

Abstract
The ubiquitin/proteasome pathway (UPP) is the major proteolytic quality control system in cells and involves tightly regulated removal of unwanted proteins and retention of those that are essential. In addition to its function in normal protein degradation, the UPP plays a critical role in the quality control process by degrading mutated or abnormally folded proteins. The proteolytic component of the UPP is a multiprotein complex known as the proteasome. Many factors, including the aging process, can cause proteasome impairment leading to formation of abnormal ubiquitin-protein aggregates that are found in most progressive neurodegenerative diseases, including Alzheimer's and Parkinson's diseases. In this chapter, we describe protocols to measure proteasome activity, evaluate its state of assembly, and assess the accumulation and aggregation of ubiquitinated proteins in two types of neuronal cultures: human neuroblastoma cells and rat primary cortical cultures. These protocols can be used with different types of neuronal cultures to estimate proteasome activity and the levels and aggregation of ubiquitinated proteins. In addition, they can be used to identify compounds potentially capable of preventing a decline in proteasome activity and formation of ubiquitin-protein aggregates associated with neurodegeneration.
Keywords: Proteasome activity, Proteasome assembly, Ubiquitinated proteins, Protein aggregates, Protein turnover, Neurodegeneration, Neuronal cell cultures
1. Introduction
In a wide variety of neurodegenerative disorders, such as Alzheimer's, Parkinson's, and Huntington's diseases as well as amyotrophic lateral sclerosis, aggregates of ubiquitinated proteins are detected in neuronal inclusions (reviewed in ref. (1)) indicating that the ubiquitin/proteasome pathway (UPP) may be deficient. The accumulation of ubiquitinated proteins in inclusions is thought to reflect a failure in proteasome activity although the mechanisms leading to inclusion formation remain unclear. There is a rising interest in the proteasome as a therapeutic target to prevent protein accumulation/aggregation and therefore delay/treat neurodegeneration in these progressive disorders (reviewed in ref. (2)).
In this chapter, we describe three different methods to measure proteasome activity in two types of neuronal cultures: (a) human neuroblastoma SK-N-SH cells and (b) rat E18 primary cortical neuronal cultures. First, the “in-gel” assay distinguishes three forms of the proteasome: the 26S with two and one cap as well as the 20S proteasome. It semiquantitatively assesses the chymotrypsin-like activity, the levels of each of the three proteasome forms and proteasome assembly. Second, the glycerol gradient centrifugation provides a means to quantitatively measure the individual activities of 26S and 20S proteasomes. Finally, the total cell lysate assay is a quick but somewhat unspecific way to measure proteasome activity without differentiating between 26S and 20S proteasomes or other enzymes that cleave the substrates. Besides the three assays to measure proteasome activity, we describe how to assess the accumulation of ubiquitinated proteins by western blot analysis, and how to detect ubiquitin-protein aggregates using the filter trap assay and immunofluorescence. To promote the accumulation of ubiquitinated proteins, we treated neuronal cultures with the irreversible proteasome inhibitor epoxomicin (3) or with the neurotoxic product of inflammation prostaglandin J2 (PGJ2) (4, 5). All of these protocols can be used to evaluate changes in proteasome activity and levels as well as accumulation/aggregation of ubiquitinated proteins under conditions that lead to neurodegeneration or prevent it.
2. Materials
2.1. Common Reagents/Equipment
Phosphate-buffered saline (PBS): Prepare 1× PBS using PBS tablets dissolved in ultrapure water. Store at 4°C.
Bradford assay for protein concentration.
Bicinchoninic acid (BCA) assay for protein concentration.
Bromophenol blue solution: 250 mg bromophenol blue dissolved in 10 mL buffer A (refer to Subheading 2.3 below). Centrifuge in a microcentrifuge at maximum speed for 5 min. Aliquot supernatant and store at 4°C.
30% acrylamide/Bis solution, 29:1. Store at 4°C.
Transfer buffer: 25 mM Trizma base; 192 mM glycine, pH 8.3; 15% (v/v) methanol. Store at room temperature.
Antibody dilution buffer: SuperBlock blocking buffer (Thermo Scientific, Rockford, IL).
ECL stock solutions: 250 mM luminol in dimethyl sulfoxide (DMSO), store at −20°C; 90 mM p-Coumaric acid in DMSO, store at −20°C. ECL solution A: 100 mM Tris–HCl, pH 8.5; 2.5 mM luminol; 0.4 mM p-Coumaric acid. ECL solution B: 100 mM Tris–HCl, pH 8.5; 0.02% H2O2. ECL reagent: Mix solution A and B (1:1) just prior to use.
