

Abstract
Gliomas are the leading cause of death among adults with primary brain malignancies. Treatment for malignant gliomas remains limited, and targeted therapies have been incompletely explored. In this study, we found that the protein expression of presenilin 2 (PS2) was significantly increased in glioma tissues, at least partially because of promoter demethylation. We further evaluated the biological functions of PS2 in U251 glioma cell proliferation, migration, invasion, and tumor growth in vivo by specific inhibition of PS2 using short hairpin RNA (shRNA). We found that PS2 depletion inhibited glioma cell growth as the result of inhibited proliferation and induced apoptosis. PS2 depletion also decreased the invasive capability of glioma cells and anchorage-independent colony formation in soft agar. Moreover, suppression of PS2 expression significantly impaired the growth of glioma xenografts in nude mice. Finally, the decrease in glioma cell growth caused by PS2 depletion seems to involve Nrg1/ErbB signaling. In summary, our data highlight the use of RNA interference (RNAi) as a tool to better understand the molecular basis of PS2 in glioma progression and to uncover new targets for the treatment of glioma.
Keywords: glioma, invasion, Nrg1/ErbB, presenilin 2, proliferation, RNAi
Malignant gliomas are the most common and most devastating primary brain tumors in adults. More than half of the primary malignant brain tumors diagnosed each year in the United States are gliomas, and these tumors are the second-most common cause of cancer-related death in the 15–44 year age group.1 The prognosis for patients with malignant gliomas is poor: median survival is 3–5 years among patients with grade III gliomas and <1 year among patients with grade IV gliomas.1 Despite continuous therapy regimens, including maximum safe resection, radiation therapy, and chemotherapy,2–4 malignant gliomas generally continue to grow. An improved understanding of the molecular pathogenesis of malignant gliomas will allow us to develop more effective adjuvant therapies.
Malignant gliomas are characterized by the sequential accumulation of genetic aberrations and the deregulation of growth-factor signaling pathways.5 The epidermal growth factor receptor (EGFR) family consists of 4 closely related receptor tyrosine kinases (human ErbB receptors 1-4, HER 1-4).6 When the EGFR family members are activated by their ligands, a variety of intracellular signaling pathways are triggered that regulate diverse cellular processes, such as cell division, survival, and invasion.7,8 Neuregulin-1 (Nrg1) is the ligand of ErbB3 and ErbB4.9 Nrg-1 activation of ErbB receptors promotes human glioma cell motility and migration.10 Presenilins are part of the γ-secretase complex, an enzymatic complex that cleaves type I transmembrane proteins implicated in cell adhesion, differentiation, survival, and proliferation.11 Mutations in the genes that encode the presenilin 1 (PS1) and presenilin 2 (PS2) proteins cause most cases of familial Alzheimer's disease.12–14 PS2 and PS1 are highly homologous transmembrane proteins, and 2 of their main roles in cellular functions are to increase the production of the amyloidogenic Aβ peptide and to regulate Notch signaling.15,16 However, the distinctly different phenotypes of PS1-deficient and PS2-deficient mice suggest that these proteins play different roles. PS1-deficient mice experience severe developmental defects and perinatal lethality,17 but PS2-deficient mice are viable and fertile and develop only mild pulmonary fibrosis and hemorrhage with increasing age.18 At the cellular level, PS1 is a negative regulator of cell proliferation.19 Loss of PS1 in murine epidermal tissues is associated with enhanced β-catenin signaling and skin tumorigenesis,20 suggesting that PS1 may play a critical role in tumor suppression. PS2 has also been shown to regulate the level of β-catenin.21 In addition, PS2 elicits a p53-dependent pro-apoptotic response that is exacerbated by FAD mutations.22 Conversely, the phosphorylation of PS2 slows caspase cleavage and retards apoptosis. Previous studies have also shown that overexpression of either wild-type or mutant PS2 did not induce apoptosis or increase susceptibility to apoptosis in different cell lines.23 Evidence suggests that PS2 alleles affect PS2 function and may potentially confer a moderate risk of susceptibility to breast cancer.24 For example, the lower efficiencies of the R71W allele of human PS-2 to rescue the Egl phenotype, in comparison with the wild-type counterpart, provided strong in vivo evidence in support of a loss-of-function effect caused by these alterations.24 A recent study has shown that the growth-promoting effects of 2-AG are associated with the activation of Notch 2 and the presence of PS2 in the γ-secretase complex.25 Considering these observations, alterations affecting PS2 might contribute to the development of cancer.
RNA interference (RNAi) is a powerful gene-silencing tool that holds great promise in the field of cancer therapy. In this study, we searched for the PS2 gene in glioma cell lines using a plasmid-delivered short-hairpin RNA (shRNA) library. To further investigate the potential role of PS2 in gliomas, we analyzed primary tumor specimens from glioma cases for the presence of sequence alterations. In addition, we explored the mechanism by which the overexpression of PS2, which emerged in the screen, supports glioma cell proliferation or invasion. We also investigated whether PS2 is coexpressed with Nrg-1 protein.
Materials and Methods
Tissue Sample Collection
Intracranial tissue samples were obtained from 38 patients who underwent surgery at the Institute of Neurosurgery of Tangdu Hospital, Fourth Military Medical University, Xi'an, China. According to the revised WHO criteria for the CNS, 7 patients were classified as having grade 2 disease (low-grade astrocytoma), 19 as having grade 3 (anaplastic astrocytoma), and 12 as having grade 4 (glioblastoma). Ten adjacent normal brain tissues were obtained from patients with glioma, and histologically confirmed to be tumor-free by frozen section. Supplementary material, Table S1 shows the clinical features of the patients. All of the tumor tissues were obtained at primary resection, and none of the patients had undergone chemotherapy or radiation therapy before resection. The samples were snap frozen in liquid nitrogen and stored at −80°C until analysis. The diagnosis of glioma was confirmed histologically in all cases. For the experimental use of the surgical specimens, informed consent was obtained from the patients according to the hospital ethical guidelines.
Cell Culture
We used human malignant glioma cell lines (U251, U87, and SHG44) obtained from the Institute of Neurosurgery of Tangdu Hospital, Fourth Military Medical University, Xi'an, China. The cells were maintained as adherent monolayer cultures in Dulbecco's modified Eagle's medium (DMEM; Gibco/BRL) and supplemented with 10% fetal bovine serum (FBS; Sigma). Cells were grown at 37°C in a humidified incubator containing 5% carbon dioxide. The medium was replaced every 2–3 days with complete medium, and cells were subcultured when confluence was reached.
To measure the effects of Nrg1, transfected cells were grown to confluence in medium containing G418 (400 μg/mL), rinsed 3 times with assay buffer (AB) consisting of serum-free medium plus 0.1% fatty acid–free bovine serum albumin, and then placed in AB with 10 ng/mL Nrg-1 and incubated for 30 min.
