of RNA and Quantitation of mRNA Levels
Total RNA was extracted from tissues and cell cultures by the guanidinium thiocyanate/phenol/chloroform method (25). The abundance of mRNA encoding FGF21 and bile salt export pump (BSEP) was measured by quantitative real time PCR analysis using the Qiagen Quantitect SYBR Green RT-PCR system. Samples of DNase I-treated RNA (100 ng) were analyzed in triplicate according to the manufacturer's instructions. PCR was performed in 96-well plates using a Bio-Rad iCycler iQ. The relative amount of mRNA was calculated using the comparative Ct method. Rat cyclophilin, human glyceraldehyde-3-phosphate dehydrogenase, and mouse 36B4 were used as reference genes. Amplification of specific transcripts was confirmed by analyzing the melting curve profile performed at the end of each run and by determining the size of the PCR products using agarose electrophoresis and ethidium bromide staining. The primer set for each gene is shown in supplemental Table S1.
Reporter Plasmids
Fragments of the rat FGF21 promoter/regulatory region were amplified by PCR and inserted into the KpnI and XhoI sites of pGL3-Basic (Promega). The FGF21 DNA fragments were named by designating the 5′- and 3′-ends of each fragment relative to the transcription start site. A mutation of the FXR response element (FXRE) at −1221 and −1220 bp was introduced into the −1316 to +68 bp FGF21 reporter plasmid using the Agilent QuikChange II XL site-directed mutagenesis kit. Structures of reporter plasmids were confirmed by nucleotide sequence analysis.
Transient Transfection
Hepatocytes were plated on 35-mm dishes and transfected with 2 μg of the −2940 to +68 bp FGF21 reporter plasmid or an equimolar amount of another reporter plasmid using Effectene reagent (Qiagen). At 12 h of incubation, the transfection medium was replaced with fresh medium with or without GW4064. At 36 h of incubation, cells were harvested, and cell extracts were prepared in 1× Cell Culture Lysis Buffer (Promega). Cell extracts were centrifuged at 14,000 × g for 1 min, and the supernatants were assayed for protein concentration and luciferase activity. Luciferase assay reagent was obtained from Promega.
Gel Mobility Shift Analysis
Complementary oligonucleotides containing the FGF21 FXRE were synthesized with biotin attached to their 3′-ends. These oligonucleotides were annealed for 2 h at room temperature. Recombinant human FXR and retinoid X receptor (RXR) α were obtained from Sigma-Aldrich. Binding reactions (20 μl) contained 200 pmol of DNA probe, 150 ng of FXR and RXRα, 10 mm Tris, pH 7.5, 50 mm KCl, 1 mm dithiothreitol, 50 μg/ml poly(dI-dC), and 2.5% (v/v) glycerol. Reactions were carried out at room temperature for 30 min. DNA and DNA-protein complexes were resolved on 6% nondenaturing polyacrylamide gels at 4 °C in 0.5× TBE (45 mm Tris, pH 8.3, 45 mm boric acid, and 1 mm EDTA) and then transferred to a positively charged nylon membrane in 0.5× TBE. Transferred DNAs were cross-linked to the membrane by UV irradiation. DNA on membranes was detected using horseradish peroxidase-conjugated streptavidin in combination with chemiluminescent substrate (LightShiftTM chemiluminescence EMSA kit, Thermo Scientific). For competition experiments, unlabeled competitor DNA was mixed with labeled oligomer prior to the addition of FXR and RXRα. For antibody supershift experiments, the reaction was incubated with antibodies against FXR or NF1 (Santa Cruz Biotechnology) for 30 min prior to the addition of the oligonucleotide probe.
Western Analyses
The culture medium (3 ml) from a single plate of cells was centrifuged to remove cellular debris. The supernatant was concentrated to 200 μl using an Amicon Ultra-4 centrifugal filter unit. Proteins contained in the concentrated supernatant were subjected to electrophoresis in 12% SDS-polyacrylamide gels and then transferred to polyvinylidene difluoride membranes (Immobilon-FL, Millipore) using an electroblotting apparatus (Bio-Rad). The blots were blocked in TBST (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.1% Tween) containing 5% nonfat dry milk for 1 h at room temperature and then incubated with primary antibody diluted 1:500 in TBST containing 5% bovine serum albumin. After incubation with primary antibody for 12 h at 4 °C, the blots were washed in TBST. Next, the blots were incubated with secondary antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories) diluted 1:2000 in TBST and 5% nonfat dry milk for 1 h at room temperature. After washing with TBST, antibody-protein complexes on blots were detected using enhanced chemiluminescence (Amersham Biosciences). Fluorescence on the blots was visualized using a Typhoon 9410 imager, and signals were quantified using ImageQuant software. Goat polyclonal antibodies against mouse FGF21 (AF3057), human FGF21 (TA303289), and albumin (sc-46293) were obtained from R&D Systems, Origene, and Santa Cruz Biotechnology, respectively.
Measurement of Plasma FGF21 Concentration
Plasma FGF21 concentration was determined using an FGF21 ELISA kit (R&D Systems).
Statistical Methods
Data were subjected to analysis of variance, and statistical comparisons were made with the Dunnett's or Student's t test.
RESULTS
Nonesterified Fatty Acids and Chylomicron Remnants Increase FGF21 Expression in Primary Rat Hepatocyte Cultures
Primary cultures of rat hepatocytes are strongly responsive to nutritional and hormonal signals and have been used successfully to screen factors that regulate hepatic gene expression (26). Prior to using this system to identify nutritional signals controlling the expression of FGF21, we wanted to verify that the nutritional regulation of rat FGF21 expression was similar to that described in other species. Feeding rats an HF-LC ketogenic diet for 6 days or starving rats for 30 h stimulated an 18–35-fold increase in hepatic FGF21 mRNA abundance compared with rats fed a standard purified diet or chow diet (Fig. 1A). The effects of these dietary manipulations on FGF21 mRNA abundance in rat liver are similar to those reported in mouse liver (4, 10, 11, 15). The stimulatory effect of starvation on FGF21 expression in rat liver is consistent with previous studies demonstrating that prolonged starvation increases circulating FGF21 levels in humans (27).
FIGURE 1.

Nonesterified unsaturated fatty acids and chylomicron remnants increase hepatic FGF21 mRNA abundance. A, nutritional regulation of hepatic FGF21 expression in rats. Sprague-Dawley rats were fed a chow diet, a standard purified diet, or an HF-LC ketogenic diet for 7 days. The composition of the diets is described under “Experimental Procedures.” A fourth group of animals was fed the standard purified diet for 6 days and then starved for 30 h. Animals were killed 6 h after the start of the dark cycle, total RNA was isolated from liver, and the abundance of FGF21 mRNA was measured by quantitative real time PCR. Hepatic FGF21 mRNA abundance was also measured in a fifth group of animals fed a standard purified diet for 6 days followed by starvation for 30 h and refeeding for 5 h. The level of FGF21 mRNA in animals fed the chow diet was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of six animals. An asterisk indicates that the mean is significantly (p ≤ 0.05) higher compared with that of animals fed the standard (Std) purified diet. B–F, effect of nutrients and hormones on FGF21 expression in primary cultures of rat hepatocytes. Rat hepatocytes were prepared as described under “Experimental Procedures” and incubated in serum-free Medium 199. B, time course of the effect of nonesterified fatty acids (250 μm) on FGF21 mRNA abundance. Hepatocytes were incubated with the indicated fatty acids complexed to BSA. Hepatocytes receiving no additions (NA) were incubated in medium containing an equivalent concentration of BSA. The level of FGF21 mRNA in cells incubated with fatty acids for 0 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. C, effect of different oleate treatment protocols on FGF21 mRNA abundance. Hepatocytes were incubated with or without oleate for 2 and 6 h without a medium change. A third treatment group (designated 4 + 2) was incubated with oleate for 6 h with a medium change 2 h prior to the end of the treatment period. The level of FGF21 mRNA in hepatocytes incubated without oleate for 2 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. Different superscript letters indicate that means are significantly (p ≤ 0.05) different. D, time course of the effect of GW7647 on FGF21 mRNA abundance. Hepatocytes were incubated with or without GW7647 (1 μm) for the indicated time periods. The level of FGF21 mRNA in cells incubated with GW7647 for 0 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells incubated in the absence of GW7647 for the same time period. E, effect of chylomicrons and chylomicron remnants on FGF21 mRNA abundance. Hepatocytes were incubated with chylomicrons and chylomicron remnants prepared from rats fed safflower oil. FGF21 mRNA was measured after 2 h of treatment. The level of FGF21 mRNA in cells incubated with no additions was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of five experiments. The asterisk indicates that the mean is significantly (p ≤ 0.05) different. F, effects of glucagon, corticosterone, GW0742, TCPOBOP, glucose, insulin, T3, and leptin on FGF21 mRNA abundance. Rat hepatocytes were incubated in serum-free Medium 199 containing low glucose (5.5 mm) or high glucose (25 mm) with or without glucagon (100 nm), corticosterone (1 μm), GW0742 (1 μm), TCPOBOP (1 μm), insulin (50 nm), T3 (150 nm), or leptin (125 nm). FGF21 mRNA abundance was measured after 6 h of treatment. The level of FGF21 mRNA incubated with 5.5 mm glucose and vehicle was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of three experiments. Different superscript letters indicate that means are significantly (p ≤ 0.05) different.