Repeater plus pipettor.
X-ray film and film developer.
Heidolph type RZR 50 benchtop homogenizer.
Spectrophotometer.
2.2. Cell Culture
SK-N-SH cells: These cells are an established human neuroblastoma cell line, which has a neuronal phenotype, synthesizes catecholamines, and expresses dopamine-β-hydroxylase. The cell line is derived from peripheral tissue (6). The cells are maintained as described in (7) and were obtained from ATCC. Culture media: Minimal essential media (MEM) with Eagle's salts containing 2 mM l-glutamine; 1 mM sodium pyruvate; 0.4% MEM vitamins; 0.4% MEM nonessential amino acids; 100 units/mL penicillin; 100 μg/mL streptomycin; and 5% normal fetal bovine serum (FBS) (see Note 1).
Rat E18 cortical neuronal cultures: These cultures are prepared from E18 embryos obtained from pregnant Sprague Dawley females following the methods described in (8). Culture media: Neurobasal media supplemented with B27 and 0.5 mM L-glutamax. Plating: Precoat 100-mm dishes with 50 μg/mL poly-d-lysine; add 6 million cells per dish.
Treatment: Treat SK-N-SH cells or neurons (DIC 8) with DMSO or PGJ2 (15 or 20 μM, PGJ2) for 16 or 24 h. PGJ2 stock: 3 or 4 mM in DMSO, respectively (see Notes 2 and 3).
2.3. In-Gel Assay
2.3.1. Cell Harvesting and Homogenization
Buffer A: 50 mM Tris–HCl, pH 7.4; 5 mM MgCl2; 10% glycerol (v/v). Store at 4°C.
ATP and DTT stock: 200 mM ATP (made in buffer A); 200 mM DTT (made in ultrapure water). Place on ice (see Note 4).
Buffer for harvesting cells and protein standards: Buffer A; 5 mM ATP; 1 mM DTT. Place on ice (see Note 4).
2.3.2. Native PAGE
Buffer B: 0.18 M boric acid; 0.18 M Trizma base; 5 mM MgCl2. Store at 4°C.
Gel buffer: Buffer B; 1 mM ATP; 1 mM DTT. For two minigels: 35 mL buffer B, 175 μL of 200 mM ATP, and 175 μL of 200 mM DTT. ATP and DTT prepared from stock (see Note 4).
Running buffer: Same as “gel buffer” and kept at 4°C until running gel.
Rhinohide polyacrylamide gel strengthener. Store at 4°C.
2.3.3. Proteasome Activity
Proteasome substrate stock solution: Succinyl-leucine-leucinevaline-tyrosine-(7-aminomethyl)-coumarine (Suc-LLVY-AMC). Make 20 mM stock reconstituted in DMSO, aliquot, and store at −80°C. This substrate is used to measure the chymotrypsin-like activity.
Proteasome activity mix: Buffer A; 1 mM ATP; 1 mM DTT. Just before measuring proteasome activity, add 300 μM of the proteasome substrate.
To make 15 mL of the proteasome activity mix: 14,625 μL buffer A; 75 μL 200 mM ATP; 75 μL 200 mM DTT; 225 μL 20 mM Suc-LLVY-AMC (see Note 4).
2.3.4. Proteasome Levels and Assembly
Blocking buffer: SuperBlock Blocking Buffer (Thermo Scientific, Rockford, IL).
Washing buffer: 0.5× PBS; 0.1% Tween 20.
PVDF membrane: Immobilon-P membrane (0.45 μm).
Primary antibodies: Rabbit anti-β5 (1:2,000); mouse anti-Rpt6 (1:2,000).
Secondary antibodies: Goat anti-rabbit IgG conjugated to horse radish peroxidase (1:10,000); goat anti-mouse IgG conjugated to horse radish peroxidase (1:10,000).
2.4. Glycerol Gradient Centrifugation
2.4.1. Cell Harvesting and Homogenization
Stock buffer: 0.1 M Tris–HCl, pH 7.5; 0.2 M ATP; 0.1 M DTT.
Harvesting buffer: 25 mM Tris–HCl, pH 7.5; 2 mM ATP; 1 mM DTT.
Epoxomicin reconstituted in DMSO to 1 mM. Aliquot and store at −80°C.
2.4.2. Glycerol Gradient Centrifugation and Fractionation
40% glycerol buffer: 25 mM Tris–HCl, pH 7.5; 2 mM ATP; 1 mM DTT; 40% glycerol (v/v).
10% glycerol buffer: 25 mM Tris–HCl, pH 7.5; 2 mM ATP; 1 mM DTT; 10% glycerol (v/v).
