

Abstract
Neuropathic pain is a common cause of pain after nerve injury, but its molecular basis is poorly understood. In a post-gene chip microarray effort to identify new target genes contributing to neuropathic pain development, we report here the characterization of a novel neuropathic pain contributor, thrombospondin-4 (TSP4), using a neuropathic pain model of spinal nerve ligation injury. TSP4 is mainly expressed in astrocytes and significantly upregulated in the injury side of dorsal spinal cord that correlates with the development of neuropathic pain states. TSP4 blockade by intrathecal antibodies, antisense oligodeoxynucleotides, or inactivation of the TSP4 gene reverses or prevents behavioral hypersensitivities. Intrathecal injection of TSP4 protein into naive rats is sufficient to enhance the frequency of EPSCs in spinal dorsal horn neurons, suggesting an increased excitatory presynaptic input, and to cause similar behavioral hypersensitivities. Together, these findings support that injury-induced spinal TSP4 may contribute to spinal presynaptic hypersensitivity and neuropathic pain states. Development of TSP4 antagonists has the therapeutic potential for target-specific neuropathic pain management.
Introduction
Neuropathic pain, or chronic pain derived from nerve injuries, is a multi-mechanism disorder affecting the quality of patients' daily lives adversely. Limited medications for neuropathic pain management are usually associated with unwanted side effects. The development of target-specific and safe medications relies on our better understanding of neuropathic pain mechanisms. In an effort to identify novel target genes that play important roles in neuropathic pain processing, our group and others have used the gene chip microarray approach to profile dysregulated genes in dorsal root ganglia and spinal cord of neuropathic pain models (Costigan et al., 2002; Wang et al., 2002; Valder et al., 2003; Yang et al., 2004; Luo and Figueroa, 2008; Kim et al., 2009). Since thrombospondin proteins have been reported recently to play a critical role in abnormal synaptogenesis (Christopherson et al., 2005; Xu et al., 2010), which may lead to spinal sensitization and pain states (Zeilhofer, 2005), we selected the gene encoding thrombospondin-4 (TSP4), among dysregulated genes, for further characterization to determine whether it is a novel player in neuropathic pain development.
Thrombospondins are a family of large oligomeric, extracellular matrix glycoproteins that mediate cell-cell and cell-matrix interactions (Adams, 2001; Bornstein, 2001). There are five members in the thrombospondin superfamily, TSP1–5, which can be subdivided into groups A (TSP1/2) and B (TSP3/4/5) with similar structure and functional domains and can be secreted as trimers or pentamers, respectively (Adams, 2001). Multiple TSPs may exist in developing tissues, but individual TSPs are often expressed by nonoverlapping cell populations, suggesting that these proteins may carry out similar functions in different tissues (Adams, 2001). As reviewed recently, TSP1/2 play roles in platelet aggregation, inflammatory response, and angiogenesis regulation during tumor growth, wound repair, and connective tissue extracellular matrix assembly. TSP5 is mainly expressed in cartilage and certain connective tissues and plays roles in chondrocyte differentiation, attachment, and cartilage extracellular matrix assembly. While multiple sites of TSP3 and TSP4 expression are detected, the biology of TSP3 and TSP4 is not well studied (Adams and Lawler, 2004). Notably, TSP4 is expressed and localized to synapses and neuromuscular junction at adulthood (Arber and Caroni, 1995). Enhanced green fluorescent protein (eGFP) expression in transgenic mice under the control of the TSP4 promoter has confirmed that TSP4 is expressed in spinal dorsal horn glial cells in adult mice (images in National Center for Biotechnology Information website for the NINDS (National Institute of Neurological Disorders and Stroke) GENSAT (The Gene Expression Nervous System Atlas) BAC (bacterial artificial chromosomes) Transgenics Project (http://www.gensat.org/login.jsp).
In this study, we examined the hypothesis that injury-induced TSP4 induces spinal neuron sensitization and neuropathic pain states with an integrative approach involving behavioral pharmacology, biochemistry, and electrophysiology analyses in a rat neuropathic pain model and genetically modified mice.
Materials and Methods
Animals
Male young adult Harlan Sprague-Dawley rats (<150 g) were from Harlan Sprague Dawley. The eGFP-labeled TSP4-expressing transgenic (TSP4-eGFP) mice were from Mutant Mouse Regional Resource Centers (Thbs4-EGFP, MMRRC, supported by the National Center for Research Resources at the National Institutes of Health). TSP4 gene knock-out (KO) mice (B6.129P2-Thbs4tm1Dgen/J) were from The Jackson Laboratory. These mice were bred internally, and only adult male mice were used for the experiments. All the genetically modified mice appeared normal with respect to grooming, social interactions, and feeding and showed no signs of abnormality or any obvious motor defects, tremor, seizure, or ataxia. All animals were exposed to a 12/12 h day/night cycle with ad libitum access to food and water. All animal care and experiments were performed according to protocols approved by the Institutional Animal Care Committees of the University of California, Irvine and Indiana University.
Intrathecal antisense oligodeoxynucleotides
We designed the antisense oligodeoxynucleotides based on a segment of the rat TSP4 gene (GenBank accession no. X89963) that has been used for producing primers in real-time PCR experiments (TaqMan Gene Expression Assay ID: Rn01494317; Applied Biosystems) and specific to the TSP4 mRNA. Mismatch oligodeoxynucleotides, which contain the same number of nucleotides but in a random order, were used as controls. The nucleotide sequences for the antisense and mismatch oligodeoxynucleotides were CCATCATTGTTGCTATCTTCC and ACCATCGTTGTTACTTTCTCC, respectively. Modification of the oligodeoxynucleotides was described previously (Li et al., 2004; Boroujerdi et al., 2008; Kim et al., 2009).
Expression and purification of TSP4 proteins
293 EBNA cells (Invitrogen) stably transfected with the rat TSP4 cDNA (GenBank accession no. X89963) with a 6-histidine (His) tag at the N-terminal were used for overexpression of the TSP4 proteins (Dunkle et al., 2007). The TSP4-His proteins were purified using an Ni-NTA column based on the manufacturer's instructions (Qiagen), concentrated with Amicon Ultra-4 Centrifugal Filter Unit (50K molecular weight cutoff; Millipore), aliquoted, and stored at −80°C until use.
Spinal nerve ligation
The surgical procedure of unilateral spinal nerve ligation (SNL) was performed as described previously (Kim and Chung, 1992). Briefly, under isoflurane anesthesia, the left L5/L6 spinal nerves of Harlan Sprague-Dawley rats or the L4 spinal nerves of mice (Rigaud et al., 2008) were exposed and ligated with a silk suture distal to DRGs and proximal to their conjunction to form the sciatic nerve. Sham operations were performed in the same way except that spinal nerves were not ligated. Rats or mice were killed at designated times after behavioral studies, and DRGs and spinal cords were isolated and either processed immediately for biochemical studies or kept at −80°C until use.
Intrathecal injection
Under light isoflurane anesthesia, the L5/6 spinal lumbar region was located, and saline, TSP4 proteins, His-tag peptides (GenScript), antisera, antisense, or mismatch oligodeoxynucleotides in a total volume of 10 μl per rat were injected intrathecally between lumbar regions L5 and L6 through a 30-gauge needle connected to a microinjector (Tritech Research). All the drug treatments in SNL animals started when the animals had developed behavioral hypersensitivities (>1-week post SNL), except for the preemptive treatments, which started before the SNL procedures, as indicated.
Behavioral tests
Behavioral tests were performed blindly. For non-SNL experiments, the average values between left and right hindpaws were used for statistical analysis between test groups.
von Frey filament test.