Antibodies and Reagents
Anti-ErbB3 and Anti-ErbB4 antibodies were obtained from Abcam, anti-neuregulin 1 and anti-GAPDH from Santa Cruz Biotechnology, polyclonal rabbit anti-PS2 protein polyclonal antibody from Cell Signaling Technology, and the protease inhibitor cocktail from Calbiochem. Cell Signaling Technology produced recombinant human Nrg-1. Penicillin/streptomycin, trypsin/EDTA, and phosphate-buffered saline (PBS) were obtained from Gibco. Protein A/G PLUS agarose beads were obtained from Calbiochem-Merck Biosciences. Dithiothreitol was purchased from Invitrogen and cycloheximide from Sigma-Aldrich. All other reagents were purchased from reputable domestic companies unless specifically stated.
Methylation Assay
Total genomic DNA was isolated from glioma and normal tissues using the DNA extraction kit according to the manufacturer's instructions (Tiangen). Genomic DNA (2 µg) was then modified with bisulfite using the EZ DNA Methylation kit (Zymo Research) according to the manufacturer's directions. Bisulfite-modified DNA was then used as template DNA for polymerase chain reaction (PCR) amplification with the external nest PCR primers corresponding to the region unaffected by the methylation status. Equal aliquots of the amplicon were then amplified with relative PCR primers. Primers were 5′-tccactccttctaaggtcgtc-3′ (sense) and 5′-agtttacgcagctggctgag-3′ (antisense) for the methylated promoter region and 5′-gatagaggtggtggttg-3′ (sense) and 5′-cctaatctattcccta-3′ (antisense) for the region unaffected by the methylation. The resulting PCR products were then electrophoresed on a 1.2% agarose gel. To determine the methylation proportion, the band of the region unaffected by the methylation status served as a loading control. For tissue samples, the intensities of the methylated and loading control bands were compared with the base pairs of the amplicons and the cycles of individual PCRs being considered. Methylation was considered to be high when the relative ratio of methylated to total DNA was >50% and to be low when the ratio was <50%. Samples with no obvious methylation bands were considered to have no methylation.
Western Blot Analysis
For Western blots, 60μg of total proteins from tissue or cell extracts was subjected to SDS-PAGE, blotted, and probed with primary antibodies. Secondary antibodies conjugated to IRDye TM 800 (1:20 000 dilution; Rockland) were detected by using an Odyssey infrared imaging system (LI-COR) to quantify Western blots using ImageJ.
Immunohistochemistry
Tumors were fixed in 4% paraformaldehyde and embedded in paraffin blocks. Sections (5 µm) were prepared for immunohistochemical examination. For immunohistochemical analysis, sections were stained and evaluated as previously described.26 Both the extent and the intensity of PS2 immunostaining were taken into consideration when analyzing the data. The intensity of staining was scored 0–3, and the extent of staining was 0%–100%. The final quantification of each staining was obtained by multiplying the 2 scores. The slides were analyzed by 2 independent pathologists.
Construction of shRNA-Expressing Plasmid and Stable Gene Transfection
A pSilencer3.1-H1neo plasmid (Ambion) was used to construct the shRNA-expressing vector. The following sequence was used for PS2: 5′-CACCGCTCACATTCATGGCCTCTGATTCAAGAGATCAGAGGCCATGAATGTGAGCTTTTTTG-3′. A non-specific siRNA, 5′-CACCGTTCTCCGAACGTGTCACGTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTG-3′ (ShanghaiGenePharma), was used as a negative control. The 2 resulting plasmids were designated as pSilencer3.1-PS2 and pSilencer3.1-NC, respectively. U251 cells were transfected with pSilencer3.1-PS2 and pSilencer3.1-NC using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, and the parental cells were used as controls. Stable cell lines were selected with G418 (800 μg/mL; Sigma), and individual clones were isolated and maintained in medium containing G418 (400 μg/mL). The stable clones were identified as U251-S, U251-S1, U251-S2 (transfected with pSilencer3.1-PS2), and U251-NC (transfected with pSilencer3.1-NC). U251-Smix was a pool of stably transfected U251 cells.
Measurement of Cell Growth by Methyl Thiazolyl Tetrazolium Assay (MTT)
U251 cells, both parental and transfected lines, were seeded at a density of 1 × 104 cells/well in 96-well plates containing 0.2 mL of DMEM (with 10% FBS) and cultured for 6 days. During this period, the cells were given fresh complete medium every 2 days. Six wells from each group were selected randomly for the MTT assay (Sigma-Aldrich; 50 μg for each well) every day. After 4 h of incubation, the reaction was stopped by adding 150 μL of dimethyl sulfoxide (DMSO; Sigma-Aldrich) to each well and incubating for 10 min. The percentage of viable cells was determined by measuring the absorbance at 490 nm on a multiscanner reader (TECAN-Spectra Mini; TECAN). With use of a mean absorbance of 490 nm from 3 independent experiments, cell growth curves were drawn. The percentage of inhibition was calculated using the formula: % inhibition = [1−(OD test/OD control)] × 100.
Annexin V-Phycoerythrin (PE)/7-Aminoactinomycin D (7-AAD) Staining Assay
Apoptosis was assessed by measuring membrane redistribution of phosphatidylserine with fluorescent annexin V. Cells were collected and washed twice with PBS and then resuspended in 500 μL of staining solution containing PE-conjugated annexin V antibody (5 μL; BD PharMingen) and 7-AAD (5 μL; BD PharMingen). After incubation for 15 min at room temperature in the dark, cells were immediately analyzed on a flow cytometer. Data acquisition analysis was performed in a FACS Calibur flow cytometer (Becton Dickinson) using CellQuest software. Apoptotic cells were double stained with annexin V and 7-AAD. The percentage of cells undergoing apoptosis was determined in 3 independent experiments.
Flow Cytometric Analysis of the Cell Cycle
Cells were seeded in 25-mL flasks in DMEM containing 10% FBS and incubated until they were 80%–85% confluent. The cells were treated with AB without or with Nrg1. Then the cells were harvested, washed twice with ice-cold PBS, fixed with 70% ethanol overnight at 4°C, washed, and resuspended in 100 μL of PBS containing a final concentration of 50 μg/mL RNase A for 30 min at room temperature. Finally, the cells were stained with 20 μg/mL propidium iodide in a final volume of 300 μL for 20 min. DNA content and the cell cycle phase were analyzed by flow cytometry (FACSCalibur, Becton Dickinson), using the software MODFIT and CELLQUEST. All of the samples were assayed in triplicate.
Monolayer Wound Healing Assay
Migration ability was determined using a wound-healing assay. U251, U251-NC, and U251-S cells were grown in 60-mm cell culture plates containing DMEM with 10% FBS. After cells reached 90% confluence, the medium was replaced with FBS-free medium for 24 h. A sterile 200-μL pipette tip was used to create a wound in the monolayer by scraping. The cells were washed with PBS and grown in FBS-free medium for 24 h. The wounds were observed under a phase contrast microscope (model BX2; Olympus). The images were analyzed by digitally drawing lines and averaging the position of the migrating cells at the wound edges using Scion Image software. The distance of cell migration was calculated by subtracting the distance between the lesion edges at 24 h from the distance measured at 0 h. The width of the scratch was measured at 0 and 24 h after treatment. The migration distance in the wound was calculated according to the following formula: cell-free area at 0 h – cell-free area at 24 h. Experiments were performed 3 times in duplicate with comparable results.