Gradient maker.
Beckman ultra-clear centrifuge tubes (14 × 89 mm).
Ultracentrifuge with SW41-rotor. Store the SW41-rotor and the Beckman tube buckets at 4°C.
Fraction recovery system for puncturing.
Fraction collector.
2.4.3. Chymotrypsin-Like Activity
Substrate: Suc-LLVY-AMC. Make 10 mM stock reconstituted in DMSO, aliquot, and store at −80°C.
Assay buffer: 0.05 M Tris–HCl, pH 7.5.
10% (w/v) trichloroacetic acid (TCA).
0.1% (w/v) sodium nitrite. Store in dark bottle.
0.5% (w/v) ammonium sulfamate.
0.05% (w/v) N-(1-naphthyl) ethylenediamine 2HCl (N-NEDA). Dissolve in 95% ethanol and store in dark bottle.
2.5. Peptidase Activities in Total Cell Lysate
2.5.1. Cell Harvesting and Homogenization
Stock buffer: 100 mM Tris–EDTA, pH 7.5; EDTA (see Note 5). Store at 4°C.
Harvesting buffer: 10 mM Tris–EDTA, pH 7.5. Store at 4°C.
Epoxomicin reconstituted in DMSO to 1 mM. Aliquot and store at −80°C.
2.5.2. Peptidase Activities
Peptidase substrates: Chymotrypsin-like activity, Suc-LLVY-AMC; trypsin-like activity, benzyloxycarbonyl-glycine-glycinearginine-β-naphthylamide (Z-GGR-bNA); caspase-like activity, benzyloxycarbonyl-leucine-leucine-glutamic acid-β-naphthyl-amide (Z-LLE-βNA). Make 10 mM stock solutions in DMSO, aliquot, and store at −20°C.
Assay buffers: Chymotrypsin-like activity, 0.05 M Tris–HCl, pH 7.5; trypsin- and caspase-like activities, 0.05 M Tris–HCl, pH 8.0.
10% (w/v) TCA.
0.1% (w/v) sodium nitrite. Store in dark bottle.
0.5% (w/v) ammonium sulfamate.
0.05% (w/v) N-NEDA. Dissolve in 95% ethanol and store in dark bottle.
2.6. SDS-PAGE and Western Blotting for Proteasome Subunits α4, β5, and Rpt6, for β-Actin, and for Ubiquitinated Proteins
2.6.1. SDS-PAGE
1% SDS lysis buffer: 10 mM Tris–EDTA, pH 7.5; 1% SDS. Store at room temperature.
Loading buffer: 50% glycerol; 50% β-mercaptoethanol; bromophenol blue. Store at room temperature.
Resolving buffer: 3 M Tris–HCl, pH 8.8. Store at 4°C.
Stacking buffer: 0.5 M Tris–HCl, pH 6.8. Store at 4°C.
Running buffer: 25 mM Trizma base; 192 mM glycine, pH 8.3; 0.1% (w/v) SDS. Store at room temperature.
Sonicator cell disruptor.
2.6.2. Western Blotting
Blocking buffer: 10 mM Tris–HCl, pH 7.3; 5% (w/v) nonfat dry milk; 10 mM NaCl; 0.1% (v/v) Tween 20 (see Note 4).
Washing buffer: 0.5× PBS; 0.1% Tween 20. Store at room temperature.
PVDF membrane: Immobilon-P membrane (0.45 μm).
Primary antibodies: Mouse monoclonal anti-α4 (1:500), rabbit polyclonal anti-β5 (1:2,000), and mouse anti-Rpt6 (1:2,000); mouse anti-β-actin (1:10,000); rabbit anti-ubiquitin antibody (1:1,500).
Secondary antibodies: Goat anti-rabbit IgG conjugated to horse radish peroxidase (1:10,000); goat anti-mouse IgG conjugated to horse radish peroxidase (1:10,000).
2.7. Filter Trap Assay for Ubiquitinated Protein Aggregates
2.7.1. Cell Harvesting and Homogenization
2× lysis buffer base: 40 mM Tris–HCl, pH 7.5; 274 mM NaCl; 2 mM EGTA; 20% glycerol (v/v). Store at 4°C.
Lysis (RIPA) buffer (5 mL): 2.5 mL of 2× lysis buffer base; 100 μL protease inhibitor cocktail; 1 mM sodium orthovanadate; 1 mM phenylmethylsulfonyl fluoride; 1 mM β-glycerophosphate; 2.5 mM sodium pyrophosphate; 50 mM sodium fluoride; 1% Nonidet P40 (see Note 4).