Previous studies have shown that the supply of nonesterified fatty acids to the liver is enhanced by HF-LC consumption and starvation (12, 28–31) and that oleate (C18:1, n-9) and linoleate (C18:2, n-6) are the primary nonesterified fatty acids found in the blood during these dietary conditions (32). Unsaturated fatty acids can stimulate gene expression by enhancing the activity of the nuclear receptor PPARα (33). These observations plus work demonstrating that nutritional regulation of FGF21 expression is dependent on the presence of PPARα (4, 10, 11) led us to investigate whether albumin-bound oleate and linoleate modulated hepatic FGF21 expression. Incubating primary rat hepatocyte cultures with oleate or linoleate stimulated an increase in FGF21 mRNA abundance (Fig. 1B). The effects of oleate and linoleate on FGF21 mRNA levels were dose-dependent (supplemental Fig. S1) and were maximal (4.3–6.1-fold) at 2 h of treatment (Fig. 1B). The stimulatory effect of oleate and linoleate on FGF21 mRNA abundance decreased at 6 and 12 h of treatment. Pawar and Jump (34) have shown that eicosapentaenoic acid (EPA) (C20:5, n-3) increases PPARα activity in rat hepatocytes and have proposed that EPA is an intracellular PPARα ligand. Incubating hepatocytes with EPA caused an increase in FGF21 mRNA abundance with a time course similar to that of oleate and linoleate (Fig. 1B). The maximal increase in FGF21 mRNA caused by EPA was 3.3-fold at 2 h of treatment. In contrast to the results for oleate, linoleate, and EPA, incubating hepatocytes with hexanoate or dodecanoate had no effect on FGF21 mRNA abundance. These results provide support for a role of nonesterified unsaturated fatty acids in mediating diet-induced changes in FGF21 gene expression.
Greater than 80% of the oleate or EPA added to the culture medium of rat hepatocyte cultures is cleared within 6 h of treatment (34). Thus, the decrease in fatty acid regulation of FGF21 mRNA abundance observed at 6 and 12 h of treatment may be caused by the hepatic metabolism and depletion of fatty acids in the culture medium. To investigate this possibility, a modification of the 6-h treatment protocol (designated 4 + 2) was used in which the treatment medium was replaced with fresh medium 2 h prior to the end of the treatment period. The ability of oleate to induce FGF21 mRNA abundance using the 4 + 2 protocol was greater than that observed using the 6-h treatment protocol without medium replacement (Fig. 1C). The extent of the stimulatory effect of oleate on FGF21 mRNA abundance using the 4 + 2 protocol was similar to that observed for 2 h of oleate treatment. The ability of linoleate and EPA to induce FGF21 mRNA abundance was also greater using the 4 + 2 protocol compared with the 6-h treatment protocol without medium replacement (data not shown). These findings are consistent with a role of fatty acid depletion in mediating the decreased oleate-, linoleate-, and EPA-induced FGF21 mRNA abundance at 6 h of treatment.
If oleate, linoleate, and EPA enhance FGF21 expression in hepatocytes by activating PPARα bound to the FGF21 gene, then treatment with the selective PPARα ligand/agonist GW7647 should mimic the effects of these fatty acids on FGF21 expression. Incubating hepatocytes with an optimal concentration of GW7647 (1 μm) stimulated a rapid (<2 h) and sustained (>24 h) increase in FGF21 mRNA abundance (Fig. 1D). Interestingly, the maximal effect of GW7647 on FGF21 mRNA abundance (2.9-fold relative to no addition at 12 h of incubation) was lower than the maximal effect of oleate on FGF21 mRNA abundance (6.1-fold at 2 h of incubation), suggesting that oleate generates endogenous ligands that are more effective than GW7647 in activating PPARα activity. Alternatively, oleate may act through additional mechanisms besides ligand activation of PPARα to enhance FGF21 expression.
In addition to nonesterified fatty acids derived from the extrahepatic hydrolysis of triacylglycerols in chylomicrons (i.e. nonesterified fatty acid spillover), dietary fatty acids are delivered to the liver in the form of chylomicron remnants. These lipoproteins contain 15–35% of the dietary triacylglycerol originally packaged in the chylomicrons (35). Triacylglycerols contained in chylomicron remnants are incorporated into hepatocytes through the action of hepatic lipase on the extracellular surface and by receptor-mediated endocytosis followed by the hydrolysis of the triacylglycerols by lysosomal enzymes (36). We asked whether chylomicron remnants obtained from rats fed safflower oil were effective in modulating FGF21 expression in rat hepatocyte cultures. The fatty acid composition of chylomicron remnants prepared from rats infused intraduodenally with safflower oil was 14.4% palmitate, 16.1% oleate, and 69.5% linoleate. Incubating chylomicron remnants (33.3 μg of triacylglycerol/ml) with rat hepatocytes for 2 h stimulated a 2.6-fold increase in FGF21 mRNA abundance (Fig. 1E). The concentration of chylomicron remnants used in this experiment would result in 115 μm fatty acid in the medium if all three fatty acids on the glycerol backbone were hydrolyzed. In contrast to the effect of chylomicron remnants on FGF21 expression, incubating hepatocytes with whole chylomicrons (33.3 μg of triacylglycerol/ml) had no effect on FGF21 mRNA levels. Thus, dietary fatty acids delivered in the form of chylomicron remnants are effective in stimulating FGF21 expression.
Effects of Glucagon, Corticosterone, GW0742, TCPOBOP, Glucose, Insulin, T3, and Leptin on FGF21 Expression
Incubating hepatocytes with nonesterified unsaturated fatty acids, chylomicron remnants, or a synthetic PPARα agonist does not quantitatively recapitulate the effects of HF-LC consumption or starvation on hepatic FGF21 expression in intact animals (Fig. 1, A–E). This observation plus previous work demonstrating that deletion of PPARα does not completely suppress the effects of HF-LC consumption and starvation on FGF21 mRNA abundance (10, 11) suggests that additional signaling pathways are involved in mediating the nutritional regulation of FGF21 gene expression. Signaling pathways whose activities are enhanced by HF-LC consumption and/or starvation include those that are activated by glucagon (37), glucocorticoids (38), PPARβ agonists (39), and constitutive androstane receptor agonists (40). Signaling pathways whose activities are suppressed by HF-LC consumption and/or starvation include those that are activated by glucose (13, 30, 41), insulin (13, 42), T3 (43), and leptin (44). The role of these pathways in the nutritional regulation of FGF21 expression was investigated by determining the effects of glucagon, corticosterone, GW0742 (PPARβ agonist), TCPOBOP (constitutive androstane receptor agonist), high glucose (25 mm), insulin, T3, and leptin on FGF21 mRNA abundance in rat hepatocyte cultures. Incubating hepatocytes with glucagon and corticosterone in the presence of 5.5 mm glucose (i.e. blood glucose concentration during starvation) caused a 50 and 33% decrease in FGF21 mRNA abundance, respectively (Fig. 1F). Treatment with glucagon and corticosterone stimulated a 10–35-fold increase in phosphoenolpyruvate carboxykinase mRNA abundance (data not shown), confirming that the glucocorticoid and glucagon signaling pathways are active in primary rat hepatocytes. Incubating hepatocytes in 25 mm glucose (i.e. blood glucose concentration in the portal vein after consumption of a high carbohydrate, low fat meal) stimulated a 3.1-fold increase in FGF21 mRNA abundance (Fig. 1F). Addition of insulin in the presence of 25 mm glucose caused a further increase in FGF21 mRNA abundance. Treatment with GW0742 and TCPOBOP in the presence of 5.5 mm glucose and treatment with leptin and T3 in the presence of 25 mm glucose had no effect on FGF21 mRNA abundance. Treatment with GW0742 caused a 3.6-fold increase in pyruvate dehydrogenase kinase 4 mRNA abundance, and treatment with TCPOBOP caused a 40% decrease in CYP7A1 mRNA abundance (data not shown), confirming that the PPARβ and constitutive androstane receptor signaling pathways are active in primary rat hepatocytes. These findings do not support a role of glucagon, glucocorticoids, PPARβ, constitutive androstane receptor, glucose, insulin, T3, and leptin in mediating the increase in FGF21 expression caused by HF-LC consumption and starvation.