Transwell Invasion Assay
The invasiveness of cells was measured by using the Matrigel invasion assay. In brief, Transwell insert chambers (Corning Life Sciences) with 8-μm pore filters were coated with a final concentration of 1 mg/mL Matrigel (BD Sciences). Cells (104/well) were seeded in the upper chambers of the wells in 200 μL of FBS-free medium, and the lower chambers were filled with 500 μL of 10% FBS medium to induce cell migration. After incubation for 24 h, the cells were treated with AB without or with Nrg1. The cells on the filter surface were fixed with 4% formaldehyde, stained with 10% Giemsa (Sigma-Aldrich), and examined under a microscope. Eight random high-power microscopic fields (100× magnification) per filter were photographed, and the numbers of cells were counted.
Plate Colony Formation Assay
For colony formation assays, cells (1 × 103) were seeded onto 60-mm dishes with 5 mL of DMEM supplemented with 10% FBS and 400 μg/mL G418. After 14 days, the resulting colonies were rinsed with PBS, fixed with methanol at −20°C for 5 min, and stained with Giemsa (Sigma-Aldrich) for 20 min. Only clearly visible colonies (diameter, >50 μm) were counted.
Soft Agar Assay
Cells (1 × 104) were added to 3 mL of DMEM (supplemented with 10% FBS) with 0.3% agar and layered onto 6 mL of 0.5% agar beds in 60-mm dishes. Cells were cultured for 2 weeks, after which colonies were photographed. Colonies larger than 50 μm in diameter were counted as positive for growth. Assays were conducted in duplicate in 3 independent experiments.
In Vivo Experiments
Thirty female BALB/c nu/nu mice weighing 14–18 g that were 5 weeks of age were purchased from the Shanghai SLAC Laboratory Animal Company. The mice were maintained under pathogen-free conditions at 26°C and 70% relative humidity and with a 12-h light/dark cycle. All animal experiments complied with international guidelines for the care and treatment of laboratory animals. The mice were assigned randomly to 1 of 3 groups. U251, U251-NC, and U251-S cells were harvested and counted. The cells (1 × 107) were suspended in 0.2 mL of normal saline and then inoculated subcutaneously into the left flank of the nude mice, which led to palpable nodules on day 4. Tumor growth was measured with calipers every 4 days through day 24. As reported previously,27 tumor volumes in mice were measured with slide calipers and recorded using the formula: volume = a × b2/2, where a is the larger of the 2 dimensions and b is the smaller.
Coimmunoprecipitation
Whole U251 cell lysates were obtained by resuspending U251 cell pellets in RIPA buffer (150 mM NaCl, 20 mM Tris-HCl pH 7.4, 5 mM EDTA, 1% NP-40, 1% Na-deoxycholate, 0.1% SDS, 1 mM PMSF, 20 μg/mL leupeptin, 20 μg/mL aprotinin, 3 μg/mL pepstatin A). Lysates were incubated overnight with Nrg1 antibody before being absorbed with protein A/G PLUS agarose beads. Precipitated immunocomplexes were released by boiling with 2 × SDS electrophoresis sample buffer and were prepared for Western blot analysis.
Statistical Analysis
All experiments were performed in triplicate, and standard deviations were calculated. All statistical analyses were performed using SPSS software (version 11.0; SPSS). Comparisons among all groups were performed using one-way analysis of variance (ANOVA). A P < .05 was considered to be statistically significant.
Results
PS2 Protein Levels in Glioma Tissue Samples and Glioma Cell Lines
To evaluate the role of PS2 in gliomas, we first analyzed the expression levels of PS2 in low-grade glioma tissue samples (grade 2, astrocytoma: n = 7), high-grade glioma samples (grade 3, anaplastic astrocytoma: n = 19; grade 4, glioblastoma: n = 12), and adjacent normal brain tissues (n = 10). Western blot demonstrated increased expression levels of PS2 in glioma tissue relative to the level of PS2 in adjacent normal brain tissue. Representative data are shown in Fig. 1B. The values in each sample were standardized for sample-to-sample variations using GAPDH as normalization. Similar results were observed through immunohistochemical analysis (Fig. 1D). Of interest, the level of PS2 expression was correlated with increasing tumor grade in brain tumors (Fig. 1C). Among these 38 glioma tissue samples, the 7 low-grade astrocytomas displayed a relatively small increase in PS2 protein levels, compared with normal brain, whereas the 31 high-grade glioma samples showed a significant increase in the amount of PS2 protein, compared with normal brain (Fig. 1E). Thus, it appears that PS2 is overexpressed in many advanced glial neoplasms, which was also confirmed in several high-grade glioma–derived cell lines, such as U87, U251, and SHG44 (Fig. 1A).
Fig. 1.

Expression pattern of PS2 in normal and glioma tissues. (A) Protein levels of PS2 in multiple glioblastoma cell lines and adjacent normal tissue were detected by Western blot. GAPDH served as an internal control to ensure equal loading. (B) Protein levels of PS2 in 10 glioma specimens and adjacent normal counterparts were detected by Western blot. GAPDH served as an internal control to ensure equal loading. (C) Quantitative analysis of the mean ratio of optical density values in 10 glioma specimens and adjacent normal counterparts. (D) The expression of PS2 in 38 cases of gliomas and 10 adjacent normal tissues was detected by immunohistochemistry. PS2 expression was much higher in most glioma tissues than in the adjacent normal tissues. Representative data are shown. Scale bars, 50 μm. (E) Box plot of PS2 expression in brain tumors with different histological grades.
Demethylation of PS2 Promoter Contributes to the Overexpression of PS2 in Human Gliomas
To determine the underlying mechanisms responsible for overexpression of PS2 in cancer cells, we analyzed the promoter region of PS2. The PS2 promoter region is rich in CpG islands, especially in the 500 base pairs upstream of the putative transcription start site, leading us to ask whether CpG demethylation accounted for the high expression of PS2 in human gliomas. Glioma tissue samples and adjacent normal counterparts of the 10 patients analyzed above were collected for methylation analysis.
The methylation-specific PCR (MSP) analysis of adjacent normal brain tissues showed that 1 sample was unmethylated, 5 samples had weak methylated promoter regions, and 4 samples had highly methylated promoter regions. Of the 10 glioma tissue samples, 9 were without methylation signal and 1 had weak methylation (Fig. 2). Taken together, these results indicate that carcinogenesis and methylation of the P2 promoter are associated with each other.
Fig. 2.

Methylation status of the PS2 promoter region in normal (N) and glioma (T) tissues. Methylated promoter and the total DNA level (served as a loading control) of each sample were amplified with specific primers.