Normalizing buffer: 2% SDS; 10 mM Tris–EDTA, pH 7.5. Store at 4°C.
Disposable cell scrapers.
2.7.2. Filter Trap
Washing buffer: 0.1% SDS; 10 mM Tris–EDTA, pH 7.5. Store at 4°C.
Trans-Blot nitrocellulose membrane, 0.2 μm.
Qualitative filter paper.
96-well minifold Dot-blot system.
Vacuum source.
2.7.3. Western Blotting for Ubiquitinated Proteins
Blocking buffer: 10 mM Tris–HCl, pH 7.3; 5% (w/v) nonfat dry milk; 10 mM NaCl; 0.1% (v/v) Tween 20 (see Note 4).
Washing buffer: 0.5× PBS; 0.1% Tween 20. Store at room temperature.
Primary antibody: Rabbit anti-ubiquitin antibody (1:1,500).
Secondary antibody: Goat anti-rabbit IgG conjugated to horse radish peroxidase (1:10,000).
2.8. Immuno fluorescence for Detecting Ubiquitinated Protein Aggregates
2.8.1. Cell Culture and Fixation
Cells are plated onto eight-well chamber slides precoated with poly-d-lysine at a density of 50,000 cells/mL.
Paraformaldehyde stock: 16% paraformaldehyde solution, EM grade.
Paraformaldehyde (4%): Dilute paraformaldehyde stock with 1× PBS. Store at 4°C.
Ice-cold methanol.
2.8.2. Immunofluorescence
Dilution buffer: 25 mM Tris–HCl, pH 7.2; 300 mM NaCl; 0.3% v/v Triton X-100; 0.5 mg/mL bovine serum albumin; 0.01% w/v thimerosal. Store at 4°C.
Blocking buffer: 5% v/v normal goat serum in dilution buffer (see Note 4).
Primary antibodies: Rabbit polyclonal anti-ubiquitin antibody (1:150); mouse monoclonal anti-β III tubulin (1:1,000). Dilute both in the same microtube in blocking buffer.
Secondary antibodies: Alexa fluor 488 goat anti-rabbit (1:500); Alexa fluor 546 goat anti-mouse (1:500). Prepare both in the same microtube in dilution buffer.
Mounting medium hard set with DAPI.
3. Methods
3.1. In-Gel Assay
3.1.1. Cell Harvesting and Homogenization
Prepare harvesting buffer: For 4 mL harvesting buffer: 100 μL of 200 mM ATP; 20 mL μL of 200 mM DTT; 3,880 μL of buffer A. Place on ice.
When ready to harvest the cells, place dishes on ice and carefully aspirate the media from the dishes.
Wash cells once with 5 mL ice-cold 1× PBS (see Note 6).
Add 100 μL of harvesting buffer to one dish and collect cells by scraping (see Note 7).
Homogenize each sample on ice for 1 min with a benchtop homogenizer at maximum speed.
Centrifuge for 15 min at 19,000 × g (4°C). Transfer supernatant to new chilled tubes and save the pellets for future SDS-PAGE analysis [see Subheading 3.4 (steps 1 and 2) Samples from “ingel” assay – for SDS-PAGE and western blotting].
Determine protein concentration with the Bradford assay.
Normalize samples to equal protein concentration with harvesting buffer. Add to each sample 2 μL of bromophenol blue solution. Mix. Samples should remain on ice at all times.
3.1.2. Native PAGE (9)
Prepare three gel mixes (3, 4, and 5%) to make a 1.5-mm thick native gradient minigel. The components and respective volumes of each mix are shown in Table 1.
Add the 5% mix first to make a 2-cm layer on the bottom of the minigel cassette and overlay it with water. Once the gel layer polymerizes, carefully remove the water, dry with filter paper by gently tapping, and add the 4% mix to make a 3-cm layer above the 5% layer. Add water, and follow the same steps as above. Finally, add the 3% mix to completely fill the cassette and quickly insert the comb. Once the gel polymerizes, carefully remove the comb and with a 3-mL Falcon transfer pipette wash the wells with ultrapure water followed by running buffer.
Prepare the running buffer. For example: 396 mL buffer B; 2 mL of 200 mM ATP; 2 mL of 200 mM DTT. Set up the gel apparatus to run the minigel and load the samples (40 μg of protein per lane) at 4°C.
Run the nondenaturing native minigel at 4°C with 150 V for 2 h.
Following electrophoresis, transfer the gel into a small plastic box containing 15 mL gel buffer without substrate; make sure that the entire gel is covered with gel buffer. Wash the gel on a rocker at room temperature for 1 min.