Hindpaw sensitivities to von Frey filament stimulation were tested as described previously (Boroujerdi et al., 2008; Kim et al., 2009). Briefly, animals were allowed to acclimatize for at least 30 min in a clear plastic, wire mesh-bottomed cage. The 50% paw withdrawal thresholds (PWTs) to von Frey filament (Stoelting Wood Dale) stimulation were determined using the up-down method of Dixon (Dixon, 1980). A series of filaments, starting with one that has a buckling weight of 2.0 g for rats and 0.41 g for mice, were applied consecutively to the plantar surface of the hindpaw with a force causing the filament to buckle. Paw lifting within 5 s indicated a positive response and prompted the use of the next weaker filament. Absence of a paw-lifting response after 5 s prompted the use of the next filament with increasing weight. This paradigm continued until four more measurements had been made after the initial change in the behavioral response or until five consecutive negative (assigned a score of 15 g for rats and 3 g for mice) or four consecutive positive (assigned a score of 0.25 g for rats and 0.01 g for mice) responses had occurred. The resulting scores were used to calculate the 50% response threshold, as described previously (Luo et al., 2001).
Hot box (Hargreaves) test.
Thermal hyperalgesia was measured as reduced hindpaw withdrawal latency (PWL) to thermal stimuli using a Hargreaves apparatus (Hargreaves et al., 1988). Free moving animals were acclimated individually for at least 15 min on the glass top of a hot box maintained at 30 ± 0.1°C. A heat stimulus from a high-intensity light bulb was applied to the hindpaw plantar surface through a small aperture below the glass surface. PWL, defined as the time between the application of heat stimulus and withdrawal of the targeted hindpaw, was measured. A cutoff time of 20 s was set to prevent the sensitization and thermal injury of the skin. The averaged value of two readings per paw was calculated and used for statistical analysis.
Paw pressure (Randall-Selitto) test.
Rats were first acclimated to human touch and holding positions for a week before the experiments. Mechanical hyperalgesia was measured with the Randall-Selitto Test using a Paw Pressure Analgesy-Meter (Ugo Basile North America) that applies an increasing force with a rate of 16 g/s to the hindpaw between a flat surface and a blunt pointer (Randall and Selitto, 1957). Paw pressure withdrawal thresholds were determined as the amount of force that induced a hindpaw withdrawal.
Motor function tests
To determine whether locomotor functions were altered in rats after antisense/antibody treatments or in genetically modified mice, the Basso, Beattie, Bresnahan Locomotor Rating Scale for rats (Basso et al., 1995) or the Basso Mouse Scale for locomotion (Basso et al., 2006) for mice was performed blindly. Briefly, animals were acclimated to their testing environment daily by handling and exposure to the open-field test apparatus for a week before testing. The locomotor function tests are based on a scale focused on consistent coordination between the frontlimbs and hindlimbs, such as paw position, toe clearance, trunk stability, and tail position (Boroujerdi et al., 2011).
Western blots
Western blot analysis was used to examine levels of proteins of interest in DRG and dorsal spinal cord samples. After electrophoresis, sample proteins were transferred to nitrocellulose membranes electrophoretically. In most cases, membranes were cut into sections for probing with primary antibodies against different target proteins concurrently. After blocking nonspecific binding sites with 5% low-fat milk in PBS, the membranes were incubated for 1 h at room temperature with primary antibodies against the proteins of interest followed by secondary antibodies conjugated with horseradish peroxidase. Primary antibodies included those against TSP1 (mouse, 1:400; Neomarkers), TSP2 (mouse, 1:500; BD Transduction Laboratories), and TSP4 (guinea pig, 1:750). The specificity of the noncommercial guinea pig TSP4 antibody against rat recombinant full-length TSP4 has been characterized previously (Sodersten et al., 2006; Dunkle et al., 2007). Antibodies against β-actin (mouse, 1:10,000; Novus Biologicals) were used for loading control analysis. The antibody–protein complexes were visualized by chemiluminescent reagents. The band densities were quantified by either imaging quantification or densitometry within the linear range of the film sensitivity curve. Ratios of the band density for the protein of interest to those for β-actin were calculated within each sample first before cross-sample comparisons to calculate the percentage changes in the levels of proteins of interest in the experimental groups compared with those from the control groups. Variations in band densities of proteins of interest in the control groups (contralateral side) were determined by comparing each band density with the mean of the band densities from at least two different control samples run in the same Western blot after its ratios to β-actin band densities were calculated.
Fluorescent histochemistry
Spinal cord samples collected from 2-week spinal nerve-ligated TSP4-eGFP animals were immersion fixed in 4% PFA overnight, followed by sedimentation in 30% sucrose PBS solution. Spinal cord samples frozen in optimal cutting tissue medium were sectioned (10 μm). Sections on slides were incubated with primary antibodies diluted in Antibody Diluent Solution (DAKO) for 2 d, followed by incubation with respective secondary antibodies with different color emissions for 2 h at room temperature. After washing, slides were mounted with Vectashield DAPI Hardmount (Vector Laboratories). Primary antibodies against Iba-1 (1:1000; Wako), MBP (1:500; Abcam), eGFP (1:500; Abcam), glial fibrillary acidic protein (GFAP, 1:500; BD Biosciences), and NF-70 (1:500; Millipore) were used. For detecting GFAP or NF-70 immunoreactivities in spinal cord sections, antigen retrieval was determined necessary and performed by incubating the sections in heated sodium citrate buffer, pH 6.0, for 5 min using a pressure cooker before incubation with primary antibodies. Fluorescent images were taken with a Zeiss LSM 780 two-photon laser scanning confocal microscope. Optical sections were line averaged and collected at 0.3 μm intervals. Stacks of seven consecutive optical sections with the best quality of immunoreactivities were then merged in Volocity (version 6.0; PerkinElmer) for data analysis. For each cell marker, staining was performed on three slices at least 100 μm apart from each of the four animals used.
For eGFP signal quantification, mirroring regions of interest spanning from the midline to both sides of the dorsal spinal cord were selected. eGFP fluorescence above the background level was selected for analysis using the Volocity (version 6.0; PerkinElmer) find object using intensity function. Data analyses were performed on six slices (minimum 50 μm intervals) per animal from four animals. Surface area and intensity of eGFP-expressing cells from the contralateral (uninjured) or ipsilateral (injured) sides of all four animals were then pooled respectively and compared. For quantifying the number of eGFP-expressing cells, eGFP-expressing cells with nuclei in the same regions of interest as described for surface area and intensity analyses were identified at a higher magnification and counted manually. Data from the injured or uninjured sides were pooled, respectively, and compared. For colocalization data, thresholded Pearson's correlation coefficient values as described by Barlow et al. (2010) were obtained using the Volocity (version 6.0; PerkinElmer) colocalization function.
Electrophysiology on spinal cord slices
Spinal cords were collected by hydraulic extrusion from animals anesthetized with ketamine-HCl (1 mg/kg, i.p.). Transverse slices (300–350 μm) were cut from the lumbar enlargement using a vibratome (VT1000S; Leica) in an ice-cold (4°C) sucrose solution containing the following (in mm): 230 sucrose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 10 MgSO4, 10 glucose, pH 7.4, 290–305 mOsm/L, equilibrated with 95% O2 and 5% CO2. The slices were maintained in artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, 295–305 mOsm/L. ACSF was continuously equilibrated with 95% O2 and 5% CO2, and slices were incubated for >1 h before recording. Recording electrodes were prepared from borosilicate glass (World Precision Instruments) using a horizontal electrode puller (P-97; Sutter Instruments). Electrodes have resistances of 2–4 MΩ when filled with an intracellular solution containing the following (in mm): 92 CsMeSO4, 43 CsCl, 1 MgCl2, 2 EGTA, 5 tetraethylammonium (TEA), 10 HEPES, and 2 Mg-ATP, pH 7.4, 295–300 mOsm/L. Oxygenated ACSF was used as bath solution, and the flow rate was adjusted to 2–3 ml/min. Neurons in the superficial dorsal horn were visualized with an infrared-differential interference contrast microscope (BX50WI; Olympus Optical) and a CCD camera.