Stable Suppression of PS2 Using shRNA in U251 Cells
To study the role of PS2 in the malignant process of glioma, we generated PS2 shRNA-stable clones. In brief, DNA oligonucleotides specific for PS2 and negative controls were generated and ligated into the pSilencer3.1-H1 neo plasmid, referred to hereafter as pSilencer3.1-PS2 and pSilencer3.1-NC, respectively. U251 cells were transfected with these constructs, and individual clones were selected by G418. Suppression of PS2 expression was detected by Western blot. Three pSilencer3.1-PS2-transfected clones (U251-S, U251-S1, and U251-S2) showed a significant decrease in PS2 expression, compared with the pSilencer3.1-NC-transfected clones (U251-NC) (Fig. 3A). We observed a similar result in another pSilencer3.1-PS2-transfected clone, U251-Smix, which was a pool of stably transfected U251 cells (Fig. 3B). Of these 3 individual clones, U251-S was used for further investigation. To eliminate the possibility that results could be attributable to a clonal artifact, the pooled clone U251-Smix was also used in the subsequent experiments. The inhibition of PS2 expression in U251-S cells was confirmed by Western blot, whereas there was no obvious difference between U251-NC and parental U251 cells (P >.05) (Fig. 3C). These results demonstrate that pSilencer3.1-PS2 successfully reduced PS2 expression in human glioma U251 cells.
Fig. 3.
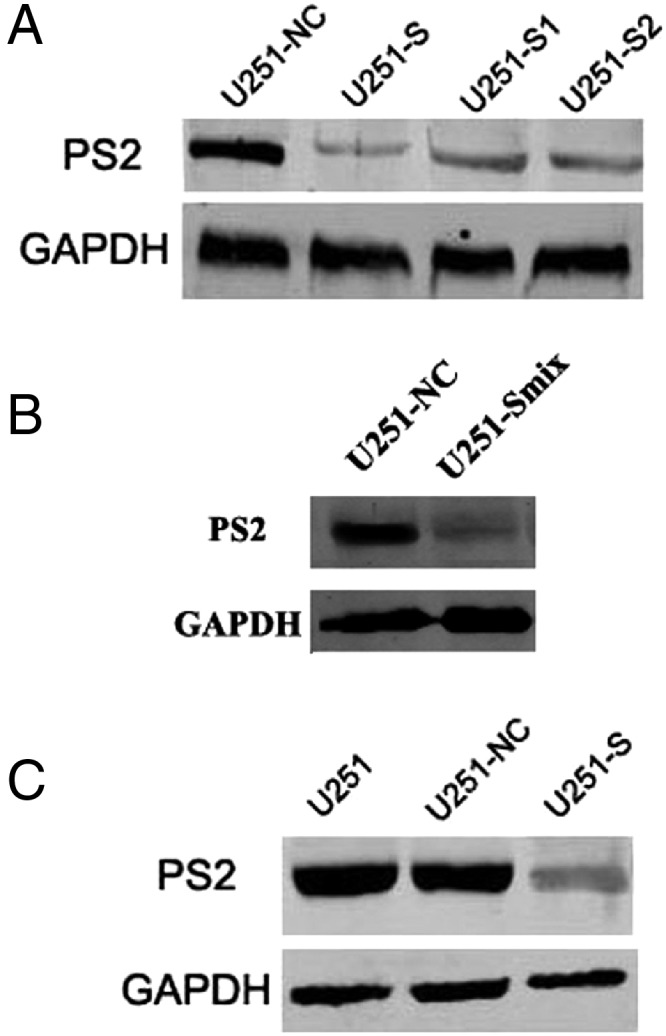
RNA interference reduced expression of PS2 in U251 cells. (A) Decrease of PS2 protein expression in clones U251-S, U251-S1, and U251-S2 compared with control-transfected clone. (B) Decrease of PS2 protein expression in clone U251-Smix. Clonal lines of stably transfected cells were pooled together to produce a mixture clone, U251-Smix. (C) Decrease of PS2 protein expression in clone U251-S was confirmed by Western blot. U251: parental cells; U251-NC: U251 cells transfected with vector pSilencer3.1-NC; U251-S, U251-S1, U251-S2, and U251-Smix: different U251 clones transfected with pSilencer3.1-PS2.
Suppression of PS2 Inhibited Growth of U251 Cells In Vitro
To investigate the possible role of PS2 in U251 cell growth, we first analyzed cellular growth of U251-S cells and U251-Smix clones. The cell growth curves showed that the growth of U251-S cells and U251-Smix was notably inhibited in a time-dependent manner (Fig. 4A). The inhibitory rates were (calculated as described in Materials and Methods) were 60.4% ± 3.5% (P < .05) for U251-S cells and 56.8% ± 3.2% (P < .05) for U251-Smix at day 6. However, there was no significant difference in cell growth between U251-NC and the parental U251 cells (P > .05; Fig. 4A). To explore the potential contribution of PS2 shRNA to cell cycle progression, we used flow cytometry to evaluate the cell cycle distribution. The results showed that U251-S cells accumulated in G0/G1 phase (78.1% ± 3.5%; P < .05), but the cell numbers in G2/M phase were reduced sharply (10.4% ± 2.3%; P < .05). A similar cell distribution was observed for U251-Smix cells, with 74.3% ± 3.2% cells in G0/G1 phase and 4.59% ± 2.2% cells in G2/M phase. There was no obvious difference in cell cycle distribution between U251-NC and parental U251 cells (P > .05) (Fig. 4B and C). To address whether the decreased cell number was attributable to apoptosis induced by PS2-targeting shRNA, we compared apoptosis in U251-S and U251-Smix cells with apoptosis occurring in U251 control cells. We found a significant difference in the apoptosis rate in U251-S and U251-Smix, compared with control cells (Fig. 4D). According to flow cytometry results, PS2 shRNA induced apoptosis in 40.3% ± 3.8% of the U251-S cells and 32.4% ± 3.2% of the U251-Smix cells. We therefore concluded that PS2 might participate in the regulation of the cell cycle, the inhibited proliferation, and the induced apoptosis in U251 cells with PS2 expression reduced by RNAi.
Fig. 4.

RNAi-mediated suppression of PS2-inhibited growth of U251 cells in vitro. (A) Cell growth curve. Parental U251 cells, vector control U251 cells (U251-NC), and PS2 shRNA-stably transfected cells (U251-S and U251-Smix) were seeded onto 96-well tissue culture plates. Cell viability was measured using the MTT assay. Cell growth curves were determined by reading the absorbance at 490 nm on a multiscanner reader. (B) Cell cycle distribution. Parental U251 cells, vector control U251 cells (U251-NC), and PS2 shRNA-stably transfected cells (U251-S and U251-Smix) were stained with propidium iodide. The DNA content of the cells was analyzed by FACS. (C) The differences in cell cycle distribution of parental U251 cells, vector control U251 cells (U251-NC), and PS2 shRNA-stably transfected cells (U251-S and U251-Smix). Cells in G0/G1, S, and G2/M of the cell cycle were sorted based on DNA content. Cells (10 000) were sorted using flow cytometry and analyzed. (D) Cell apoptosis in shRNA-stably transfected U251 cells. Parental U251 cells, vector control U251 cells (U251-NC), and PS2 shRNA-stably cells (U251-S and U251-Smix) were stained by annexinV/7-ADD and analyzed by FACS to detect apoptosis. Values are shown as the mean ± SD of 3 independent experiments.