3.1.3. Proteasome Activity
Remove the gel buffer and add newly prepared proteasome activity mix (15 mL buffer A; 1 mM ATP; 1 mM DTT; 300 μM Suc-LLVY-AMC; see Note 4) into the box making sure that the entire gel is covered. Incubate the gel on a rocker for 5–10 min at 37°C.
Remove the gel from the box and place it on a UV transilluminator with the proteasome activity mix. Three proteasome bands, including 26S with two caps, 26S with one cap, and 20S, should be visible (see Fig. 1a).
Photograph the gel with a NIKON Cool Pix 8700 camera with a 3-4219 fluorescent green filter. For a better image, remove all bubbles on top of the gel.
Semiquantification of proteasome bands is done by image analysis with the ImageJ program (Rasband, W.S., ImageJ, U.S. NIH, Maryland, http://rsb.info.nih.gov/ij/, 1997–2006).
3.1.4. Proteasome Levels and Assembly
Following proteasome activity, gently wash gel with transfer buffer.
Transfer proteins from the minigel onto a PVDF membrane for 2 h at 110 mA per gel at 4°C.
Block the membrane with blocking buffer for 30 min at 37°C.
Incubate the membrane with a primary antibody in blocking buffer overnight at 4°C. The anti-β5 antibody reacts with a subunit of the 20S core particle, thus detecting both the 26S and 20S proteasomes (see Fig. 1b). The anti-Rpt6 antibody reacts with a subunit of the regulatory particle, thus detecting 26S proteasomes only (see Fig. 1c).
The next day, remove the primary antibody (can be reused) and wash the membrane three times (10 min each) with washing buffer on a rocker at room temperature.
Incubate the membrane with the appropriate secondary antibody in blocking buffer on a rocker at room temperature for 45 min.
Wash the membrane again three times (10 min each) with washing buffer on a rocker at room temperature.
Develop the blot by a chemiluminescent horseradish peroxidase method with the ECL reagent. Wrap the blot in a thin plastic bag and with tape attach it to an X-ray film-developing cassette.
In the darkroom, place an X-ray film in the cassette for the desired time and develop the film in an X-ray developer.
Three proteasome bands, including 26S with two caps, 26S with one cap, and 20S, should be visible depending on which antibody was used to probe the blots (see Fig. 1b, c).
Semiquantification of proteasome bands is done by image analysis with the ImageJ program.
3.2. Glycerol Gradient Centrifugation
3.2.1. Cell Harvesting and Homogenization
SK-N-SH cells are treated at 75–80% confluence with 25 nM epoxomicin (irreversible proteasome inhibitor) or DMSO (control) for 24 h (see Note 8).
When ready to harvest the cells, place dishes on ice and carefully aspirate the media from the dishes.
Wash cells twice with 5 mL ice-cold 1× PBS (see Note 6).
Add 100 μL of harvesting buffer to each dish and collect cells by scraping. Transfer to 1.5-mL chilled microtubes.
Centrifuge for 1 min at 19,000 × g (4°C).
Homogenize each sample on ice for 1 min with a benchtop homogenizer at maximum speed.
Centrifuge for 10 min at 19,000 × g (4°C). Transfer supernatant to new chilled microtubes and discard the pellet.
Determine protein concentration with the Bradford assay. Adjust protein concentration to 2 mg/sample in 500 μL total volume containing 10% glycerol. Samples should remain on ice at all times.
3.2.2. Glycerol Gradient Centrifugation and Fractionation (10, 11)
The gradient maker contains two chambers with a connecting valve centered between them that controls the generation of the gradient. Gradient flow rate is controlled by an outflow valve fitted on one of the chambers with attached tubing to deliver the gradient buffer (see Fig. 2).
Before starting to pour the gradient, both valves need to be closed and a magnetic spin bar should be placed into the chamber attached to the outflow valve (chamber 1).
Slowly dispense 6 mL of 40% glycerol buffer into chamber 1.
Slowly dispense 6 mL of 10% gradient buffer into chamber 2.
Turn on magnetic stirrer to gently mix the glycerol buffer.
Open the connecting valve first and then the outflow valve to allow pouring through gravity into each of the Beckman centrifuge tubes. The outflow rate should be slow.
Once all Beckman centrifuge tubes are filled, carefully load 500 μL of the cell lysate (2 mg of protein) onto the top of the gradient in each tube. Label each tube accordingly to the respective treatment. Transfer centrifuge tubes carefully into a chilled centrifuge bucket.