To record α-amino-3-hydroxyl-5-methylisoxazole-4-propionic acid (AMPA) receptor-mediated miniature EPSCs (mEPSCs), bath application of tetrodotoxin (TTX, 1 μm; Sigma), strychnine (1 μm; Sigma), bicuculline (30 μm; Research Biochemicals), and APV (50 mm; Research Biochemicals) were used during recording to block TTX-sensitive Na+, glycinergic, GABAergic, and N-methyl-d-aspartate (NMDA) currents, respectively. All recordings were performed at 25 ± 1°C with an Axopatch 200B amplifier (Molecular Devices). Membrane holding potential was −70 mV for measuring AMPA receptor-mediated mEPSCs. At this holding potential, >90% of the mEPSCs from dorsal spinal cord neurons were mediated by AMPA receptors because they could be blocked by the AMPA receptor antagonist CNQX (Nguyen et al., 2009), and NMDA receptor-mediated currents were not activated due to voltage-dependent Mg2+ blockade (Shimoyama et al., 2000). NMDA receptor-mediated mEPSCs were not examined since they only represented <10% of the mEPSCs even at a depolarizing potential to remove the Mg2+ blockade (Nguyen et al., 2009). During whole-cell recordings, series resistance (8–15 MΩ) was monitored periodically, and cells with >15% changes were excluded from the analysis. Signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a data acquisition program (Axograph 4.6; Molecular Devices). To compare the frequency of mEPSCs between control and TSP4 treatment groups, the Kolmogorov–Smirnov test was used, and percentage cumulative frequency distributions were constructed based on the average frequency of each individual cell.
Statistics
Student's t tests were performed for pairwise comparisons, and one-way or two-way ANOVA with post-test was performed for multigroup comparisons, as indicated in the figure legends. Significance was indicated by a two-tailed p < 0.05.
Results
Peripheral nerve injury induced TSP4 upregulation in dorsal spinal cord that correlated with the development of behavioral hypersensitivities
We hypothesized that if TSP4 plays a critical role in neuropathic pain development after nerve injury, the development of neuropathic pain states should have a temporal correlation with injury-induced TSP4 dysregulation. Accordingly, we examined TSP4 expression levels in DRG and spinal cord from a rat neuropathic pain model derived from unilateral L5/6 SNL (Kim and Chung, 1992) at designated time points correlated with different stages of behavioral hypersensitivity development. SNL, but not sham (data not shown), rats displayed behavioral hypersensitivities to different stimuli in the injury side compared with basal sensitivities in the contralateral side. The behavioral hypersensitivities included reduced PWTs to von Frey filament stimulation (tactile allodynia), pressure stimulation (mechanical hyperalgesia), and reduced PWL to thermal stimulation (thermal hyperalgesia); all were followed by a gradual recovery (Fig. 1a–c). Western blot data indicated that TSP4 protein levels were increased in the injury side of L5/6 dorsal spinal cord at days 2 and 14 postinjury, near the onset and correlating with behavioral hypersensitivities, respectively, but were similar to those in the noninjury side at 10 weeks (and 20 weeks) (data not shown) postinjury when the injured animals recovered from the hypersensitive states (Fig. 1d). In contrast, TSP4 protein levels in L5/6 DRGs were reduced in the injury side after SNL at all the time points measured, independent of the presence or absence of behavioral hypersensitivities (Fig. 2a). While the role of uninjured L4 DRG in the injury side to neuropathic pain processing is an active topic of discussion (Fukuoka et al., 2012), it has been reported that SNL also induces changes in uninjured L4 DRG at the injury side that could have contributed to pain processing (Schafers et al., 2003; Obata et al., 2004; Katsura et al., 2006; Xu et al., 2007; Jeon et al., 2009; Shibasaki et al., 2010). Therefore, we also examined TSP4 protein levels in adjacent, uninjured L4 DRG at time points correlating with different developmental stages of neuropathic pain states. Our data indicated that TSP4 protein levels in adjacent, uninjured L4 DRG at the injury side were not altered significantly at all the time points examined (Fig. 2b). Since only TSP4 dysregulation in the injury side of dorsal spinal cord correlated temporarily with behavioral hypersensitivities, which may underlie a central mechanism of pain processing, we focused our subsequent investigation at the dorsal spinal cord level.
Figure 1.

Nerve injury-induced TSP4 upregulation correlated temporally with the development of behavioral hypersensitivities. a–c, Unilateral L5/6 SNL injury caused a reduction in PWT to light touch (tactile allodynia) (a), PWL to thermal stimulation (thermal hyperalgesia) (b), and hindpaw pressure withdrawal thresholds (PPT) (mechanical hyperalgesia) (c) in the injury side compared to those in the noninjury side. In the paw pressure testing, there was a time-dependent conditioning adaptive increase in PPT in both groups. All behavioral hypersensitive states recovered gradually after ∼7–10 weeks post-ligation injury. Data presented are the mean ± SEM of at least five rats in each group. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the pretreatment levels as determined by two-way ANOVA with Bonferroni post-tests. d, Western blot data indicated that L5/6 SNL injury caused TSP4 protein upregulation in dorsal spinal cord (DSC) of the injury side at time points that correlated with the initiation and maintenance of behavioral hypersensitivities. The injury-induced TSP4 upregulation returned to a level similar to that in the noninjury side when injured animals recovered from the behavioral hypersensitive states. Representative Western blot bands are shown on top of each bar group, and estimated molecular weights (kDa) are shown on the left. Data shown are the mean ± SEM of 5–6 rats in each group. *p < 0.05 compared with the contralateral side by Student's t test. C, Contralateral; Ip, ipsilateral.
Figure 2.
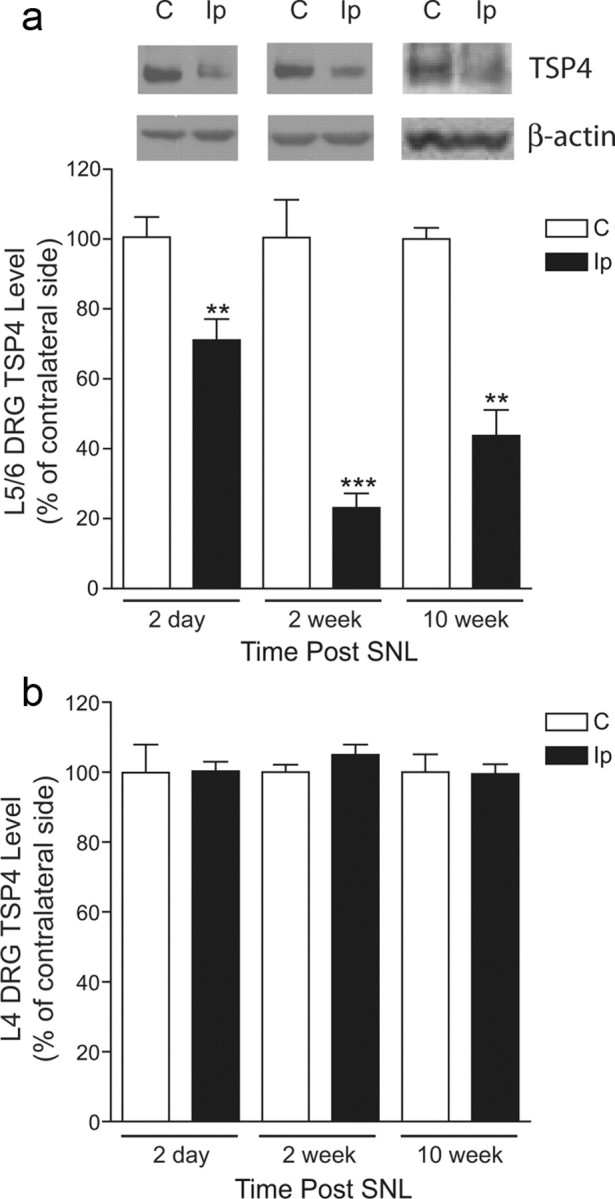
Spinal nerve injury decreased TSP4 protein expression in injured, but not in adjacent uninjured, DRGs. a, Western blot data indicated that L5/6 spinal nerve ligation injury caused the downregulation of TSP4 proteins in L5/6 DRGs of the injury side at all the time points examined. Representative Western blot bands are shown on top of each bar group in the same order as the summarized bar graph data, which are the mean ± SEM of ≥9 rats in each group. **p < 0.01 and ***p < 0.001 compared with the contralateral side by Student's t test. b, In contrast, L5/6 spinal nerve ligation injury did not change TSP4 protein levels in uninjured, adjacent L4 DRG from the injury side. Data presented are the mean ± SEM of nine rats in each group. C, Contralateral; Ip, ipsilateral.