Bile Acids and FGF19 Increase FGF21 Secretion in Rat and Human Hepatocytes
Other signaling factors that may contribute to the nutritional regulation of FGF21 expression include bile acids, amphipathic molecules that are synthesized in the liver and secreted into the small intestine where they facilitate the absorption of dietary lipids. As bile acids are efficiently reabsorbed in the small intestine and transported to the liver, they can also function as signaling molecules communicating nutritional status to the liver (45). Previous studies have shown that the enterohepatic circulation of bile acids is enhanced in animals fed a high fat, low carbohydrate diet relative to animals fed a high carbohydrate, low fat diet (46). These observations prompted us to investigate whether bile acids modulated the expression of FGF21 in rat hepatocyte cultures. Incubating hepatocytes with chenodeoxycholic acid (CDCA), a key end product of the bile acid synthesis pathway, stimulated a dose-dependent increase in FGF21 mRNA abundance (Fig. 2A). The stimulatory effect of CDCA on FGF21 mRNA abundance was maximal at 2 h of CDCA incubation (25-fold) and then declined at 6 and 12 h of CDCA incubation (Fig. 2B). Treatment of rat hepatocytes with taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA), and taurodeoxycholic acid (TDCA) also increased FGF21 mRNA abundance, whereas treatment with tauro-β-muricholic acid had no effect on FGF21 mRNA abundance. Ingestion of cholic acid (CA) in the diet was also effective in stimulating FGF21 mRNA abundance in livers of intact rats (Fig. 2C). In addition, treatment with CDCA increased FGF21 mRNA abundance in human HepG2 cells (Fig. 2D). These results demonstrate that bile acids activate FGF21 gene expression in liver cells from rats and humans.
FIGURE 2.

Bile acids increase hepatic FGF21 mRNA abundance. A, effect of different concentrations of CDCA on FGF21 mRNA abundance in primary rat hepatocyte cultures. Cells were incubated with the indicated concentrations of CDCA in serum-free Medium 199. After 2 h of treatment, total RNA was isolated, and FGF21 mRNA abundance was measured. The level of FGF21 mRNA in cells incubated with 0 μm CDCA was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of three experiments. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells incubated with 0 μm CDCA. B, time course of the effect of different bile acids (100 μm) on FGF21 mRNA abundance in primary rat hepatocyte cultures. The level of FGF21 mRNA in cells incubated with bile acids for 0 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. C, effect of consuming a diet containing cholate on hepatic FGF21 mRNA abundance. Rats were starved for 24 h and then fed a standard (Std) purified diet supplemented with or without 1% cholate for 5 h. FGF21 mRNA abundance was measured in total RNA isolated from liver. The level of FGF21 mRNA in animals fed the cholate-free diet was set at 1, and the other value was adjusted proportionately. Values are means of six animals. The asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of animals fed the cholate-free diet. D, time course of the effect of CDCA (100 μm) on FGF21 mRNA abundance in human HepG2 cells. The level of FGF21 mRNA in cells incubated with CDCA for 0 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of three experiments. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells treated without CDCA for the same time period. E, effect of different CDCA treatment protocols on FGF21 mRNA abundance. Primary rat hepatocyte cultures were incubated with or without CDCA (100 μm) for 2 and 6 h without a medium change. A third treatment group (designated 4 + 2) was incubated with CDCA for 6 h with a medium change 2 h prior to the end of the treatment period. The level of FGF21 mRNA in hepatocytes incubated without CDCA for 2 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. Different superscript letters indicate that means are significantly (p ≤ 0.05) different. F, interaction between CDCA and oleate in the regulation of FGF21 mRNA abundance. Primary rat hepatocyte cultures were incubated with or without oleate (250 μm), CDCA (100 μm), or oleate plus CDCA for 2 or 6 h. All treatments contained 62.5 μm BSA. The level of FGF21 mRNA in cells incubated with no additions (NA) for 2 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. *, mean is significantly (p ≤ 0.05) higher than that of cells incubated without oleate and CDCA for the same time period. **, mean is significantly (p ≤ 0.05) higher than that of any other treatment of the same time period. Tβ-MCA, tauro-β-muricholic acid.
Previous studies have shown that exogenous CDCA is rapidly metabolized to β-muricholic acid in rat hepatocyte cultures (47). To investigate whether the decrease in CDCA regulation of FGF21 mRNA abundance observed at 6 and 12 h of treatment (Fig. 2B) was caused by a depletion of CDCA in the culture medium, experiments were carried out in which the CDCA treatment medium was replaced with fresh medium 2 h prior to the end of a 6-h treatment period (designated 4 + 2). The ability of CDCA to induce FGF21 mRNA abundance using the 4 + 2 protocol was greater than that observed for the 6-h treatment protocol without medium replacement (Fig. 2E). The extent of the stimulatory effect of CDCA on FGF21 mRNA abundance using the 4 + 2 protocol was similar to that observed for 2 h of CDCA treatment. These findings are consistent with a role of CDCA depletion in mediating the decrease in CDCA-induced FGF21 mRNA abundance at 6 h of treatment.
We next investigated the interaction of bile acids with unsaturated fatty acids in the regulation of FGF21 mRNA abundance. Treatment of rat hepatocytes with CDCA plus oleate stimulated a greater increase in FGF21 mRNA abundance than treatment with CDCA or oleate alone (Fig. 2F). This finding is consistent with the observation that both PPARα-dependent and PPARα-independent mechanisms contribute to the induction of FGF21 gene expression caused by consumption of an HF-LC ketogenic diet (10, 11).
To further characterize the regulation of FGF21 expression by bile acids, we determined the effects of bile acids on the secretion of FGF21 protein into the culture medium. Treatment of rat hepatocyte cultures with CDCA, TCDCA, TCA, and TDCA stimulated an increase in FGF21 secretion into the culture medium, whereas treatment with tauro-β-muricholic acid had no effect on FGF21 secretion (Fig. 3A). In cells treated with CDCA, TCDCA, TCA, and TDCA for 12 h, FGF21 levels in the culture medium were increased by 14.8-, 5.1-, 3.6-, and 5.8-fold, respectively, relative to FGF21 levels of cells incubated without bile acids for 12 h. Treatment with CDCA was also effective in increasing FGF21 secretion in human HepG2 cells (Fig. 3B). Treatment of rat hepatocytes and HepG2 cells with bile acids had no effect on the secretion of albumin into the culture medium. Ingestion of CA in the diet increased plasma FGF21 concentration in intact rats (Fig. 3C). Thus, bile acids that induce FGF21 mRNA abundance (i.e. CDCA, TCDCA, TCA, TDCA, and CA) also increase hepatic FGF21 secretion or plasma FGF21 concentration.
FIGURE 3.