Equilibrate each centrifuge bucket/tube group by placing it in a small Erlenmeyer flask on a scale.
The weight difference between two opposing bucket/tube groups in the rotor should not be more than 0.1 g. Equilibrate the opposing bucket/tube groups by adding 10% glycerol buffer to the tube in the lighter group.
Carefully place the bucket/tube groups on the chilled rotor according to their numbers.
Centrifuge for 24 h at 83,000 × g at 4°C in the Beckman ultracentrifuge.
The fraction collection in the following day should be done at 4°C (cold room).
Prepare 25 labeled 2.0-mL microtubes per sample (treatment) with caps removed (cut with scissors) to minimize any errors during fraction collection. Place tubes on the fraction collector.
Carefully place the first Beckman centrifuge tube on the fraction recovery system for puncturing. The fraction recovery system is attached to the fraction collector by a long tube (see Fig. 2). Puncture the bottom of the centrifuge tube and allow a few seconds for the flow to start.
Set the fraction collector to collect 500 μL of gradient mix per 2-mL microtube at 4°C. Monitor the flow and fraction collection throughout.
3.2.3. Chymotrypsin-Like Activity
Label new 1.5-mL microtubes (with caps) to correspond to each collected fraction.
Prepare the master mix to measure the chymotrypsin-like activity: 400 μM Suc-LLVY-AMC (substrate from 10 mM stock) in 0.05 M Tris–HCl, pH 7.5. Prepare enough master mix to add 50 μL per microtube (see Note 4). To save time, distribute the master mix with a repeater plus pipettor.
Add 50 μL of each fraction to the corresponding microtube and close the cap. Vortex.
Incubate at 37°C overnight on a rocker.
The next day, stop the reaction in each microtube with 100 μL of 10% TCA.
Vortex and to develop the color, add to each microtube in sequence 200 μL of 0.1% sodium nitrite; 200 μL of 0.5% ammonium sulfamate, and 400 μL 0.05% N-NEDA. Vortex before adding the next reagent. Upon addition of the last reagent, notice color development in the tubes where the substrate was cleaved.
Read absorbance in spectrophotometer at 560 nm.
Average the OD of every two consecutive tubes (example: average OD for tubes 1 and 2, then 3 and 4, and 5 and 6 to the end) to obtain the OD for a total of 13 fractions (tube #25 = fraction #13). Use the 13 averaged OD values (#1 the heaviest; #13 the lightest) to generate activity graphs for each condition (see Fig. 3a).
3.3. Peptidase Activities in Total Cell Lysate
3.3.1. Cell Harvesting and Homogenization
SK-N-SH cells are treated at 75–80% confluence for different time points with 25 nM epoxomicin (irreversible proteasome inhibitor) or DMSO (control) (see Note 9).
When ready to harvest the cells, place dishes on ice and carefully aspirate the media from the dishes.
Wash cells twice with 5 mL ice-cold 1× PBS (see Note 6).
Add 100 μL of harvesting buffer to each dish and collect cells by scraping. Transfer samples to 1.5-mL chilled microtubes.
Centrifuge for 1 min at 19,000 × g (4°C).
Homogenize each sample on ice for 1 min with a benchtop homogenizer at a maximum speed.
Centrifuge for 10 min at 19,000 × g (4°C). Transfer supernatant to new chilled microtubes and discard the pellet.
Determine protein concentration with the BCA kit. Protein samples should remain on ice at all times.
Normalize samples to 2 μg/μL of protein, an optimal concentration to carry out this assay.
3.3.2. Peptidase Activities (12)
Label new microtubes in duplicate for each condition for three peptidase activities: chymotrypsin-, trypsin-, and caspase-like activities.
- Prepare three master mixes for chymotrypsin-, trypsin-, and caspase-like activities (see Note 4):
- Chymotrypsin-like activity: 400 μM Suc-LLVY-AMC (substrate from stock 10 mM) in 0.05 M Tris–HCl, pH 7.5
- Tryspin-like activity: 400 μM of Z-GGR-βNA (substrate from stock 10 mM) in 0.05 M Tris–HCl, pH 8.0
- Caspase-like activity: 400 μM of Z-LLE-βNA (substrate from stock 10 mM) in 0.05 M Tris–HCl, pH 8.0
Prepare enough master mix for each activity to add 75 μL per microtube. To save time, distribute the master mix with a repeater plus pipettor.
Add 25 μL (50 μg) of sample per microtube and close caps. Vortex.
Incubate at 37°C overnight on a rocker.
The next day, stop the reaction in each microtube with 100 μL of 10% TCA.