Blocking injury-induced spinal TSP4 upregulation reversed behavioral hypersensitivities in the neuropathic pain model
To determine whether injury-induced TSP4 upregulation contributed to the maintenance of behavioral hypersensitivities, we examined whether blocking injury-induced TSP4 upregulation could reverse established neuropathic pain states. Intrathecal bolus injection of TSP4 antibodies (chicken polyclonal) into the lumbar L5/6 region 2 weeks postinjury could dose dependently reverse established allodynia in the injury side (Fig. 3a). The effects of TSP4 antisera (80 μg/rat) in allodynia reversal had an onset time of ∼4 h and a duration of >6 h (Fig. 3b). Similar treatment also caused a reversal of thermal hyperalgesia (Fig. 3c) and mechanical hyperalgesia (Fig. 3d) in the SNL rats without affecting the behavioral sensitivity at the noninjury side. The specificity of the noncommercial chicken TSP4 antibody against recombinant rat full-length TSP4 has been characterized previously (Dunkle et al., 2007). Intrathecal injection of the guinea pig TSP4 antibody used in the biochemical studies also caused a similar reversal of behavioral hypersensitivity (data not shown). Antibodies against another protein, peripherin (chicken polyclonal), chicken IgY, or heated TSP4 antibodies did not cause significant changes in behavioral sensitivity over the pretreatment level (Fig. 3b–d). It is not clear why intrathecal antibodies at the peak effect time point caused a complete reversal in thermal hyperalgesia (Fig. 3c) and mechanical hyperalgesia (Fig. 3d), which are mediated by high-threshold nociceptors such as myelinated Aδ and nonmyelinated C fibers, but a partial reversal in tactile allodynia (Fig. 3a,b), which are mediated by low-threshold myelinated Aβ fibers. In addition, intrathecal injection for 4 d with TSP4 antisense (but not mismatch) oligodeoxynucleotides, at a dose (50 μg/rat/d) that is effective in knocking down other target gene mRNAs (Li et al., 2004; Boroujerdi et al., 2008; Kim et al., 2009), caused a time-dependent reversal of established allodynia (Fig. 3e), thermal hyperalgesia (Fig. 3f), and mechanical hyperalgesia (Fig. 3g) at the injury side. The antisense effects had an onset time of 1–3 d, peaked approximately 3–5 d after the initiation of the treatments, and lasted >2–5 d after the last injection, depending on the modality tested. It seemed that thermal hyperalgesia was more sensitive to TSP4 antibody and antisense oligodeoxynucleotide treatments with an earlier onset time of reversal than tactile allodynia and mechanical hyperalgesia. Neither the antisense nor the mismatch oligodeoxynucleotides altered behavioral thresholds at the noninjury side (Fig. 3e–g). Western blot data indicated that the antisense, but not the mismatch, treatment blocked injury-induced dorsal spinal cord TSP4 upregulation (Fig. 3h). Neither the antibody nor the antisense oligodeoxynucleotide treatments affected locomotor functions significantly (Fig. 4a,b, respectively). Together, these data support that injury-induced spinal TSP4 upregulation plays a critical role in the maintenance of neuropathic pain states.
Figure 3.
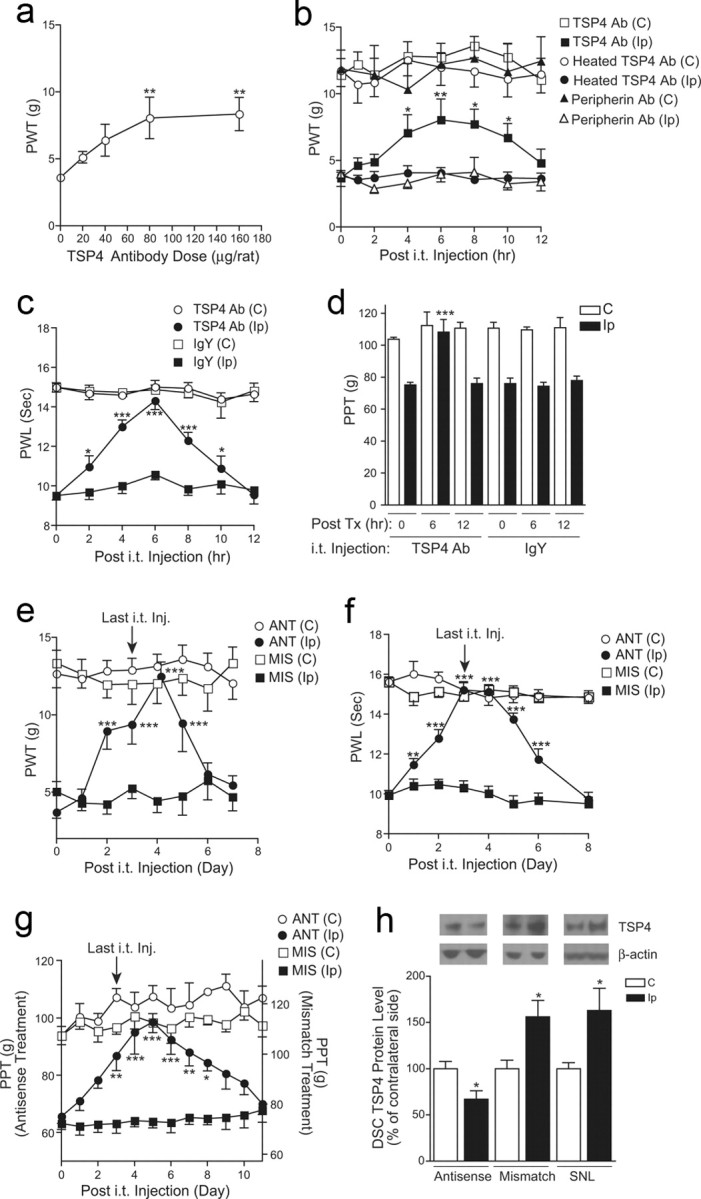
Blocking injury-induced spinal TSP4 resulted in reversal of behavioral hypersensitivities. a, Bolus intrathecal injection of TSP4 antibodies dose dependently reversed injury-induced tactile allodynia in SNL rats. Behavioral test on the injury side started 6 h after the bolus injection of TSP4 antibodies. Data shown are the mean ± SEM of 3–6 rats in each group. **p < 0.01 compared with the pretreatment level, as determined by one-way ANOVA with Dunnett's multiple comparison tests. b, Bolus intrathecal injection of TSP4 antibodies (80 μg/rat) reversed injury-induced tactile allodynia in SNL rats without affecting the behavioral thresholds on the noninjury side. The anti-allodynic effect of TSP4 antibodies had an onset time of 4 h, peaked at 6 h, and had a duration of >6 h. Neither heated TSP4 antibodies nor antibodies against another protein, peripherin, showed anti-allodynia effects. Data shown are the mean ± SEM of 6 rats in each group. *p < 0.05 and **p < 0.01 compared with the pretreatment level as determined by two-way ANOVA with Bonferroni post-tests. c, d, Bolus intrathecal injection of TSP4 antibodies (80 μg/rat) reversed injury-induced thermal hyperalgesia (c) and mechanical hyperalgesia (d) in SNL rats without affecting the behavioral thresholds on the noninjury side. The same dose of chicken IgY was used as control. To avoid sensitization of the hindpaw in paw pressure testing, mechanical hyperalgesia was tested in a 6 h interval as shown (d). Data shown are the mean ± SEM of 5–6 rats in each group. *p < 0.05 and ***p < 0.001 compared with the pretreatment level, as determined by two-way ANOVA with Bonferroni post-tests. e–g, Daily intrathecal injection of TSP4 antisense (ANT), but not mismatch (MIS), oligodeoxynucleotides (50 μg/rat/d) for 4 d reversed injury-induced tactile allodynia (e), thermal hyperalgesia (f), and mechanical hyperalgesia (g) in SNL rats. Behavioral tests were performed before each daily injection and daily after the last injection. Depending on the modality tested, the anti-nociceptive effect of TSP4 antisense oligodeoxynucleotides had an onset time of 1–3 d, peaked at 3–5 d, and lasted for >2–5 d after the last injection. The baseline PPT value for the mismatch treatment (g, right y-axis) was higher than that in the antisense treatment due to the conditioning adaptive increase in PPT (as shown in Fig. 1c) in the same group of rats that was used for the antisense treatment first, followed by the mismatch treatment at least 48 h apart. Data shown are the mean ± SEM of ≥5 rats in each group. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the pretreatment level as determined by two-way ANOVA with Bonferroni post-tests. h, Intrathecal TSP4 antisense treatment (50 μg/rat/d for 4 d) blocked injury-induced TSP4 expression in dorsal spinal cord. L5/6 dorsal spinal cord (DSC) samples were collected 1 d after the last oligodeoxynucleotide treatment from SNL rats and were subjected to Western blot analyses. Representative Western blot bands are shown on top of each bar group. Data shown are the mean ± SEM of ≥7 rats in each group. SNL, 6-week SNL rats without treatment; C, Contralateral; Ip, ipsilateral.