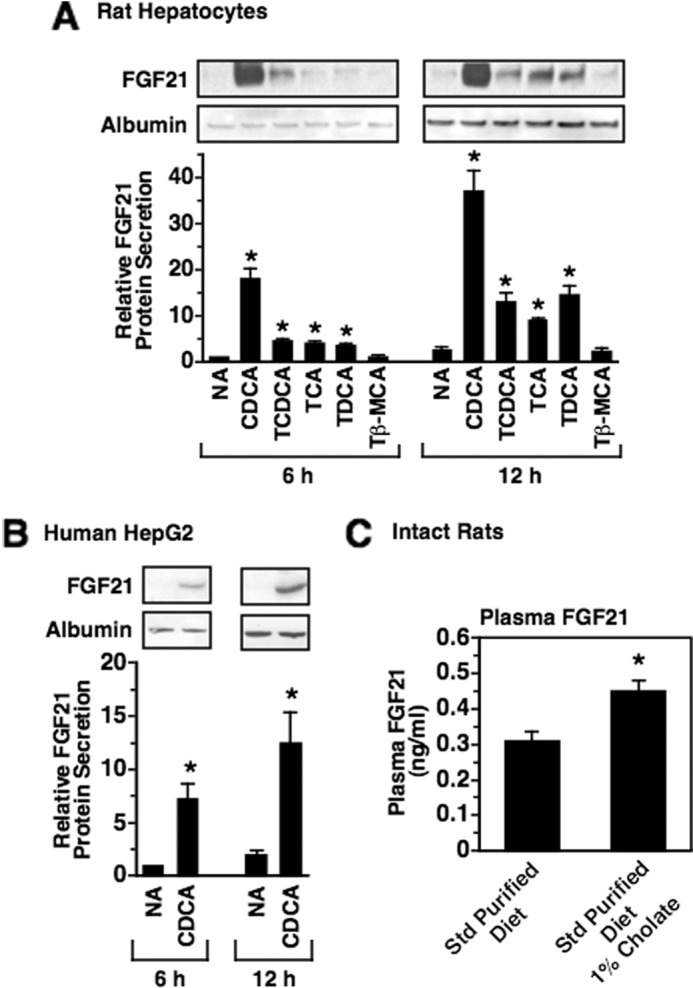
Bile acids increase hepatic FGF21 secretion and plasma FGF21 concentration. The effect of bile acids on FGF21 accumulation in the culture medium was investigated using primary rat hepatocyte cultures (A) and human HepG2 cells (B). Cells were incubated with or without the indicated bile acids (100 μm) in serum-free medium. After 6 and 12 h of treatment, the level of FGF21 and albumin in the culture medium was measured by Western analysis. Top panels, Western analysis of FGF21 from a representative experiment. Bottom panels, signals for FGF21 protein were quantitated. The level of FGF21 protein in the medium of cells incubated with no additions (NA) for 6 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. C, effect of consuming a diet containing cholate on plasma FGF21 concentration. Rats were starved for 24 h and then fed a standard (Std) purified diet supplemented with or without 1% cholate for 5 h. The plasma FGF21 concentration was measured using an ELISA. Values are the means of six animals. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells or animals treated without of bile acid for the same time period. Tβ-MCA, tauro-β-muricholic acid.
FGF19 and glucagon-like peptide-1 (GLP-1) are gut hormones whose production is induced by intestinal bile acids and high fat consumption (48–51). As FGF19 and GLP-1 regulate metabolic processes in liver (52–54), we investigated the effects of FGF19 and GLP-1 on hepatic FGF21 expression and secretion. Treatment of rat hepatocytes with recombinant GLP-1 for 2 and 6 h had no effect on FGF21 mRNA abundance and FGF21 secretion (Fig. 4A). Treatment of rat hepatocytes with recombinant FGF19 also had no effect on FGF21 mRNA abundance but stimulated an increase in FGF21 secretion (Fig. 4A). The magnitude of the effect of FGF19 on FGF21 secretion was 10.8- and 12.6-fold at 2 and 6 h of treatment, respectively (Fig. 4A). Treatment with FGF19 also increased FGF21 secretion in the absence of changes in FGF21 mRNA abundance in human HepG2 cells (Fig. 4B). Intravenous injection of mice with recombinant FGF19 stimulated a 1.8-fold increase in plasma FGF21 concentration (Fig. 4C). This effect was associated with a decrease in hepatic FGF21 mRNA abundance. These results identify FGF19 as another enterohepatic signal that activates hepatic FGF21 production.
FIGURE 4.

FGF19 increases hepatic FGF21 secretion and plasma FGF21 concentration. A and B, effect of FGF19 and GLP-1 on FGF21 mRNA abundance and FGF21 secretion. Primary rat hepatocyte cultures (A) and human HepG2 cells (B) were incubated with or without FGF19 (100 ng/ml) or GLP-1 (100 nm) in serum-free medium for the indicated treatment times. Cells and culture medium were harvested, total RNA was isolated, and FGF21 mRNA abundance and FGF21 protein levels were measured as described under “Experimental Procedures.” Left panels, effect of FGF19 and GLP-1 on FGF21 mRNA abundance. The level of FGF21 mRNA in cells incubated with vehicle for 2 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. Right panels, effect of FGF19 and GLP-1 on FGF21 secretion into the culture medium. The FGF21 protein level in the culture medium of cells incubated with vehicle for 1 (rat hepatocytes) or 2 h (HepG2) was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells incubated with vehicle for the same time period. C, effect of FGF19 on plasma FGF21 concentration. Mice were injected intravenously with vehicle or recombinant FGF19 (0.4 mg/kg). Animals were killed 4 h after injection. Hepatic FGF21 mRNA abundance (left panel) and plasma FGF21 concentration (right panel) were measured. D, interaction between FGF19 and CDCA in the regulation of FGF21 mRNA abundance and FGF21 secretion. Primary rat hepatocyte cultures were incubated with or without CDCA (100 μm), FGF19 (100 ng/ml), or CDCA plus FGF19 for 6 h. The level of FGF21 mRNA (left panel) and the level of FGF21 protein in the culture medium (right panel) of cells incubated with vehicle was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of three experiments. *, mean is significantly (p ≤ 0.05) different from that of cells or mice treated with vehicle for the same time period. **, mean is significantly (p ≤ 0.05) higher than that of any other treatment of the same time period.
The observation that FGF19 increased hepatic FGF21 secretion and plasma FGF21 concentration in the absence of an elevation in hepatic FGF21 mRNA abundance (Fig. 4, A, B, and C) indicated that FGF19 regulated FGF21 secretion at a translational and/or posttranslational step. In contrast, stimulatory effects of bile acids on FGF21 secretion were associated with an increase in FGF21 mRNA abundance (Figs. 2 and 3), suggesting that bile acids controlled FGF21 secretion at a pretranslational step. The observation that FGF19 and bile acids regulated FGF21 secretion through translational/posttranslational and pretranslational mechanisms, respectively, led us to investigate the interaction between FGF19 and CDCA in the regulation of FGF21 secretion. Treatment of rat hepatocytes with FGF19 plus CDCA caused a greater increase in FGF21 levels (20.6-fold) in the culture medium than treatment with FGF19 (12.3-fold) or CDCA (8.4-fold) alone (Fig. 4D). FGF21 mRNA levels in cells treated with FGF19 plus CDCA were similar to those observed in cells treated with CDCA alone. The additive effects of FGF19 and CDCA on FGF21 secretion are consistent with data demonstrating that FGF19 and CDCA regulate FGF21 secretion through different mechanisms.
Activation of the Farnesoid X Receptor Increases FGF21 Expression
The FXR is a nuclear bile acid receptor that regulates the transcription of genes involved in bile acid, carbohydrate, and lipid metabolism (55). CDCA, TCA, TCDCA, TDCA, and CA function as activating ligands of the FXR, whereas tauro-β-muricholic acid is not effective in activating FXR (56, 57). This pattern of bile acid activation of FXR is similar to the pattern of bile acid activation of FGF21 mRNA abundance (Fig. 2), suggesting that FXR plays a role in mediating the stimulatory effects of bile acids on FGF21 gene expression. To investigate whether ligand-activated FXR increased FGF21 expression, we determined the effect of the selective FXR-activating ligand GW4064 on FGF21 expression. Incubating rat hepatocytes with an optimal concentration of GW4064 (3 μm) caused a rapid (≤1 h) and sustained (≥24 h) increase in the abundance of FGF21 mRNA (Fig. 5A). The GW4064-induced increase in FGF21 mRNA abundance was maximal (3.2-fold) at 6 h of incubation and was accompanied by a similar elevation in FGF21 secretion into the culture medium (Fig. 5A). Treatment with GW4064 also increased FGF21 mRNA abundance and FGF21 secretion in human HepG2 cells (Fig. 5B). We also determined the effect of GW4064 administration on FGF21 mRNA abundance in intact mice. Oral administration of GW4064 stimulated a 2.2–2.7-fold increase in FGF21 and BSEP mRNA levels in livers of wild-type mice (Fxr+/+) but had no effect on FGF21 and BSEP mRNA levels in livers of FXR knock-out mice (Fxr−/−) (Fig. 5C). These results indicate that activation of FXR enhances hepatic FGF21 expression.
FIGURE 5.