Vortex and to develop the color, add to each microtube in sequence 200 μL of 0.1% sodium nitrite; 200 μL of 0.5% ammonium sulfamate, and 400 μL 0.05% N-NEDA. Vortex before adding the next reagent. Upon addition of the last reagent, notice color development in the tubes where the substrate was cleaved.
Read absorbance in spectrophotometer at 560 nm for chymotrypsin-like activity, and 580 nm for trypsin- and caspase-like activities.
Export data to Excel and generate activity graphs (see Fig. 4).
3.4. SDS-PAGE and Western Blotting for Proteasome Subunits α4, β5, and Rpt6, for β-Actin, and for Ubiquitinated Proteins
3.4.1. Samples from “In-Gel” Assay (Rat E18 Cortical Neuronal Cultures)
Supernatant remaining from “in-gel” assay (see Subheading 3.1 step 6): Adjust to 1% SDS lysis buffer.
Pellet remaining from “in-gel” assay (see Subheading 3.1 step 6): Resuspended in 1% SDS lysis buffer and sonicate until no particles are visible.
Heat all samples (supernatant and pellet) at 100°C for 5 min.
Determine protein concentration with BCA kit. Normalize samples with 1% SDS lysis buffer.
Add loading buffer to each sample (1:12.5, v/v).
Heat samples again at 100°C for 5 min. Samples are ready to load for SDS-PAGE and western blotting (see Fig. 5) or can be stored at −80°C until ready to load.
3.4.2. Samples from Glycerol Gradient Centrifugation (Human Neuroblastoma SK-N-SH Cells)
To the remaining 450 μL of each of the 25 original fractions collected in 2-mL microtubes (see Subheading 3.2 step 15), add 3 volumes (1,350 μL) of ice-cold acetone.
Mix well by vortexing and keep at −20°C overnight.
Next day, spin the 2-mL microtubes for 15 min at 4°C at a maximum speed. Carefully discharge supernatant and retain the pellet.
Dry tubes by inversion on tissue paper (see Note 10).
Resuspend protein pellets in 25 μL of 1% SDS lysis buffer per sample.
Heat all samples at 100°C for 5 min.
Combine the samples of every two consecutive tubes (example: combine tubes 1 and 2, then 3 and 4, and 5 and 6 to the end) to obtain a total of 13 fractions (tube #25 = fraction #13). Fraction #1 is the heaviest and #13 the lightest. Each fraction now contains 50 μL of sample.
Add loading buffer to each fraction (1:12.5, v/v).
Heat samples again at 100°C for 5 min. Samples are ready to load (20 μL per lane) for SDS-PAGE and western blotting (see Fig. 3b) or can be stored at −80°C until ready to load.
3.4.3. Samples for Detecting Ubiquitinated Proteins (Rat E18 Cortical Neuronal Cultures)
Prepare hot 1% SDS lysis buffer in a 10-mL glass tube to harvest the cells (see Note 11).
When ready to harvest the cells, place dishes at room temperature and carefully aspirate the media from the dishes.
Wash the cells once with 5 mL PBS (room temperature, see Note 6).
Add 100 μL of hot 1% SDS lysis buffer to one dish and collect cells by scraping (see Note 7).
Heat samples at 100°C for 5 min.
Sonicate samples until no particles are visible.
Determine protein concentration with BCA kit. Normalize samples with 1% SDS lysis buffer.
Add loading buffer to each sample (1:12.5, v/v).
Heat samples again at 100°C for 5 min. Samples are ready to load for SDS-PAGE and western blotting (see Fig. 6) or can be stored at −80°C until ready to load.
3.4.4. SDS-PAGE
Prepare gel mix (8% for ubiquitinated proteins; 10% for proteasome subunits and actin) to make a 1.0-mm thick SDS-polyacrylamide minigel. The components and respective volumes are shown in Table 2.
Pour the resolving gel leaving space for the stacking gel and overlay with water. The gel should polymerize in about 30 min.
Prepare the stacking gel mix as shown in Table 2.
Once the resolving gel polymerizes, carefully remove the water, dry with filter paper by gently tapping, add the stacking gel mix to completely fill the cassette, and quickly insert the comb.
Once the stacking gel polymerizes, carefully remove the comb and with a 3-mL Falcon transfer pipette wash the wells with ultrapure water followed by running buffer.
Set up the electrophoresis apparatus at room temperature. Add the running buffer into the upper and lower chambers of one gel unit. Load each sample (40 μg of protein per lane) and the molecular weight markers.
Run the SDS-polyacrylamide gel at room temperature with 150 V until the loading dye reaches the bottom of the gel.