Figure 4.

Locomotor functions were not impaired by intrathecal treatments with TSP4 antibody or antisense oligodeoxynucleotides. a, b, Locomotor functions were analyzed with the Basso, Beattie, Bresnahan (BBB) locomotor rating scale in SNL rats before and after SNL surgery and treatments with antibodies (a) or oligodeoxynucleotides (b) as described. The post-treatment time points were 6 h after bolus antibody injections (a) or overnight after the last intrathecal injection of the 4 d antisense treatments (b); both were correlated with reversals of behavioral hypersensitivities shown in Figure 3b–d and 3e–g, respectively. Data presented are the mean ± SEM of ≥5 rats in each group. Tx, Treatment.
Blocking injury induction of TSP4 prevented the development of behavioral hypersensitivities
To determine whether TSP4 induction by SNL was required for the initiation of neuropathic pain states, we treated SNL rats preemptively with daily intrathecal injection of TSP4 antisera (80 μg/rat/d) for 8 d, started immediately before the SNL surgery, followed by daily behavioral testing before injection. Preemptive TSP4 antibody treatment prevented allodynia onset at the injury side without altering the baseline thresholds significantly at the noninjury side. The TSP4 effects lasted for 3 d after the last injection (Fig. 5a). These data support that injury-induced TSP4 is critical in neuropathic pain initiation. To confirm this, we examined whether allodynia was diminished or abolished in SNL TSP4 KO mice compared with sex- and age-matched wild-type (WT) littermates. We confirmed that TSP4 gene null expression did not affect the appearance and physical activity, including locomotor functions (Fig. 5b inset), of the mice, as described (B6.129P2-Thbs4tm1Dgen/J; (The Jackson Laboratory), and there was no significant difference in the body weights between the adult WT and KO mice (WT, 27.1 ± 1.9 g; KO, 26.9 ± 1.0 g; mean ± SEM) or in their basal sensory sensitivities at the time of SNL surgeries. SNL induced allodynia in the injury side of WT but not of TSP4 null mice and not at the noninjury side of WT and TSP4 null mice (Fig. 5b).
Figure 5.

Blocking injury induction of spinal TSP4 prevented neuropathic allodynia initiation. a, Intrathecal preemptive treatment with TSP4 antibodies prevented allodynia onset in spinal nerve-ligated rats in a reversible manner. TSP4 antibodies (80 μg/rat/d) were injected directly into L5/6 spinal region immediately before and daily after SNL for 8 d. Behavioral tests were performed before and daily after SNL and before each daily injection. Data presented are the mean ± SEM of 6 rats in each group. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the saline-injected group. #p < 0.05 and ###p < 0.001 compared with the pretreatment level using two-way ANOVA with Bonferroni post-tests. b, TSP4 null mice had diminished tactile allodynia post SNL. Unilateral L5 SNL was performed in adult male TSP4 null mice (KO) and their age- and sex-matched WT littermates. Behavioral tests were performed blindly before and after SNL. Data presented are the mean ± SEM of ≥7–12 mice in each group. TSP4 null expression did not impair the locomotor functions of the TSP4 KO mice compared with their WT littermates, as analyzed with the Basso Mouse Scale (BMS) for locomotion from ≥8 mice in each group (inset, mean ± SEM). **p < 0.01 and ***p < 0.001 compared with pre-SNL level using two-way ANOVA with Bonferroni post-tests. C, Contralateral; Ip, ipsilateral.
To determine whether SNL might also cause dysregulation of other TSP proteins in dorsal spinal cord, which could have contributed to behavioral hypersensitivity and could have been affected by TSP4 null expression, we examined TSP1 and TSP2 protein levels in dorsal spinal cords of SNL WT and SNL TSP4 KO mice. Western blot data indicated that TSP1 and TSP2 protein levels in dorsal spinal cord were similar between injury and noninjury sides of WT and TSP4 KO mice (Fig. 6) when allodynia was evident in the injury side of SNL WT, but not TSP4 KO, mice (Fig. 5b). Thus, SNL in TSP4 KO mice does not seem to induce a compensatory increase of other TSP proteins, nor does SNL in WT mice seem to induce dysregulation of other TSP proteins, which could have contributed to the development of abnormal sensations (data not shown). Together, these findings support that injury induction of TSP4 at the spinal level plays a critical role in the initiation of neuropathic pain states.
Figure 6.
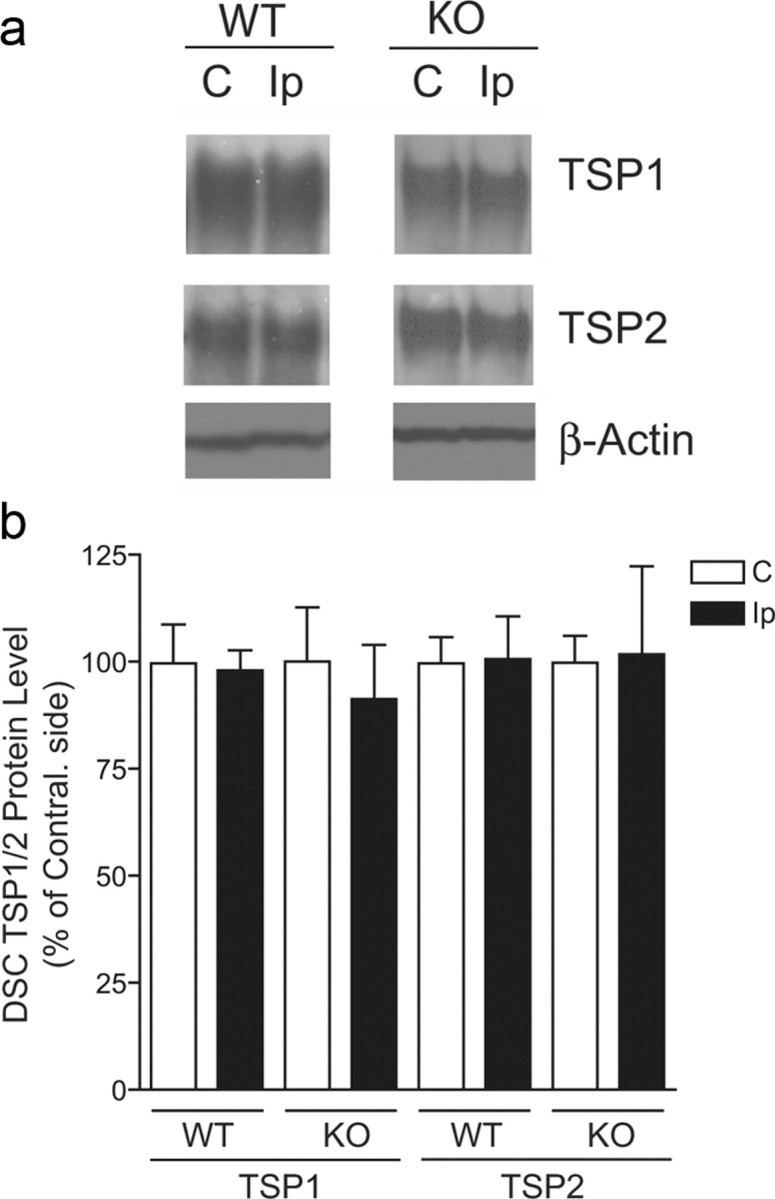
SNL did not alter TSP1 and TSP2 protein levels in dorsal spinal cords of WT and TSP4 KO mice. Western blot data indicated that L5 SNL injury (4 weeks) did not alter dorsal spinal cord (DSC) TSP1/2 protein levels significantly in WT and TSP4 KO mice when allodynia was observed in SNL WT but not SNL TSP4 KO mice, as shown in Figure 5b. a, Representative Western blot data. b, Summarized Western blot data presented as the mean ± SEM of 3–4 independent experiments in each group.