Activation of FXR increases hepatic FGF21 mRNA abundance and FGF21 secretion. A and B, effect of the FXR-selective agonist GW4064 on FGF21 mRNA abundance and FGF21 secretion in primary rat hepatocyte cultures (A) and human HepG2 cells (B). Cells were incubated with or without GW4064 (3 μm) for the indicated time periods. Cells and culture medium were harvested, total RNA was isolated, and FGF21 mRNA abundance and FGF21 protein levels were measured as described under “Experimental Procedures.” Left panels, FGF21 mRNA abundance in cells incubated with GW4064 for 0 h was set at 1, and the other values were adjusted proportionately. Right panels, the FGF21 protein level in the culture medium of cells incubated with vehicle (DMSO) for 6 h was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of four experiments. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of cells incubated with vehicle for the same time period. C, effect of GW4064 administration on FGF21 and BSEP mRNA levels in intact mice. Wild-type mice (Fxr+/+) and FXR knock-out mice (Fxr−/−) were orally administered GW4064 (30 mg/kg twice a day) or vehicle (2-hydroxypropyl-β-cyclodextrin) for 7 days, and FGF21 and BSEP mRNA levels were measured in liver. An asterisk indicates that the mean is significantly (p ≤ 0.05) different compared with that of animals of the same genotype treated with vehicle.
To investigate the mechanism by which FXR activation regulates FGF21 gene expression, transient transfection experiments were performed to identify cis-acting elements mediating the effect of GW4064 on FGF21 gene transcription. Hepatocytes were transfected with a series of DNA constructs containing 5′-deletions of the rat FGF21 promoter linked to the luciferase gene. In cells transfected with the construct containing the longest FGF21 fragment (−2940 to +68 bp), treatment with GW4064 caused a 4-fold increase in luciferase activity (Fig. 6A). Deletion of FGF21 sequences to −2074, −1656, and −1316 bp had no effect on GW4064 responsiveness. Deletion of FGF21 sequences from −1316 to −1164 bp suppressed FGF21 promoter activity in both the absence and presence of GW4064. Because the extent of the decrease in promoter activity was greater in cells incubated in the presence of GW4064, GW4064 responsiveness was decreased by 59%. Further deletion of FGF21 sequence to −103 bp had no effect on residual promoter activity in the absence or presence of GW4064. These data suggest that the region between −1316 and −1164 bp contains a sequence that mediates the effect of FXR activation on FGF21 gene transcription.
FIGURE 6.

Identification of an FXRE in the FGF21 gene. A, hepatocytes were transiently transfected with a series of plasmids containing fragments of the rat FGF21 gene linked to the luciferase (Luc) gene as described under “Experimental Procedures.” After transfection, cells were treated with or without GW4064 for 24 h. Cells were harvested, extracts were prepared, and luciferase assays were performed. Left, the constructs used in these experiments. The number at the left of each construct is the 5′-end of FGF21 DNA in nucleotides relative to the transcription initiation site. The 3′-end of each construct is +68 bp. The location of the FXRE (−1222 to −1210 bp) is indicated by a vertical line. A mutation of the FXRE (FXRE Mut) is indicated by an X through the vertical line. Right, luciferase activity of cells transfected with the −2949 to +68 bp FGF21 construct and treated with vehicle was set at 1, and all other activities were adjusted proportionately. The -fold stimulation by GW4064 was calculated by dividing the luciferase activity for cells treated with GW4064 by that for cells treated with vehicle. The -fold responses were calculated for individual experiments and then averaged. The results are the means ± S.E. (error bars) of three experiments. B, the sequence of the rat FGF21 gene between −1231 and −1196 bp. The hexameric half-sites comprising the FXRE are indicated by arrows. The sequence of a mutation of the FXRE (FXRE Mut) is shown underneath. Mutated sequences are boxed. C, gel mobility shift assays were performed using recombinant FXR and/or RXRα and an oligonucleotide probe containing the FGF21 FXRE (−1231 to −1196 bp). In lanes 4–10, competition analyses were performed with a 2.5-, 5-, and 10-fold molar excess of unlabeled competitor DNA. The sequences of the probe and competitor DNAs are shown in B. In lanes 12 and 14, the receptor preparations were incubated with antibodies against FXR or nuclear factor 1 (NF1) prior to the addition of the probe. Positions of FXR/RXR and supershifted complexes are indicated by arrows.
Sequence comparison analysis (MatInspector, Genomatix) of the region between −1316 and −1164 bp revealed the presence of a sequence (−1222 to −1210 bp) that resembled an FXRE (Fig. 6B). This sequence comprised two hexameric half-sites arranged as inverted repeats with 1 bp separating the half-sites. In transient transfection assays, mutation of this putative FXRE (FXRE Mut) in the context of the −1316 to +68 bp FGF21 fragment abolished the ability of GW4064 to stimulate FGF21 promoter activity (Fig. 6A). In gel mobility shift assays, an oligonucleotide probe containing the FGF21 FXRE (−1231 to −1196 bp) bound to FXR/RXR heterodimers (Fig. 6C). Results of competition experiments with unlabeled DNA fragments indicated that the binding affinity of FXRE Mut for FXR/RXR heterodimers was markedly reduced compared with that of the wild-type FGF21 FXRE. Thus, the elimination of GW4064 regulation of FGF21 promoter activity caused by FXRE Mut was associated with a decrease in FXR/RXR binding activity. These findings indicate that ligand-bound FXR binds directly to the FGF21 gene and activates FGF21 gene transcription.
Role of FXR in the Nutritional Regulation of FGF21 Expression
Previous studies demonstrating that PPARα deletion does not completely suppress diet-induced changes in hepatic FGF21 mRNA abundance suggest that other signaling pathways besides PPARα play a role in mediating the nutritional regulation of FGF21 expression (4, 10, 11). This observation plus results of the present study demonstrating that ligand-activated FXR stimulates FGF21 mRNA abundance led us to investigate the role of FXR in mediating diet-induced changes in FGF21 expression. FXR knock-out mice (Fxr−/−) and wild-type mice (Fxr+/+) were divided into three groups. One group was fed a standard purified diet for 7 days. A second group was fed an HF-LC ketogenic diet for 7 days. A third group was fed a standard purified diet for 6 days and then starved for 24 h. Hepatic FGF21 mRNA levels were similar in Fxr−/− mice and Fxr+/+ mice fed a standard purified diet (Fig. 7A). Feeding Fxr+/+ mice an HF-LC ketogenic diet stimulated a 7.9-fold increase in hepatic FGF21 mRNA abundance relative to Fxr+/+ mice fed a standard purified diet. Feeding Fxr−/− mice an HF-LC ketogenic diet also increased hepatic FGF21 mRNA abundance, but the extent of this effect (3.7-fold relative to Fxr−/− mice fed a standard purified diet) was less than that observed in Fxr+/+ mice. The ability of the HF-LC ketogenic diet to increase plasma FGF21 concentration was also attenuated in Fxr−/− mice relative to Fxr+/+ mice (Fig. 7B). These results indicate that FXR plays a role in mediating the stimulatory effect of HF-LC consumption on hepatic FGF21 expression. In contrast to the results for HF-LC consumption, starvation (24 h) stimulated a similar increase in hepatic FGF21 mRNA abundance and plasma FGF21 concentration in Fxr−/− mice and Fxr+/+ mice (Fig. 7, A and B). These findings indicate that FXR is not involved in mediating the stimulatory effect of starvation on FGF21 expression.
FIGURE 7.