Loss of PS2 Expression Abrogated Anchorage-Dependent and Anchorage-Independent Colony Formation in U251 Cells
To determine the effects of PS2 suppression on tumorigenic processes in U251 cells, we examined the anchorage-dependent plate colony formation of U251-S cells and the control U251 cells. Compared with control cells, 2.1-fold fewer clones were observed in U251-S cells (Fig. 5A and B). We then evaluated the effect of PS2 suppression on anchorage-independent colony formation in soft agar as an additional assessment of tumorigenicity in vitro. Suppression of PS2 significantly impaired anchorage-independent colony growth, with both colony number and size significantly decreased. In contrast, no loss of colony-forming ability was observed in U251-NC cells, compared with parental U251cells (Fig. 5C and D). Collectively, our results indicate that inhibition of PS2 expression impaired the colony-formation ability of U251 cells in vitro.
Fig. 5.

Suppression of PS2 suppressed both anchorage-dependent and anchorage-independent colony formation. (A) Equal numbers of U251-S cells, parental U251 cells, and vector control U251 cells (U251-NC) were seeded onto 60-mm dishes. After 14 days, the cells were fixed and stained with Giemsa. (B) The average number of colonies formed in 3 independent experiments was quantified. (C and D) Equal numbers of U251-S cells, parental U251 cells, and vector control U251 cells (U251-NC) were plated in 0.3% soft agar and cultured for 14 days. Colony formation was photographed under a microscope (C) and scored (D). Colony numbers are shown as mean ± SD for 3 independent experiments. *P < .05, **P < .01 (compared with U251 and U251-NC control cells).
Effects of PS2 Suppression on Migration and Invasion of U251 Cells
Wound healing involves a number of processes, including migration and the establishment of cell polarity. We performed a wound-healing assay to study whether shRNA-mediated inhibition of PS2 could influence migration of U251 cells. As shown in Fig. 6A, the migration of U251-S cells notably decreased. In U251, U251-NC, and U251-S cells, the migration distances were 678 ± 28 μm, 691 ± 31 μm, and 464 ± 23 μm, respectively. There were no obvious differences in migration between U251-NC and parental U251 cells (Fig. 6B).
Fig. 6.

Suppression of PS2-modulated migration and invasion in U251 cells. (A) Effect of suppression of PS2 on cell migration. Monolayers of U251, U251-NC, and U251-S cells were mechanically wounded with a pipette tip. Repair of the lesion by cell migration was photographed 24 h later. (B) Quantification of cell migration. The distance of cell migration was calculated as described in Materials and Methods. Data were obtained from 3 independent experiments and are presented as mean ± SD. (C) PS2 shRNA diminished cell invasion of U251 cells. Each of the indicated U251 cell types was assayed for cell invasion using Transwell tissue culture dishes (8-μm pore size). (D) The mean cell counts of invading cells from 8 high-power fields. Values presented are the mean ± SD obtained from 3 independent experiments. *P < .05, **P < .01 (compared with U251 and U251-NC control cells).
Because tumor cell invasion is an important feature of glioma cells, we next examined whether stable shRNA expression decreases cell invasion. Results for the Matrigel invasion assay showed that inhibition of PS2 expression significantly suppressed the invasiveness of U251 cells (Fig. 6C). The mean count of cells that crossed a Matrigel-coated membrane in 8 high-power fields was 86.0 ± 6.4 for the parental U251 group, 82.0 ± 7.8 for the U251-NC group, and 36.0 ± 3.9 for the U251-S group (Fig. 6D). The number of invaded cells was notably lower for U251-S cells (P < .01) than for the control groups, between which no obvious difference in invasion was observed (P > .05). These findings demonstrate that inhibition of PS2 suppresses cell migration and invasion.
Stable Suppression of PS2 Inhibited Tumor Growth In Vivo
To test the effect of decreased PS2 levels in glioma cells in vivo, we extended our study to evaluate the effect of PS2 RNAi on tumor growth. Parental U251, U251-NC, and U251-S cells were injected into nude mice (Fig. 7A). Figure 7B shows the time course of the growth of tumors initiated by different U251 transfectants. By day 24, mice injected with U251-S cells showed a statistically significant decrease in mean tumor size, compared with all other groups (Fig. 7C). Tumors in each mouse were removed on day 24 and weighed. PS2 RNAi significantly decreased the solid tumor mass, compared with the control groups (Fig 7D). These data demonstrate that inhibition of PS2 expression suppresses tumor formation in vivo.
Fig. 7.

PS2 shRNA decreased tumorigenicity in nude mice. (A) BALB/c nu/nu mice were injected subcutaneously with 1 × 107 of the indicated control cells or U251-S cells. Representative tumor formation was photographed 24 days after injection. (B) Tumor size was determined by measuring the tumor volume every 4 days from day 4 through day 24 after injection. (C) Tumors were excised and photographed 24 days after injection. (D) Tumor weights in mice 24 days after injection. Values presented as mean ± SD were obtained from 3 independent experiments. **P < .01 compared with U251 and U251-NC control cells.