Dorsal spinal cord location of injury-induced TSP4
To determine the location of injury-induced spinal cord TSP4 upregulation, we took advantage of a transgenic mouse line in which eGFP is expressed under the control of a TSP4 promoter so that TSP4-expressing cells can be identified by green fluorescence. Data from confocal microscopy indicated that SNL injury (2 weeks) led to increased eGFP fluorescence in the injury side of the spinal cord, mainly at the dorsal spinal cord and the dorsal column (Fig. 7a,b), which correlated with behavioral hypersensitivity in SNL animals (Fig. 5b). Summarized data indicated that SNL injury induced significant increases in the surface area (Fig. 7c) and intensity (Fig. 7d) of eGFP fluorescence in TSP4-eGFP-expressing cells but not in the number of TSP4-eGFP-expressing cells (Fig. 7e), in the spinal cord, suggesting that peripheral nerve injury causes TSP4 upregulation in TSP4-expressing cells but not an increase in the number of TSP4-expressing cells. The sum of percentage increases from surface area and the intensity of eGFP signals in TSP4-eGFP-expressing cells after SNL injury was larger than the amplitude of TSP4 upregulation in dorsal spinal cord of SNL rats detected by Western blots (Fig. 1d), probably reflecting a signal amplification by eGFP fluorescence.
Figure 7.

eGFP immunoreactivity in dorsal spinal cords of SNL TSP4-eGFP mice. a, Overview of an L5 dorsal spinal cord section from a 2-week SNL TSP4-eGFP mouse (representative image from 4 mice). The dotted line outlines the dorsal horn (DH) and dorsal column (DC) areas in both sides. Scale bar, 200 μm. b, Enlarged view of dorsal spinal cord presented in a showing the structure of eGFP-positive cells. Scale bar, 200 μm. c–e, SNL injury induced significant increases in the surface area (c) and intensity (d) of eGFP signals in TSP4-eGFP expressing cells in dorsal spinal cord without altering the total number of eGFP-expressing cells (e). Data presented are the mean ± SEM of a total of 24 sections, 6 from each of the 4 SNL mice. ***p < 0.001 compared with contralateral side by Student's t test. C, Contralateral; Ip, ipsilateral.
To determine the type or types of TSP4-expressing cells in dorsal spinal cord, we performed fluorescent immunostaining on injured spinal cord sections with antibodies against different cell type markers and examined the colocalization of cell marker immunoreactivity with eGFP. Our data indicated that eGFP signals were detected mainly in cells immunoreactive to antibodies against GFAP, a marker for activated astrocytes (Fig. 8a), but not in cells immunoreactive to antibodies against NF-70, a marker for neurofilaments (Fig. 8b), Iba-1, a marker for microglia (Fig. 8c), or MBP (Fig. 8d), a marker for myelin sheath of myelinated nerve fibers. This was supported by Pearson's correlation coefficient analysis (Barlow et al., 2010) for colocalization of cell marker fluorescence with eGFP fluorescence from pooled data (1 = perfect positive correlation; −1 = perfect inverse correlation; 0 = random distribution) (Fig. 8e).
Figure 8.

TSP4-eGFP signals in dorsal spinal cord were mainly in astrocytes. a–d, Fluorescent immunoreactivity to antibodies of commonly used cell type-specific markers was superimposed with TSP4-eGFP signals in ipsilateral dorsal spinal cord sections from 2-week SNL TSP4-eGFP mice. Representative images from 4 mice are shown. TSP4 promoter-driven eGFP expression signals (green) mainly colocalized with immunoreactivity to antibodies against GFAP, a marker for activated astrocytes (red, a), but not with immunoreactivities to antibodies against NF-70, a marker for neurofilaments (red, b), Iba-1, a marker for microglial cells (red, c), or MBP (red, d). WM, white matter; GM, gray matter. Scale bars, 20 μm. Blue, nuclei. (inset) Enlarged images in each panel showing morphology of each type of immunoreactive cells, respectively. Scale bars: 10 μm. e, Possibility of colocalization of fluorescent signals from eGFP and immunoreactivity to each cell marker antibody was analyzed as described previously (Barlow et al., 2010). Data are presented as Pearson's correlation coefficients from the mean ± SEM derived from pooled data of 12 sections of 4 mice.
Increased spinal TSP4 protein alone was sufficient to induce behavioral hypersensitivities that correlated with dorsal horn neuron sensitization
To confirm a contributory role of TSP4 to the development of behavioral hypersensitivities without the influence of other injury factors that are present in the nerve injury model, we examined whether increased spinal TSP4 alone is sufficient to induce behavioral hypersensitivity in naive rats. Intrathecal bolus injection of full-length recombinant rat TSP4 fusion proteins with His-tags (for easy purification) into the L5/6 spinal level of naive rats, at a dose of 45 μg/rat, but not 20 μg/rat, resulted in the development of reversible tactile allodynia (Fig. 9a), thermal hyperalgesia (Fig. 9b), and mechanical hyperalgesia (Fig. 9c), similar to that seen in the SNL model (Fig. 1a–c). The behavioral hypersensitivities peaked approximately 3–4 d and lasted 4–6 d (Fig. 9a–c). Heat inactivation of TSP4 abolished its pro-nociceptive effect (Fig. 9a). Similarly injected equal molar doses of the His-tag peptide did not cause behavioral hypersensitivities or alter baseline behavioral sensitivities (Fig. 9a–c).
Figure 9.

Increased spinal TSP4 is sufficient to induce dorsal horn neuron sensitization and behavioral hypersensitivities through a chronic mechanism. a–c, Bolus intrathecal injection of 45 μg/rat TSP4 into L5/6 spinal regions of naive rats at time 0 led to long-lasting and reversible hindpaw hypersensitivities (averaged from both sides) assessed by blind daily behavioral test, including tactile allodynia (a), thermal hyperalgesia (b), and mechanical hyperalgesia (c). Intrathecal injection of 20 μg/rat TSP4 proteins, heated (45 μg/rat) TSP4 proteins (a), equal molar dose of His-tag peptides (a–c) did not cause behavioral hypersensitivities or alter baseline behavioral thresholds. Western blot showing purified TSP4-His fusion proteins detected by TSP4 antibodies (a, inset). Data presented are mean ± SEM of 5–6 rats in each group. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with pretreatment level by two-way ANOVA with Bonferroni post-tests. d, e, Intrathecal injection of TSP4 proteins into L5/6 spinal regions induced spinal neuron sensitization in naive rats that correlated with behavioral hypersensitivities. (d) Representative currents of AMPA receptor-mediated mEPSCs from superficial dorsal horn neuron whole-cell patch recording in L5 spinal cord slices of rats injected intrathecally with equal molar doses of His-tag peptides (Control) or TSP4-His fusion proteins (TSP4; 45 μg/rat) 4 d earlier. Intrathecal TSP4 proteins induced an increase in the average frequency (e1) as well as cumulative frequency distribution (e2), but not amplitude (e3), of mEPSCs. Data presented are summarized mean ± SEM of a total of 17–20 dorsal horn neurons recorded from spinal cord slices of 5 rats in each group. f, g, Acute application of TSP4 proteins to spinal cord slices did not alter mEPSCs in dorsal horn neurons. mEPSCs from naive rat L5 superficial dorsal horn neurons were recorded under identical conditions for spinal cord slice recording (d, e1–e3) except that TSP4 proteins were added to the recording bath for 15 min. Representative traces of mEPSC before and after bath application of 15 μg/mL TSP4 (f), a concentration that is similar to the spinal TSP4 concentration in vivo at the first injection day that caused behavioral and dorsal horn neuron sensitization later as shown in a–e, assuming the rate of CSF formation is 2.2 μl/min, approximately 3 mL/d/rat (Cserr, 1965) without considering the turnover of CSF and TSP4 proteins. Summarized graphs showing the lack of acute TSP4 effects on mEPSC average frequency (g1) and cumulative frequency distribution (g2) from rat superficial dorsal horn neurons. Data presented are the mean ± SEM of a total of 12 dorsal horn neurons recorded from spinal cord slices of 3 rats in each group. n.s., Not significant.