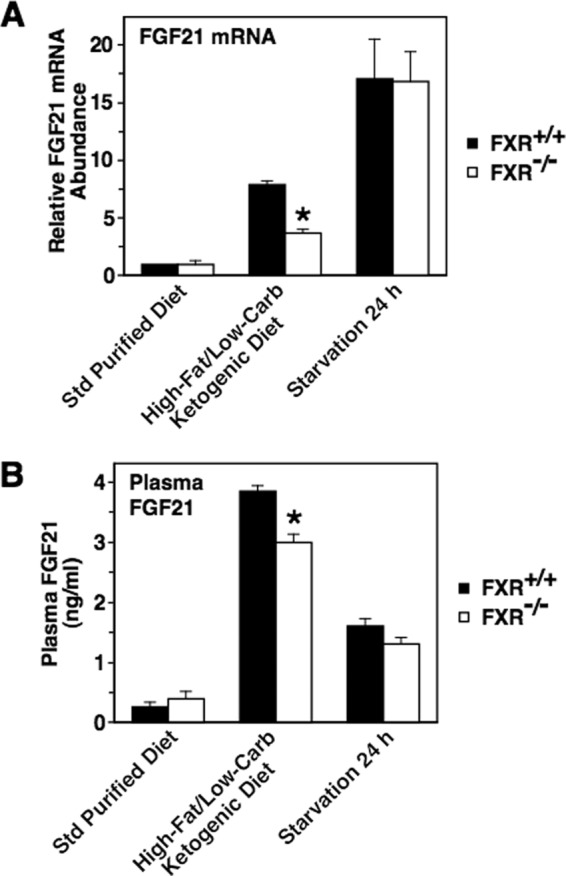
Deletion of FXR suppresses the stimulatory effect of HF-LC consumption on hepatic FGF21 mRNA abundance and plasma FGF21 concentration. FXR knock-out mice (Fxr−/−) and wild-type mice (Fxr+/+) were fed a standard (Std) purified diet or an HF-LC ketogenic diet for 7 days. A third group was fed a standard purified diet for 6 days and then starved for 24 h. Animals were killed 6 h after the start of the dark cycle, and the FGF21 mRNA abundance (A) and plasma FGF21 concentration (B) were measured. The level of FGF21 mRNA in Fxr+/+ mice fed the standard purified diet was set at 1, and the other values were adjusted proportionately. Values are means ± S.E. (error bars) of seven animals. The asterisk indicates that the mean is significantly (p ≤ 0.05) lower compared with that of Fxr+/+ mice fed the HF-LC ketogenic diet. Carb, carbohydrate.
DISCUSSION
The results of the present study identify a new signaling pathway that stimulates hepatic FGF21 expression. Ligand activation of FXR induces FGF21 gene expression and secretion. We also show that FXR plays a role in mediating the stimulatory effect of HF-LC consumption on FGF21 expression. As CDCA, TCDCA, TCA, TDCA, and CA are potent activators of FXR activity (56, 57) and FGF21 expression (Fig. 2) and bile acid flux in the enterohepatic system is elevated by HF-LC consumption (46), we propose that bile acids function as signals mediating the increase in FGF21 expression caused by HF-LC consumption (Fig. 8). Another nutritional condition that induces FGF21 expression is starvation. In contrast to HF-LC consumption, starvation decreases the circulation of bile acids in the enterohepatic system (59). Thus, the lack of involvement of FXR in mediating starvation-induced changes in FGF21 mRNA abundance (Fig. 7) is consistent with our model that bile acids play a role in the physiological regulation of FGF21 expression.
FIGURE 8.

Proposed model for how consumption of an HF-LC ketogenic diet increases hepatic FGF21 gene expression and secretion. Consumption of an HF-LC ketogenic diet enhances the hepatic delivery of multiple signaling molecules that stimulate FGF21 secretion. These signaling factors include dietary unsaturated fatty acids derived from the hepatic hydrolysis of triacylglycerols in chylomicron remnants and the extrahepatic hydrolysis of triacylglycerols in chylomicrons (i.e. nonesterified fatty acid spillover). HF-LC consumption also increases the enterohepatic circulation of bile acids and intestinal secretion of FGF19. Unsaturated fatty acids and bile acids increase hepatic FGF21 gene transcription and secretion by activating FXR and PPARα, respectively. Unsaturated fatty acids and bile acids may also act through FXR- and PPARα-independent pathways to increase FGF21 gene expression. FGF19 activates fibroblast growth factor receptor 4 (FGFR4)/β-Klotho causing an increase in FGF21 secretion via a translational and/or posttranslational mechanism. NEFA, non-esterified fatty acid; LPL, lipoprotein lipase.
Another key finding of the present study is that FGF19 induces FGF21 secretion in liver. Previous studies have shown that FGF19 is an endocrine factor that mediates the inhibitory effects of intestinal bile acids on hepatic bile acid synthesis (54). In the ileum, bile acids induce the expression and secretion of FGF19 via an FXR-dependent mechanism (48, 54). At the liver, FGF19 increases the expression of small heterodimer partner (60), a transcriptional repressor of the cholesterol 7α-hydroxylase gene, a key control point of the bile acid synthetic pathway. As HF-LC consumption enhances the intestinal recycling of bile acids (61), we propose that FGF19 is another enterohepatic signal mediating the stimulatory of effect HF-LC consumption on hepatic FGF21 secretion (Fig. 8). In support of the model, consumption of a high fat meal causes an increase in circulating FGF19 levels in humans (50). The additive effects of FGF19 and CDCA on hepatic FGF21 secretion (Fig. 4D) are consistent with a role of both of these signals in mediating diet-induced changes in circulating FGF21 levels. Interestingly, FGF19 enhances FGF21 secretion via a mechanism that does not involve changes in FGF21 mRNA abundance (Fig. 4). To our knowledge, these are the first data demonstrating that FGF21 expression/secretion is regulated by a posttranscriptional mechanism. Recent studies have shown that FGF19 increases small heterodimer partner expression by inhibiting the ubiquitination and intracellular degradation of small heterodimer partner (60). It is tempting to speculate that the mechanism mediating the stimulatory effect of FGF19 on FGF21 secretion involves alterations in the intracellular degradation of FGF21.
The results of the present study also demonstrate that nonesterified unsaturated fatty acids and unsaturated fatty acid-enriched chylomicron remnants induce hepatic FGF21 expression. The effects of these agents on FGF21 expression are likely mediated at least in part by the activation of PPARα as the FGF21 gene contains a PPAR response element and unsaturated fatty acids enhance PPARα activity (4, 33). We propose that dietary unsaturated fatty acids delivered to the liver in the form of chylomicron remnants or nonesterified fatty acids (i.e. spillover from extrahepatic triacylglycerol hydrolysis) constitute a third signal mediating the stimulatory effect of HF-LC consumption on hepatic FGF21 secretion (Fig. 8). The additive effects of oleate and CDCA on FGF21 mRNA abundance (Fig. 2F) are consistent with a role of both PPARα and FXR agonists in mediating the stimulatory effect of HF-LC consumption on FGF21 gene expression.
Although glucose and insulin are key humoral factors signaling the high carbohydrate, low fat fed state to various metabolic processes in liver (13, 30, 41, 42), our findings indicate that glucose and insulin are not involved in mediating the decrease in hepatic FGF21 expression caused by high carbohydrate, low fat consumption. Both glucose and insulin caused an increase FGF21 mRNA abundance in rat hepatocytes (Fig. 1F). Ma et al. (62) have also reported that glucose enhances hepatic FGF21 mRNA abundance. We postulate that glucose and insulin function to maintain basal levels of FGF21 expression during non-ketotic conditions. In support of this proposal, hepatic FGF21 expression is markedly decreased in chow-fed mice lacking hepatic expression of the insulin receptor substrate-1 and insulin receptor substrate-2, key components of the insulin signal transduction pathway (63). Our observation that signals of starvation and/or high fat consumption (i.e. unsaturated fatty acids and bile acids) cause a greater induction of FGF21 expression than signals of high carbohydrate consumption (i.e. glucose and insulin) is also consistent with this proposal (Figs. 1 and 2).
Previous studies have shown that administration of natural and synthetic FXR agonists attenuates insulin resistance, hypertriglyceridemia, and hepatic steatosis in animal models of metabolic syndrome (64). These effects of FXR agonists on metabolic syndrome are mediated at least in part by a decrease in hepatic fatty acid synthesis resulting in a reduction in hepatic triacylglycerol accumulation and very low density lipoprotein production (65). The mechanisms by which FXR agonists inhibit hepatic fatty acid synthesis are not clear. Previous studies have shown that transgenic expression of FGF21 or administration of recombinant FGF21 mimics the effects of FXR agonists on insulin resistance, hypertriglyceridemia, hepatic steatosis, and hepatic lipogenic enzyme expression in obese/diabetic animals (2, 3, 6, 8). These observations plus our finding that natural and synthetic FXR agonists induce hepatic FGF21 expression provide support for a role of FGF21 in mediating the beneficial effects of FXR agonists on metabolic syndrome. FXR agonists also induce the expression of FGF19, another factor that attenuates metabolic syndrome in obese/diabetic animals (66). Our observation that FGF19 induces FGF21 secretion suggests a novel interaction between FGF19 and FGF21 in the regulation of lipid metabolism by FXR agonists.