Nrg1/ErbB3 Signaling Correlates with PS2 Expression in U251 Cells
To further explore the molecular mechanisms of growth inhibition caused by PS2 suppression, we analyzed the effect of PS2 on the expression of several genes, including Nrg1, ErbB3, and ErbB4, which play an important role in glioma cell growth and invasion. Western blot results demonstrated decreased expression levels of Nrg1, ErbB3, and ErbB4 in U251-S cells, compared with control cells (Fig. 8A). To investigate whether Nrg1 protein expression level is consistent with the expression level of PS2, we measured Nrg1 protein expression in human glioma specimens using Western blot. We found that the level of Nrg1 was significantly higher in the glioma group than in the control group (Supplementary material, Fig. S1A). The level of Nrg1 expression was also correlated with increasing tumor grade in brain tumors (Supplementary material, Fig. S1B). In addition, we tested the effects of treatment of U251-S cells with the ErbB3 and EerB4 ligand Nrg1. To investigate the possible role of Nrg1 in U251-S cell growth, we first analyzed cellular growth of U251-NC, U251-S, U251-S-Nrg1, and the parental U251 cells. The cell growth curves showed that cell growth did not differ significantly among U251-NC, U251-S-Nrg1, and parental cells (Fig. 8C). Flow cytometry was used to evaluate the potential role of Nrg1 on cell cycle distribution. The results showed that U251-S-Nrg1 cells accumulated in G0/G1 phase (61.4% ± 3.2%) and that the cell numbers in G2/M phase were reduced sharply (21.5% ± 2.7%). There was no obvious difference in cell cycle distribution among U251-NC, U251-S-Nrg1, and the parental U251 cells (P > .05; Fig. 8D and E). Similar results were found for cell invasion. The mean count of cells that crossed a Matrigel-coated membrane in 8 high-power fields was 78.0 ± 6.3 for the parental U251 group, 75.0 ± 6.1 for the U251-NC group, 29.0 ± 2.7 for the U251-S group, and 56.0 ± 4.1 for the U251-S-Nrg1 group (Fig. 8F and G). The number of the invaded cells was greater for U251-S-Nrg1 cells than for U251-S cells (P < .05). No obvious difference in invasion was observed among U251-NC, U251-S-Nrg1, and parental U251 cells (P > .05). Furthermore, the interaction between PS2 and Nrg1 was investigated in the parental U251 cells by coimmunoprecipitation experiments. As shown in Fig. 8B, PS2 was coimmunoprecipitated with Nrg1 when anti-Nrg1 was used to pull down Nrg1 protein and its associated proteins. In contrast, no PS2 signal was observed in cell lysates when we used the reference serum IgG. In addition, we examined whether the decrease in the Nrg1 protein level for U251-S cells correlated with a decrease in its stability (Supplementary Material). After 1 h of cycloheximide treatment of U251-NC and U251-S cells, protein expression levels of Nrg1 changed. However, a significant decrease in Nrg1 protein expression was observed after 3 h of treatment (Supplementary material, Fig. S2). Comparison of Nrg1 protein stability in U251-NC cells with the protein stability in U251-S cells, which was stable for at least 3 h, suggests that PS2 suppression by RNAi in U251 cells can have deleterious effects on Nrg1 protein stability. These data may implicate Nrg-1 in the mechanism of involvement of PS2 in glioma cell growth and invasion.
Fig. 8.

PS2 shRNA inhibits cell growth and invasion involved in the regulation of Nrg1/ErbB signaling. (A) A decrease of Nrg1, ErbB3, and ErbB4 protein expressions in clone U251-S cells. (B) Cell lysates of U251 cells were subjected to immunoprecipitation using anti-Nrg1 antibody or control IgG. PS2 protein was detected from immunoprecipitates by Western blotting. (C) Cell growth curve. Parental U251 cells, vector control U251 cells (U251-NC), PS2 shRNA-stably transfected cells (U251-S), and Nrg1-treated PS2 shRNA-stably transfected cells (U251-S-Nrg1) were seeded onto 96-well tissue culture plates. Cell viability was measured using the MTT assay. Cell growth curves were determined by reading the absorbance at 490 nm on a multiscanner reader. (D) Cell cycle distribution. Parental U251 cells, vector control U251 cells (U251-NC), PS2 shRNA-stably transfected cells (U251-S), and Nrg1-treated PS2 shRNA-stably transfected cells (U251-S-Nrg1) were stained with propidium iodide. The DNA content of the cells was analyzed by FACS. (E) The differences in cell cycle distribution of parental U251 cells, vector control U251 cells (U251-NC), PS2 shRNA-stably transfected cells (U251-S), and Nrg1-treated PS2 shRNA-stably transfected cells (U251-S-Nrg1). (F) Nrg1 treatment induced cell invasion of U251-S cells. Each of the indicated U251 cell types was assayed for cell invasion using Transwell tissue culture dishes (8-μm pore size). (G) The average cell counts of invading cells from 8 high-power fields. Values are means ± SD from 3 independent experiments. **P < .01 compared with U251 and U251-NC control cells. *P < .05 compared with U251-S-Nrg1 cells.
Discussion
In the current study, we investigated the role of PS2 in cellular growth inhibition and cellular malignant progression of glioma cells by suppressing PS2 with a specific RNAi in U251 cells (Fig. 4). We demonstrated that suppression of PS2 inhibited U251 glioma cell growth and invasion. We also investigated the molecular mechanism of the effect of PS2 on cell growth and invasion.
Our data demonstrated that PS2 was widely distributed in glioma tissue, with very low expression in adjacent normal brain tissue (Fig. 1). Demethylation of the promoter region of oncogenes may be associated with transcriptional activation and tumor progression. Fuso et al.28 showed that PS1 (PSEN1) demethylation could be responsible for gene overexpression. In this study, we analyzed whether increased expression of PS2 in glioma tissue is attributable to promoter demethylation. Methylation-specific PCR results demonstrated that increased PS2 expression in glioma tissue is linked to its promoter DNA demethylation. This demethylation was correlated with high PS2 transcripts and protein levels in glioma tissue, compared with adjacent normal brain tissue. Therefore, PS2 expression and its promoter demethylation should be of potential value in predicting glioma carcinogenesis. Additional studies on a much larger scale are needed to draw more complete conclusions.
PS2 is a ubiquitous intramembrane protein that helps process proteins that transmit chemical signals from the cell membrane into the nucleus. Once in the nucleus, these signals turn on genes that are important for cell growth and maturation. In our study, we found that specifically reducing PS2 by RNAi negatively affected tumor cell growth in vitro (Fig. 4A). PS2 is best known for its role in processing amyloid precursor protein. Research suggests that PS2 works together with other enzymes to cut amyloid precursor protein into smaller segments (peptides).29 One of these peptides, soluble amyloid precursor protein (sAPP), shows similarities with growth factors and increases the in vitro proliferation of embryonic neural stem cells.30 Furthermore, PS/γ-secretase mediates Notch signaling and inhibits keratinocyte proliferation by promoting differentiation.31 However, Notch 1 and Notch 2 have been shown to exert opposing actions on embryonal brain tumor growth32 and have contrasting roles in breast cancer tumor differentiation.33 Recent findings suggest that 2 PS2 mutations found in breast tumors increase cell proliferation and reduce Notch signaling, indicating that they act through a loss-of-function mechanism.24 More recently, Frampton et al.25 demonstrated that the antiproliferative effects of anandamide are associated with the PS1-dependent proteolytic cleavage of Notch 1 and that the growth-promoting effects of 2-AG are associated with the PS2-dependent activation of Notch 2 signaling. These studies suggest that PS2 is involved in cell proliferation. Moreover, previous study showed that the PS2 protein is also located in the nuclear membrane, interphase kinetochores, and centrosomes and that its function is related to mitosis and chromosome organization/segregation.34 Zampese et al.35 showed that PS2 modulates endoplasmic reticulum–mitochondria interactions and Ca2+ cross-talk. Of interest, Behbahani et al.36 showed that mitochondrial functions are reduced significantly in cells from PS2-knockout animals. This energetic deficit could depend on the reduction of endoplasmic reticulum–mitochondria Ca2+ transfer, as elegantly demonstrated recently by Cardenas et al.37 Undoubtedly, there is cross-talk between the cell cycle transitions and energetic metabolism.38 For example, a decrease in ATP following mitochondrial dysfunction may result in cell cycle arrest. In addition, mitochondrial dysfunction is directly responsible for the pro-apoptotic effects on mammalian cells.39–41 As shown in Fig. 4D, we demonstrated that suppression of PS2 induced U251 cell apoptosis.