Since thrombospondin proteins play a critical role in abnormal synaptogenesis (Christopherson et al., 2005; Xu et al., 2010), which could lead to spinal sensitization and pain states (Zeilhofer, 2005), we studied whether increased spinal TSP4 caused behavioral hypersensitivity through a mechanism involving enhanced excitatory synaptic neurotransmission in the dorsal horn. AMPA receptor-mediated mEPSCs of dorsal horn neurons in spinal cord slices from TSP4-injected rats with peak behavioral hypersensitivities (4 d after TSP4 injection) were examined. mEPSCs in these neurons are induced by glutamate release from presynaptic terminals, and changes in its frequency or amplitude reflect a presynaptic or postsynaptic mechanism, respectively (Nguyen et al., 2009; Brittain et al., 2011). Our data indicated that the average frequency (Fig. 9d,e1) and the cumulative frequency distributions (Fig. 9d,e2), but not amplitudes (Fig. 9e3), of mEPSCs in dorsal horn neurons of TSP4-treated rats were increased at this time point compared with that from His-tag peptide-injected control rats (Fig. 9d,e1–e3). However, acute bath application of recombinant TSP4 proteins to the spinal cord slices for 15 min at a concentration similar to the pro-nociceptive spinal TSP4 concentration at the first day of intrathecal injection did not alter mEPSC frequency (or amplitudes) (data not shown) in L5 dorsal horn neurons (Fig. 9f,g1,g2). These data support that increased spinal TSP4 is sufficient to induce spinal neuron sensitization and behavioral hypersensitivity highly likely by enhancing presynaptic excitatory input through a chronic mechanism.
Discussion
Our results indicate that peripheral nerve injury induces TSP4 upregulation in dorsal spinal cord that correlates with initiation/maintenance of behavioral hypersensitivities mediated by different sensory fibers. Blocking injury-induced TSP4 with intrathecal antisense oligodeoxynucleotides or TSP4 gene KO as well as blocking its activity with intrathecal antibodies prevent the onset and/or reverse established neuropathic pain states. Intrathecal injection of TSP4 proteins alone is sufficient to induce behavioral hypersensitivities that correlate with dorsal horn neuron sensitization through enhanced frequency of mEPSCs. Together, these findings support our hypothesis that injury-induced spinal TSP4 contributes to the initiation and maintenance of neuropathic pain states through a yet unidentified mechanism involving, most likely, enhanced presynaptic excitatory neurotransmitter release.
The detailed mechanism underlying injury-induced spinal TSP4 in neuropathic pain processing is under active investigation. Our data support that TSP4 protein levels are not altered in uninjured, adjacent L4 DRGs from the injury side of SNL rats with neuropathic pain states, which is consistent with the latest evaluation that L4 DRG is less likely contributing to neuropathic pain processing in this model (Fukuoka et al., 2012). Therefore, it is likely that dysregulated TSP4 in the injury segment plays a critical role in the development of neuropathic pain states. It is interesting that injury leads to an increase of TSP4 protein levels in the injured spinal cord segment, but a reduction in the associated DRG, suggesting that peripheral nerve injury induces differential TSP4 regulations in the PNS and the CNS, respectively, which may underlie distinct mechanisms in neuropathic pain processing.
Our data confirm that peripheral nerve injury induces TSP4 expression mainly in GFAP immunoreactive cells in the spinal cord. The profiles of eGFP/GFAP-positive cells in the injury side closely resemble the morphology of immature astrocytes in dorsal spinal cord, as shown in Figure 2G in Edwards et al. (1990). However, we cannot exclude the possibility that some of them are radial glial cells, an embryonic precursor of astrocytes (Edwards et al., 1990), which can also be detected by GFAP antibodies (Choi, 1988) (also see Pixley and de Vellis, 1984; Edwards et al., 1990). It is possible that peripheral nerve injury reactivates these cells that otherwise are mostly silent in the adult stage of development. The presence of these embryonic astrocytes is known to correlate with the peak periods of neurogenesis (Edwards et al., 1990), suggesting a potential role of these cells in nerve regeneration after peripheral nerve injury.
TSP is considered a member of the endoneurial extracellular matrix molecules, and TSP expression is increased dramatically along the path of regenerating axons postinjury; it has been hypothesized that TSP may play a role in cell signaling during nerve regeneration postinjury (Hoffman and O'Shea, 1999; Dubovy et al., 2002). Our data support that SNL-induced TSP4 expression is mainly distributed along fiber profiles in the dorsal spinal cord and the dorsal column of the injury side (Fig. 7a,b). Since both high-threshold nociceptive fibers (Aδ and C fibers), which transmit painful signals, and low-threshold sensory fibers (Aβ fibers), which transmit activity induced by innocuous stimuli, are sending their axons to the dorsal spinal cord at the segment of entry (Yaksh and Luo, 2007), enhanced TSP4 expression in immature astrocytes surrounding these fibers in dorsal spinal cord may play a critical role in mediating behavioral hypersensitivities such as thermal/mechanical hyperalgesia (by Aδ and C fibers) and tactile allodynia (by Aβ fibers). In addition, myelinated Aβ fibers are also sending axons collaterally into the dorsal column (Yaksh and Luo, 2007), and enhanced TSP4 expression in immature astrocytes surrounding these axons in the dorsal column may play a critical role in mediating tactile allodynia.
Recently, it has been reported that astrocyte-secreted TSP induces excitatory synaptogenesis in the CNS (Christopherson et al., 2005) through interactions with its neuronal receptor, the calcium channel α-2-δ-1 subunit proteins (Eroglu et al., 2009). In addition, trigeminal nerve injury is shown to induce astrocyte activation in the brain stem sensory trigeminal nuclei, which leads to reactive synaptogenesis presumably involving thrombospondins (Lo et al., 2011). TSP1 is shown to increase the speed of synaptogenesis in young, but not mature, hippocampal neurons (Xu et al., 2010). Even though intrathecal injection of TSP1 proteins also induces behavioral hypersensitivity in rats (data not shown), our data indicated that TSP1/2 protein levels in the dorsal spinal cord remain similar between SNL WT mice with allodynia and SNL TSP4 KO mice without allodynia (Fig. 6b), suggesting that peripheral nerve injury-induced behavioral hypersensitivity is not mediated by dysregulation of other TSP proteins.
It is possible that injury-induced TSP4 overexpression in immature astrocytes surrounding the injured sensory fibers provides a TSP4-rich perisynaptic environment, which may lead to abnormal excitatory synaptogenesis in the spinal cord. This plasticity can contribute to enhanced dorsal horn neuron sensitization, wider spreading of sensory signals, and evoked behavioral hypersensitivities. This is supported by our data showing that intrathecal TSP4 injection into naive rats can lead to sensitization of dorsal horn neurons through a mechanism most likely involving enhanced excitatory neurotransmitter release from presynaptic terminals and behavioral hypersensitivities similar to those observed after peripheral nerve injury (Fig. 9a–e). Since acute bath application of TSP4 failed to elicit dorsal horn neuron sensitization (Fig. 9f,g1,g2), our data support that the pronociceptive effects of TSP4 in the injury model are likely mediated through a chronic mechanism, which may include, but is not limited to, de novo TSP4 synthesis and/or altered excitatory synaptogenesis that requires days to complete (Christopherson et al., 2005). Our data supporting this conclusion include the slow onset of behavioral hypersensitivity after preemptive TSP4 antibody treatment in SNL rats (Fig. 5a) and the slow onset and long-lasting effects of TSP4-induced behavioral hypersensitivity in naive animals (Fig. 9a–c) and of TSP4 antisense reversal in behavioral hypersensitivities in SNL animals (Fig. 3e,f). Neutralizing TSP4 secreted from astrocytes in dorsal spinal cord postinjury by antibody treatment (Figs. 3a–d, 5a) may present an alternative intervention that is faster in onset (h) but shorter in duration (h) in reversing neuropathic pain states.