The synthetic FXR agonist GW4064 has been shown to bind to FXR with an affinity that is >100-fold higher than that of CDCA, TCDCA, TCA, and TDCA (56, 57, 67). Interestingly, GW4064 is less effective than CDCA, TCDCA, TCA, and TDCA in stimulating FGF21 mRNA abundance in rat hepatocytes and human HepG2 cells (Figs. 2 and 5). This observation suggests that other mechanisms besides ligand activation of FXR play a role in mediating the bile acid regulation of FGF21 expression. Previous studies have shown that bile acids increase the activity of protein kinase B (Akt) (68), extracellular signal-regulated kinases 1 and 2 (ERK1/2) (68), and c-Jun N-terminal kinase (JNK) (69) in liver. Other work has shown that bile acid regulation of expression of small heterodimer partner, CYP7A1, and the low density lipoprotein receptor is dependent in part on the presence of Akt, ERK1/2, and/or JNK (69, 70). Interestingly, muscle-specific overexpression of Akt1 induces FGF21 expression in skeletal muscle of mice (71). Future studies will investigate the role of Akt, ERK1/2, and JNK in mediating the bile acid regulation of FGF21 expression.
In conclusion, bile acids, FGF19, nonesterified unsaturated fatty acids, and chylomicron remnants induce FGF21 secretion in hepatocytes, and FXR plays a role in mediating the increase in hepatic FGF21 expression caused by consumption of an HF-LC ketogenic diet. We propose that consumption of an HF-LC ketogenic diet increases FGF21 secretion by enhancing the delivery of bile acids, FGF19, and dietary unsaturated fatty acids to the liver (Fig. 8). Bile acids and unsaturated fatty acids activate FXR and PPARα, respectively, causing an increase in FGF21 gene transcription and secretion. Bile acids and unsaturated fatty acids may also act through FXR- and PPARα-independent pathways to increase FGF21 expression. FGF19 activates FGF21 secretion via a posttranscriptional mechanism.
Supplementary Material
This work was supported by American Diabetes Association Basic Science Award 1-12-BS-75 (to F. B. H.) and United States Department of Agriculture National Institute of Food and Agriculture National Research Initiative Competitive Grant 2010-65206-20618 (to F. B. H.). This work was also supported, in whole or in part, by National Institutes of Health Grants R01-HL103227 (to Y. Z.) and R01-DK46897 (to L. M. S.).

This article contains supplemental Fig. S1 and Table S1.
- HF-LC
- high fat, low carbohydrate
- FXR
- farnesoid X receptor
- FXRE
- FXR response element
- PPAR
- peroxisome proliferator-activated receptor
- CDCA
- chenodeoxycholic acid
- TCDCA
- taurochenodeoxycholic acid
- TCA
- taurocholic acid
- TDCA
- taurodeoxycholic acid
- CA
- cholic acid
- GLP-1
- glucagon-like peptide-1
- T3
- 3,5,3′-triiodothyronine
- EPA
- eicosapentaenoic acid
- BSEP
- bile salt export pump
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- RXR
- retinoid X receptor.
REFERENCES
- 1. Nishimura T., Nakatake Y., Konishi M., Itoh N. (2000) Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 1492, 203–206 [DOI] [PubMed] [Google Scholar]
- 2. Kharitonenkov A., Shiyanova T. L., Koester A., Ford A. M., Micanovic R., Galbreath E. J., Sandusky G. E., Hammond L. J., Moyers J. S., Owens R. A., Gromada J., Brozinick J. T., Hawkins E. D., Wroblewski V. J., Li D. S., Mehrbod F., Jaskunas S. R., Shanafelt A. B. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Investig. 115, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coskun T., Bina H. A., Schneider M. A., Dunbar J. D., Hu C. C., Chen Y., Moller D. E., Kharitonenkov A. (2008) Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 149, 6018–6027 [DOI] [PubMed] [Google Scholar]
- 4. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 5. Potthoff M. J., Inagaki T., Satapati S., Ding X., He T., Goetz R., Mohammadi M., Finck B. N., Mangelsdorf D. J., Kliewer S. A., Burgess S. C. (2009) FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. U.S.A. 106, 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu J., Lloyd D. J., Hale C., Stanislaus S., Chen M., Sivits G., Vonderfecht S., Hecht R., Li Y. S., Lindberg R. A., Chen J. L., Jung D. Y., Zhang Z., Ko H. J., Kim J. K., Véniant M. M. (2009) Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kharitonenkov A., Larsen P. (2011) FGF21 reloaded: challenges of a rapidly growing field. Trends Endocrinol. Metab. 22, 81–86 [DOI] [PubMed] [Google Scholar]
- 8. Kharitonenkov A., Wroblewski V. J., Koester A., Chen Y. F., Clutinger C. K., Tigno X. T., Hansen B. C., Shanafelt A. B., Etgen G. J. (2007) The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology 148, 774–781 [DOI] [PubMed] [Google Scholar]
- 9. Fon Tacer K., Bookout A. L., Ding X., Kurosu H., John G. B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D. J., Kliewer S. A. (2010) Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 24, 2050–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., Maratos-Flier E. (2007) Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 11. Oishi K., Uchida D., Ohkura N., Horie S. (2010) PPARα deficiency augments a ketogenic diet-induced circadian PAI-1 expression possibly through PPARγ activation in the liver. Biochem. Biophys. Res. Commun. 401, 313–318 [DOI] [PubMed] [Google Scholar]
- 12. Bielohuby M., Menhofer D., Kirchner H., Stoehr B. J., Müller T. D., Stock P., Hempel M., Stemmer K., Pfluger P. T., Kienzle E., Christ B., Tschöp M. H., Bidlingmaier M. (2011) Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am. J. Physiol. Endocrinol. Metab. 300, E65–E76 [DOI] [PubMed] [Google Scholar]
- 13. Jornayvaz F. R., Jurczak M. J., Lee H. Y., Birkenfeld A. L., Frederick D. W., Zhang D., Zhang X. M., Samuel V. T., Shulman G. I. (2010) A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am. J. Physiol. Endocrinol. Metab. 299, E808–E815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Badman M. K., Koester A., Flier J. S., Kharitonenkov A., Maratos-Flier E. (2009) Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 150, 4931–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lundåsen T., Hunt M. C., Nilsson L. M., Sanyal S., Angelin B., Alexson S. E., Rudling M. (2007) PPARα is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 360, 437–440 [DOI] [PubMed] [Google Scholar]
- 16. Berglund E. D., Li C. Y., Bina H. A., Lynes S. E., Michael M. D., Shanafelt A. B., Kharitonenkov A., Wasserman D. H. (2009) Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 150, 4084–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang X., Yu C., Jin C., Yang C., Xie R., Cao D., Wang F., McKeehan W. L. (2006) Forced expression of hepatocyte-specific fibroblast growth factor 21 delays initiation of chemically induced hepatocarcinogenesis. Mol. Carcinog. 45, 934–942 [DOI] [PubMed] [Google Scholar]
- 18. Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., Wahli W. (1999) Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 103, 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinal C. J., Tohkin M., Miyata M., Ward J. M., Lambert G., Gonzalez F. J. (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 [DOI] [PubMed] [Google Scholar]
- 20. Stabile L. P., Klautky S. A., Minor S. M., Salati L. M. (1998) Polyunsaturated fatty acids inhibit the expression of the glucose-6-phosphate dehydrogenase gene in primary rat hepatocytes by a nuclear posttranscriptional mechanism. J. Lipid Res. 39, 1951–1963 [PubMed] [Google Scholar]
- 21. Mooney R. A., Lane M. D. (1981) Formation and turnover of triglyceride-rich vesicles in the chick liver cell. Effects of cAMP and carnitine on triglyceride mobilization and conversion to ketones. J. Biol. Chem. 256, 11724–11733 [PubMed] [Google Scholar]
- 22. Tso P., Karlstad M. D., Bistrian B. R., DeMichele S. J. (1995) Intestinal digestion, absorption, and transport of structured triglycerides and cholesterol in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 268, G568–G577 [DOI] [PubMed] [Google Scholar]
- 23. Kohan A. B., Qing Y., Cyphert H. A., Tso P., Salati L. M. (2011) Chylomicron remnants and nonesterified fatty acids differ in their ability to inhibit genes involved in lipogenesis in rats. J. Nutr. 141, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Florén C. H., Nilsson A. (1977) Degradation of chylomicron remnant cholesteryl ester by rat hepatocyte monolayers. Inhibition by chloroquine and colchicine. Biochem. Biophys. Res. Commun. 74, 520–528 [DOI] [PubMed] [Google Scholar]
- 25. Chomczynski P., Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 [DOI] [PubMed] [Google Scholar]