Glioma cell invasion and migration are characteristic features that distinguish these tumor cells from normal cells. We therefore propose that PS2 might contribute to these tumor-specific behaviors in glioma cells. Of interest, cell migration and invasion are inhibited in PS2-depleted glioma cells (Fig. 6). Our results also show that suppression of PS2 affected the transformed morphology of the glioma cells. Inhibition of both anchorage-dependent and anchorage-independent colony growth was observed clearly in U251-S cells, compared with control cells, suggesting that PS2 could enhance mitogenic or survival signals (Fig. 5).
These results, together with the high levels of PS2 expression found in glioma tissue and glioma cells (Fig. 1), demonstrate the function of PS2 in cancer development. Because we showed that PS2 is important in different malignant processes involved in glioma progression in vitro, we further investigated the ability of PS2 to affect the growth of glioma tumor xenografts in vivo. Suppression of PS2 did impair the growth of glioma tumor xenografts. Because a decrease in PS2 specifically inhibited growth in vitro and in vivo (Figs. 4 and 7), the inhibition of glioma growth, resulting from suppression of PS2 in vivo, was the result of inhibited proliferation and induced apoptosis. Next, we sought to define the potential molecular mechanisms of the inhibition of glioma cell growth and invasion caused by suppression of PS2. We found reduced expression of Nrg1, ErbB3, and ErbB4 after suppression of PS2 in U251 cells. Nrg1 transcripts and protein expression have been detected in human gliomas42 and in glial cells and neurons throughout the brain.43–45 Nrg-1 acts as an autocrine or paracrine signal, and there is an Nrg1/ErbB3 autocrine growth-promoting loop in a subset of primary ovarian cancers and ovarian cancer cell lines.46 Similarly, Ritch et al. suggested that a functional Nrg1/ErbB receptor autocrine or paracrine signaling network plays a significant role in growth modulation of human astrocytic glioma cells.47 Our studies indicate that Nrg-1 was sufficient to effectively promote the growth of U251-S cells (Fig. 8). In addition, Nrg-1 treatment markedly reduced PS2 RNAi-mediated G1 phase cell cycle arrest in U251 cells. Nrg-1 also plays an important modulatory role in glioma cell invasion.10 We demonstrated that Nrg-1 treatment could significantly increase the invasion of U251-S cells. In addition, we were able to coimmunoprecipitate PS2 and Nrg-1 in U251 cells (Fig. 8). A comparison of the expressions of Nrg1 and PS2 demonstrated that Nrg1 expression in human glioma specimens was similarly associated with PS2 expression (Supplementary material, Fig. S1). Cycloheximide treatment revealed that PS2 binding Nrg1 could stabilize Nrg1 expression in U251 cells (Supplementary material, Fig. S2). Taken together, these results indicate that PS2 suppression may alter Nrg1/ErbB signaling. However, the precise mechanism still needs further study.
Because PS2 is increased in glioma tumors and that depletion of PS2 in U251 cells suppressed multiple tumor-specific behaviors, PS2 could be an intriguing candidate as a molecular target in glioma therapy. In this study, we specifically suppressed expression of PS2 by shRNA and consequently impaired all the malignant processes involved in glioma progression. These results provide the biological basis for inhibition of PS2 using RNAi as a novel therapeutic approach for glioma.
In conclusion, we showed that targeted PS2 inhibition by specific RNAi prevents cell growth and suppresses multiple tumorigenic properties of glioma cells both in vitro and in vivo. The inhibition of cell growth and invasion caused by PS2 RNAi is associated with Nrg1/ErbB signaling. These results indicate great potential for the RNAi-mediated suppression of PS2 in therapeutic applications for the treatment of glioma.
Supplementary Material
Funding
This work was supported by the National Natural Science Foundation of China (grant 30772221). The funding organization had no influence on the contents or on any other aspect of this article.
Supplementary Material
Acknowledgments
We thank the patients and their families, for their participation in this study, and Dr. Xiao-Ying Liu, for her expert help with the U251 cell line. B.L., L.W., L.-L.S., and M.-Z.S. contributed equally to this work.
Conflict of interest statement. None declared.
References
- 1.Chang SM, Parney IF, Huang W, et al. Patterns of care for adults with newly diagnosed malignant glioma. JAMA. 2005;293:557–564. doi: 10.1001/jama.293.5.557. doi:10.1001/jama.293.5.557. [DOI] [PubMed] [Google Scholar]
- 2.Frappaz D, Chinot O, Bataillard A, et al. Summary version of the standards, options and recommendations for the management of adult patients with intracranial glioma (2002) Br J Cancer. 2003;89(Suppl 1):S73–S83. doi: 10.1038/sj.bjc.6601087. doi:10.1038/sj.bjc.6601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parney IF, Chang SM. Current chemotherapy for glioblastoma. Cancer J. 2003;9:149–156. doi: 10.1097/00130404-200305000-00003. doi:10.1097/00130404-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Laperriere N, Zuraw L, Cairncross G. Radiotherapy for newly diagnosed malignant glioma in adults: a systematic review. Radiother Oncol. 2002;64:259–273. doi: 10.1016/s0167-8140(02)00078-6. doi:10.1016/S0167-8140(02)00078-6. [DOI] [PubMed] [Google Scholar]
- 5.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. doi:10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 6.Riese DJ, 2nd., Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. doi:10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Hunter T. The role of tyrosine phosphorylation in cell growth and disease. Harvey Lect. 1998;94:81–119. [PubMed] [Google Scholar]
- 8.Mustelin T, Hunter T. Meeting at mitosis: cell cycle-specific regulation of c-Src by RPTPα. Sci STKE. 2002;2002:pe3. doi: 10.1126/stke.2002.115.pe3. [DOI] [PubMed] [Google Scholar]
- 9.Carraway KL, 3rd., Burden SJ. Neuregulins and their receptors. Curr Opin Neurobiol. 1995;5:606–612. doi: 10.1016/0959-4388(95)80065-4. doi:10.1016/0959-4388(95)80065-4. [DOI] [PubMed] [Google Scholar]
- 10.Ritch PA, Carroll SL, Sontheimer H. Neuregulin-1 enhances motility and migration of human astrocytic glioma cells. The Journal of biological chemistry. 2003;278:20971–20978. doi: 10.1074/jbc.M213074200. doi:10.1074/jbc.M213074200. [DOI] [PubMed] [Google Scholar]
- 11.Koo EH, Kopan R. Potential role of presenilin-regulated signaling pathways in sporadic neurodegeneration. Nat Med. 2004;10(Suppl):S26–33. doi: 10.1038/nm1065. doi:10.1038/nm1065. [DOI] [PubMed] [Google Scholar]