In summary, our findings provide a large body of evidence to support that injury-induced TSP4 expression in presumably immature astrocytes in spinal cord plays a critical role in the initiation and maintenance of neuropathic pain states. Blocking the initiation of this pathological pathway may serve as a new approach in developing the next generation of target-specific medications for neuropathic pain management.
Footnotes
This work was supported in part by National Institutes of Health Grants DE14545 (Z.D.L.), NS064341 (Z.D.L.), DE021847 (Z.D.L.), and DA15043 (B.B.) and by a pilot grant from the Department of Anesthesiology and Perioperative Care, University of California Irvine (Z.D.L.). We thank May Song and Tim Yolo for their excellent technical assistance in some initial studies and Douglas Annis and Dr. Deane Mosher for their helpful information on TSP antibodies.
References
- Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995;131:1083–1094. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow AL, Macleod A, Noppen S, Sanderson J, Guérin CJ. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of Pearson's correlation coefficient. Microsc Microanal. 2010;16:710–724. doi: 10.1017/S143192761009389X. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroujerdi A, Kim HK, Lyu YS, Kim DS, Figueroa KW, Chung JM, Luo ZD. Injury discharges regulate calcium channel alpha-2-delta-1 subunit upregulation in the dorsal horn that contributes to initiation of neuropathic pain. Pain. 2008;139:358–366. doi: 10.1016/j.pain.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroujerdi A, Zeng J, Sharp K, Kim D, Steward O, Luo ZD. Calcium channel alpha-2-delta-1 protein upregulation in dorsal spinal cord mediates spinal cord injury-induced neuropathic pain states. Pain. 2011;152:649–655. doi: 10.1016/j.pain.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2) channel complex. Nat Med. 2011;17:822–829. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BH. Prenatal gliogenesis in the developing cerebrum of the mouse. Glia. 1988;1:308–316. doi: 10.1002/glia.440010503. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Costigan M, Befort K, Karchewski L, Griffin RS, D'Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 2002;3:16. doi: 10.1186/1471-2202-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr H. Potassium exchange between cerebrospinal fluid, plasma, and brain. Am J Physiol. 1965;209:1219–1226. doi: 10.1152/ajplegacy.1965.209.6.1219. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dubový P, Klusáková I, Svízenská I. A quantitative immunohistochemical study of the endoneurium in the rat dorsal and ventral spinal roots. Histochem Cell Biol. 2002;117:473–480. doi: 10.1007/s00418-002-0411-5. [DOI] [PubMed] [Google Scholar]
- Dunkle ET, Zaucke F, Clegg DO. Thrombospondin-4 and matrix three-dimensionality in axon outgrowth and adhesion in the developing retina. Exp Eye Res. 2007;84:707–717. doi: 10.1016/j.exer.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Edwards MA, Yamamoto M, Caviness VS., Jr Organization of radial glia and related cells in the developing murine CNS: an analysis based upon a new monoclonal antibody marker. Neuroscience. 1990;36:121–144. doi: 10.1016/0306-4522(90)90356-9. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Yamanaka H, Kobayashi K, Okubo M, Miyoshi K, Dai Y, Noguchi K. Re-evaluation of the phenotypic changes in L4 dorsal root ganglion neurons after L5 spinal nerve ligation. Pain. 2012;153:68–79. doi: 10.1016/j.pain.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hoffman JR, O'Shea KS. Thrombospondin expression in nerve regeneration, II: comparison of optic nerve crush in the mouse and goldfish. Brain Res Bull. 1999;48:421–427. doi: 10.1016/s0361-9230(99)00022-2. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Cho HJ. Expression of monocyte chemoattractant protein-1 in rat dorsal root ganglia and spinal cord in experimental models of neuropathic pain. Brain Res. 2009;1251:103–111. doi: 10.1016/j.brainres.2008.11.046. [DOI] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Sakagami M, Noguchi K. Antisense knock down of TRPA1, but not TRPM8, alleviates cold hyperalgesia after spinal nerve ligation in rats. Exp Neurol. 2006;200:112–123. doi: 10.1016/j.expneurol.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Kim DS, Figueroa KW, Li KW, Boroujerdi A, Yolo T, Luo ZD. Profiling of dynamically changed gene expression in dorsal root ganglia post peripheral nerve injury and a critical role of injury-induced glial fibrillary acidic protein in maintenance of pain behaviors [corrected] Pain. 2009;143:114–122. doi: 10.1016/j.pain.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Li CY, Song YH, Higuera ES, Luo ZD. Spinal dorsal horn calcium channel α2δ-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci. 2004;24:8494–8499. doi: 10.1523/JNEUROSCI.2982-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo FS, Zhao S, Erzurumlu RS. Astrocytes promote peripheral nerve injury-induced reactive synaptogenesis in the neonatal CNS. J Neurophysiol. 2011;106:2876–2887. doi: 10.1152/jn.00312.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Figueroa KW. Multilevel genomic approach in pain research: basic science and clinical implications. Rev Analg. 2008;10:45–58. [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Deng P, Matthews EA, Kim DS, Feng G, Dickenson AH, Xu ZC, Luo ZD. Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel alpha-2-delta-1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol Pain. 2009;5:6. doi: 10.1186/1744-8069-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pixley SK, de Vellis J. Transition between immature radial glia and mature astrocytes studied with a monoclonal antibody to vimentin. Brain Res. 1984;317:201–209. doi: 10.1016/0165-3806(84)90097-x. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, Hogan QH. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfers M, Sorkin LS, Geis C, Shubayev VI. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett. 2003;347:179–182. doi: 10.1016/s0304-3940(03)00695-5. [DOI] [PubMed] [Google Scholar]
- Shibasaki M, Sasaki M, Miura M, Mizukoshi K, Ueno H, Hashimoto S, Tanaka Y, Amaya F. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain. 2010;149:514–521. doi: 10.1016/j.pain.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Shimoyama M, Shimoyama N, Hori Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain. 2000;85:405–414. doi: 10.1016/S0304-3959(99)00283-3. [DOI] [PubMed] [Google Scholar]
- Södersten F, Ekman S, Schmitz M, Paulsson M, Zaucke F. Thrombospondin-4 and cartilage oligomeric matrix protein form heterooligomers in equine tendon. Connect Tissue Res. 2006;47:85–91. doi: 10.1080/03008200600584124. [DOI] [PubMed] [Google Scholar]
- Valder CR, Liu JJ, Song YH, Luo ZD. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem. 2003;87:560–573. doi: 10.1046/j.1471-4159.2003.02016.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun H, Della Penna K, Benz RJ, Xu J, Gerhold DL, Holder DJ, Koblan KS. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546. doi: 10.1016/s0306-4522(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Xu JT, Tu HY, Xin WJ, Liu XG, Zhang GH, Zhai CH. Activation of phosphatidylinositol 3-kinase and protein kinase B/Akt in dorsal root ganglia and spinal cord contributes to the neuropathic pain induced by spinal nerve ligation in rats. Exp Neurol. 2007;206:269–279. doi: 10.1016/j.expneurol.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci. 2010;13:22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]
- Yaksh T, Luo ZD. Anatomy of the pain processing system. In: Waldman SD, editor. Pain management. Philadelphia: Saunders Elsevier; 2007. pp. 11–20. [Google Scholar]
- Yang L, Zhang FX, Huang F, Lu YJ, Li GD, Bao L, Xiao HS, Zhang X. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. Eur J Neurosci. 2004;19:871–883. doi: 10.1111/j.0953-816x.2004.03121.x. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU. Synaptic modulation in pain pathways. Rev Physiol Biochem Pharmacol. 2005;154:73–100. doi: 10.1007/s10254-005-0043-y. [DOI] [PubMed] [Google Scholar]