- 26. Ichihara A., Nakamura T., Tanaka K., Tomita Y., Aoyama K., Kato S., Shinno H. (1980) Biochemical functions of adult rat hepatocytes in primary culture. Ann. N.Y. Acad. Sci. 349, 77–84 [DOI] [PubMed] [Google Scholar]
- 27. Gälman C., Lundåsen T., Kharitonenkov A., Bina H. A., Eriksson M., Hafström I., Dahlin M., Amark P., Angelin B., Rudling M. (2008) The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARα activation in man. Cell Metab. 8, 169–174 [DOI] [PubMed] [Google Scholar]
- 28. Barrows B. R., Parks E. J. (2006) Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 91, 1446–1452 [DOI] [PubMed] [Google Scholar]
- 29. Cahill G. F., Jr. (1970) Starvation in man. N. Engl. J. Med. 282, 668–675 [DOI] [PubMed] [Google Scholar]
- 30. Kennedy A. R., Pissios P., Otu H., Roberson R., Xue B., Asakura K., Furukawa N., Marino F. E., Liu F. F., Kahn B. B., Libermann T. A., Maratos-Flier E. (2007) A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 292, E1724–E1739 [DOI] [PubMed] [Google Scholar]
- 31. Miles J. M., Nelson R. H. (2007) Contribution of triglyceride-rich lipoproteins to plasma free fatty acids. Horm. Metab. Res. 39, 726–729 [DOI] [PubMed] [Google Scholar]
- 32. Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk G. D., Muoio D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 33. Lefebvre P., Chinetti G., Fruchart J. C., Staels B. (2006) Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J. Clin. Investig. 116, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pawar A., Jump D. B. (2003) Unsaturated fatty acid regulation of peroxisome proliferator-activated receptor α activity in rat primary hepatocytes. J. Biol. Chem. 278, 35931–35939 [DOI] [PubMed] [Google Scholar]
- 35. Redgrave T. G. (1970) Formation of cholesteryl ester-rich particulate lipid during metabolism of chylomicrons. J. Clin. Investig. 49, 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mahley R. W., Huang Y. (2007) Atherogenic remnant lipoproteins: role for proteoglycans in trapping, transferring, and internalizing. J. Clin. Investig. 117, 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Longuet C., Sinclair E. M., Maida A., Baggio L. L., Maziarz M., Charron M. J., Drucker D. J. (2008) The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 8, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vegiopoulos A., Herzig S. (2007) Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 275, 43–61 [DOI] [PubMed] [Google Scholar]
- 39. Sanderson L. M., Degenhardt T., Koppen A., Kalkhoven E., Desvergne B., Müller M., Kersten S. (2009) Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) but not PPARα serves as a plasma free fatty acid sensor in liver. Mol. Cell. Biol. 29, 6257–6267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maglich J. M., Watson J., McMillen P. J., Goodwin B., Willson T. M., Moore J. T. (2004) The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J. Biol. Chem. 279, 19832–19838 [DOI] [PubMed] [Google Scholar]
- 41. Collier J. J., Scott D. K. (2004) Sweet changes: glucose homeostasis can be altered by manipulating genes controlling hepatic glucose metabolism. Mol. Endocrinol. 18, 1051–1063 [DOI] [PubMed] [Google Scholar]
- 42. Saltiel A. R., Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 43. Hillgartner F. B., Salati L. M., Goodridge A. G. (1995) Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev. 75, 47–76 [DOI] [PubMed] [Google Scholar]
- 44. Ahima R. S., Prabakaran D., Mantzoros C., Qu D., Lowell B., Maratos-Flier E., Flier J. S. (1996) Role of leptin in the neuroendocrine response to fasting. Nature 382, 250–252 [DOI] [PubMed] [Google Scholar]
- 45. Hylemon P. B., Zhou H., Pandak W. M., Ren S., Gil G., Dent P. (2009) Bile acids as regulatory molecules. J. Lipid Res. 50, 1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Juste C., Demarne Y., Corring T. (1983) Response of bile flow, biliary lipids and bile acid pool in the pig to quantitative variations in dietary fat. J. Nutr. 113, 1691–1701 [DOI] [PubMed] [Google Scholar]
- 47. Hylemon P. B., Gurley E. C., Kubaska W. M., Whitehead T. R., Guzelian P. S., Vlahcevic Z. R. (1985) Suitability of primary monolayer cultures of adult rat hepatocytes for studies of cholesterol and bile acid metabolism. J. Biol. Chem. 260, 1015–1019 [PubMed] [Google Scholar]
- 48. Holt J. A., Luo G., Billin A. N., Bisi J., McNeill Y. Y., Kozarsky K. F., Donahee M., Wang D. Y., Mansfield T. A., Kliewer S. A., Goodwin B., Jones S. A. (2003) Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 17, 1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu W. J., Yang Q., Sun W., Woods S. C., D'Alessio D., Tso P. (2007) The regulation of the lymphatic secretion of glucagon-like peptide-1 (GLP-1) by intestinal absorption of fat and carbohydrate. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G963–G971 [DOI] [PubMed] [Google Scholar]
- 50. Schreuder T. C., Marsman H. A., Lenicek M., van Werven J. R., Nederveen A. J., Jansen P. L., Schaap F. G. (2010) The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G440–G445 [DOI] [PubMed] [Google Scholar]
- 51. Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., Pellicciari R., Auwerx J., Schoonjans K. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abu-Hamdah R., Rabiee A., Meneilly G. S., Shannon R. P., Andersen D. K., Elahi D. (2009) Clinical review: the extrapancreatic effects of glucagon-like peptide-1 and related peptides. J. Clin. Endocrinol. Metab. 94, 1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhatnagar S., Damron H. A., Hillgartner F. B. (2009) Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 284, 10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., Gerard R. D., Repa J. J., Mangelsdorf D. J., Kliewer S. A. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225 [DOI] [PubMed] [Google Scholar]
- 55. Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191 [DOI] [PubMed] [Google Scholar]
- 56. Wang H., Chen J., Hollister K., Sowers L. C., Forman B. M. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 [DOI] [PubMed] [Google Scholar]
- 57. Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 58.Deleted in proof
- 59. Dumaswala R., Berkowitz D., Setchell K. D., Heubi J. E. (1994) Effect of fasting on the enterohepatic circulation of bile acids in rats. Am. J. Physiol. 267, G836–842 [DOI] [PubMed] [Google Scholar]
- 60. Miao J., Xiao Z., Kanamaluru D., Min G., Yau P. M., Veenstra T. D., Ellis E., Strom S., Suino-Powell K., Xu H. E., Kemper J. K. (2009) Bile acid signaling pathways increase stability of small heterodimer partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 23, 986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Corring T., Juste C., Lhoste E. F. (1989) Nutritional regulation of pancreatic and biliary secretions. Nutr. Res. Rev. 2, 161–180 [DOI] [PubMed] [Google Scholar]
- 62. Ma L., Robinson L. N., Towle H. C. (2006) ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 281, 28721–28730 [DOI] [PubMed] [Google Scholar]
- 63. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. (2008) Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 [DOI] [PubMed] [Google Scholar]
- 65. Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 113, 1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fu L., John L. M., Adams S. H., Yu X. X., Tomlinson E., Renz M., Williams P. M., Soriano R., Corpuz R., Moffat B., Vandlen R., Simmons L., Foster J., Stephan J. P., Tsai S. P., Stewart T. A. (2004) Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145, 2594–2603 [DOI] [PubMed] [Google Scholar]
- 67. Maloney P. R., Parks D. J., Haffner C. D., Fivush A. M., Chandra G., Plunket K. D., Creech K. L., Moore L. B., Wilson J. G., Lewis M. C., Jones S. A., Willson T. M. (2000) Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 43, 2971–2974 [DOI] [PubMed] [Google Scholar]
- 68. Dent P., Fang Y., Gupta S., Studer E., Mitchell C., Spiegel S., Hylemon P. B. (2005) Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology 42, 1291–1299 [DOI] [PubMed] [Google Scholar]
- 69. Gupta S., Stravitz R. T., Dent P., Hylemon P. B. (2001) Down-regulation of cholesterol 7α-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem. 276, 15816–15822 [DOI] [PubMed] [Google Scholar]
- 70. Nakahara M., Fujii H., Maloney P. R., Shimizu M., Sato R. (2002) Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J. Biol. Chem. 277, 37229–37234 [DOI] [PubMed] [Google Scholar]
- 71. Izumiya Y., Bina H. A., Ouchi N., Akasaki Y., Kharitonenkov A., Walsh K. (2008) FGF21 is an Akt-regulated myokine. FEBS Lett. 582, 3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.