- 12.Campion D, Flaman JM, Brice A, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer's disease. Hum Mol Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. doi:10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 13.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. doi:10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 14.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. doi:10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 15.Wakabayashi T, De Strooper B. Presenilins: members of the γ-secretase quartets, but part-time soloists too. Physiology. 2008;23:194–204. doi: 10.1152/physiol.00009.2008. doi:10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- 16.Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. doi:10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 17.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. doi:10.1016/S0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 18.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA. 1999;96:11872–11877. doi: 10.1073/pnas.96.21.11872. doi:10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soriano S, Kang DE, Fu M, et al. Presenilin 1 negatively regulates β-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of β-amyloid precursor protein and Notch processing. J Cell Biol. 2001;152:785–794. doi: 10.1083/jcb.152.4.785. doi:10.1083/jcb.152.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia X, Qian S, Soriano S, et al. Loss of presenilin 1 is associated with enhanced β-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. doi:10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janicki SM, Stabler SM, Monteiro MJ. Familial Alzheimer's disease presenilin-1 mutants potentiate cell cycle arrest. Neurobiol Aging. 2000;21:829–836. doi: 10.1016/s0197-4580(00)00222-0. doi:10.1016/S0197-4580(00)00222-0. [DOI] [PubMed] [Google Scholar]
- 22.Alves da Costa C, Paitel E, Mattson MP, et al. Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc Natl Acad Sci USA. 2002;99:4043–4048. doi: 10.1073/pnas.062059899. doi:10.1073/pnas.062059899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamliel A, Teicher C, Hartmann T, Beyreuther K, Stein R. Overexpression of wild-type presenilin 2 or its familial Alzheimer's disease-associated mutant does not induce or increase susceptibility to apoptosis in different cell lines. Neuroscience. 2003;117:19–28. doi: 10.1016/s0306-4522(02)00830-8. doi:10.1016/S0306-4522(02)00830-8. [DOI] [PubMed] [Google Scholar]
- 24.To MD, Gokgoz N, Doyle TG, et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25:3557–3564. doi: 10.1038/sj.onc.1209397. doi:10.1038/sj.onc.1209397. [DOI] [PubMed] [Google Scholar]
- 25.Frampton G, Coufal M, Li H, Ramirez J, DeMorrow S. Opposing actions of endocannabinoids on cholangiocarcinoma growth is via the differential activation of Notch signaling. Exp Cell Res. 2010;316:1465–1478. doi: 10.1016/j.yexcr.2010.03.017. doi:10.1016/j.yexcr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Wang T, Wang L, Wang C, Zhang H, Gao GD. Up-regulation of major vault protein in the frontal cortex of patients with intractable frontal lobe epilepsy. J Neurol Sci. 2011;308:88–93. doi: 10.1016/j.jns.2011.05.047. doi:10.1016/j.jns.2011.05.047. [DOI] [PubMed] [Google Scholar]
- 27.Jin N, Chen W, Blazar BR, Ramakrishnan S, Vallera DA. Gene therapy of murine solid tumors with T cells transduced with a retroviral vascular endothelial growth factor–immunotoxin target gene. Hum Gene Ther. 2002;13:497–508. doi: 10.1089/10430340252809793. doi:10.1089/10430340252809793. [DOI] [PubMed] [Google Scholar]
- 28.Fuso A, Nicolia V, Cavallaro RA, Scarpa S. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer's disease models. J Nutr Biochem. 2011;22:242–251. doi: 10.1016/j.jnutbio.2010.01.010. doi:10.1016/j.jnutbio.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Palmert MR, Podlisny MB, Witker DS, et al. The β-amyloid protein precursor of Alzheimer disease has soluble derivatives found in human brain and cerebrospinal fluid. Proc Natl Acad Sci USA. 1989;86:6338–6342. doi: 10.1073/pnas.86.16.6338. doi:10.1073/pnas.86.16.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caille I, Allinquant B, Dupont E, et al. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development. 2004;131:2173–2181. doi: 10.1242/dev.01103. doi:10.1242/dev.01103. [DOI] [PubMed] [Google Scholar]
- 31.Lefort K, Dotto GP. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol. 2004;14:374–386. doi: 10.1016/j.semcancer.2004.04.017. doi:10.1016/j.semcancer.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 32.Fan X, Mikolaenko I, Elhassan I, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64:7787–7793. doi: 10.1158/0008-5472.CAN-04-1446. doi:10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- 33.Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14:779–786. doi: 10.3892/ijmm.14.5.779. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. doi:10.1016/S0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- 35.Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci USA. 2011;108:2777–2782. doi: 10.1073/pnas.1100735108. doi:10.1073/pnas.1100735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Behbahani H, Shabalina IG, Wiehager B, et al. Differential role of presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J Neurosci Res. 2006;84:891–902. doi: 10.1002/jnr.20990. doi:10.1002/jnr.20990. [DOI] [PubMed] [Google Scholar]
- 37.Cardenas C, Miller RA, Smith I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. doi:10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguilar V, Fajas L. Cycling through metabolism. EMBO Mol Med. 2010;2:338–348. doi: 10.1002/emmm.201000089. doi:10.1002/emmm.201000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. doi:10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 40.Ferri KF, Kroemer G. Mitochondria–the suicide organelles. Bioessays. 2001;23:111–115. doi: 10.1002/1521-1878(200102)23:2<111::AID-BIES1016>3.0.CO;2-Y. doi:10.1002/1521-1878(200102)23:2<111::AID-BIES1016>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 41.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. doi:10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 42.Westphal M, Meima L, Szonyi E, et al. Heregulins and the ErbB-2/3/4 receptors in gliomas. J Neurooncol. 1997;35:335–346. doi: 10.1023/a:1005837122181. doi:10.1023/A:1005837122181. [DOI] [PubMed] [Google Scholar]
- 43.Pollock GS, Franceschini IA, Graham G, Marchionni MA, Barnett SC. Neuregulin is a mitogen and survival factor for olfactory bulb ensheathing cells and an isoform is produced by astrocytes. Eur J Neurosci. 1999;11:769–780. doi: 10.1046/j.1460-9568.1999.00484.x. doi:10.1046/j.1460-9568.1999.00484.x. [DOI] [PubMed] [Google Scholar]
- 44.Raabe TD, Clive DR, Wen D, DeVries GH. Neonatal oligodendrocytes contain and secrete neuregulins in vitro. J Neurochem. 1997;69:1859–1863. doi: 10.1046/j.1471-4159.1997.69051859.x. doi:10.1046/j.1471-4159.1997.69051859.x. [DOI] [PubMed] [Google Scholar]
- 45.Pinkas-Kramarski R, Eilam R, Spiegler O, et al. Brain neurons and glial cells express Neu differentiation factor/heregulin: a survival factor for astrocytes. Proc Natl Acad Sci USA. 1994;91:9387–9391. doi: 10.1073/pnas.91.20.9387. doi:10.1073/pnas.91.20.9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheng Q, Liu X, Fleming E, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010;17:298–310. doi: 10.1016/j.ccr.2009.12.047. doi:10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritch PS, Carroll SL, Sontheimer H. Neuregulin-1 enhances survival of human astrocytic glioma cells. Glia. 2005;51:217–228. doi: 10.1002/glia.20197. doi:10.1002/glia.20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.