3.4.5. Western Blotting
Remove the minigel from the cassette, place it in a small box, and wash it with transfer buffer.
Transfer proteins from the minigel onto a PVDF membrane for 2 h at 110 mA per gel at room temperature.
Block the membrane with blocking buffer for 30 min at 37°C on a rocker.
Incubate the membrane with primary antibody in SuperBlock blocking buffer overnight at 4°C.
The next day, remove the primary antibody (recycle) and wash the membrane three times (10 min each) with washing buffer on a rocker at room temperature.
Incubate the membrane with the appropriate secondary antibody in SuperBlock blocking buffer on a rocker at room temperature for 45 min.
Wash the membrane again three times (10 min each) with washing buffer on a rocker at room temperature.
Develop the blot by a chemiluminescent horseradish peroxidase method with the ECL reagent. Rap the blot in a thin plastic bag and with tape attach it to an X-ray film-developing cassette.
In the darkroom, place an X-ray film in the cassette for the desired time and develop the film in an X-ray developer.
After developing the film, wash the membrane for 10 min in washing buffer on the rocker. If desired, block the membrane again at 37°C for 30 min, and incubate it with another primary antibody overnight at 4°C for developing the next day. Follow the same steps as for the first primary antibody. Follow the same steps for incubating the membrane with yet another primary antibody and to develop the blot.
The molecular masses for Rpt6, α4, β5, and β-actin are approximately 46 kDa, 28 kDa, 22 kDa, and 42 kDa, respectively (see Figs. 3 and 5). Ubiquitinated proteins are detected as a high-molecular-weight smear starting at the top of the blot (see Fig. 6).
Semiquantification of protein bands is done by image analysis with the ImageJ program.
3.5. Filter Trap Assay for Ubiquitinated Protein Aggregates
3.5.1. Cell Harvesting and Homogenization
Prepare ice-cold lysis (RIPA) buffer to harvest the cells.
Label two 1.5-mL microtubes for each treatment and place them on ice.
When ready to harvest the cells, place dishes on ice and carefully aspirate the media from the dishes.
Wash the cells once with 5 mL 1× PBS (ice cold, see Note 6).
Add 100 μL of lysis (RIPA) buffer to one dish and collect cells by scraping. Transfer cell lysate into a microtube.
Incubate samples at −80°C for 15 min to ensure total cell lysis.
Centrifuge for 10 min at 19,000 × g (4°C). Transfer supernatant to a new chilled microtube. Discard pellet.
Determine protein concentration with the BCA kit. Protein samples should remain on ice at all times.
Normalize samples to 1 μg/μL of protein with normalizing buffer. Save remaining samples at −80°C.
3.5.2. Filter Trap Preparation
Cut the nitrocellulose membrane and Whatman filter paper to completely cover the chamber of the 96-well Dot-blot system.
Pre-wet the nitrocellulose membrane and Whatman filter paper with normalizing buffer for at least 5 min.
Set up the two bottom parts of the manifold.
Place two Whatman filter papers above the manifold. Place the nitrocellulose membrane over the top filter paper. Place the manifold top over the nitrocellulose membrane and seal tightly.
Establish a template for sample loading. Close tightly all the wells that are not going to be used with clear packing tape making sure to leave one empty row all around your samples. Do not load samples in wells at the edge of the apparatus; cover them with tape instead.
Set up vacuum, but DO NOT connect it to the manifold yet (see Note 12).
3.5.3. Filter Trap Running (13, 14)
Load 100 μg of each sample (in the same volume for all samples) into the respective wells.
Load 100 μl of washing buffer to the row of empty wells surrounding the samples.
Connect the vacuum line to the manifold and turn the vacuum on. All the samples should flow through the membrane and the wells are empty. Complete flow takes a few seconds.
Turn off the vacuum and disconnect its line from the manifold to release the vacuum.
Wash each untapped well two times with 100 μL of washing buffer. Connect the vacuum/turn it on each time and turn it off/disconnect it after each wash to release the vacuum.
Remove the top of the manifold. Mark the nitrocellulose membrane with a pencil to remember the sample loading order.
Place the membrane in a plastic box and proceed as for western blotting (see Subheading 3.4, steps 3–9), including blocking and probing the membrane with the anti-ubiquitin antibody. A positive signal appears as a dark dot (see Fig. 7b).
Semiquantification of protein dots is done by image analysis with the ImageJ program.
3.6. Immunoflu orescence for Detecting Ubiquitinated Protein Aggregates
3.6.1. Cell Culture and Fixation
Prepare the fixation solution (4% paraformaldehyde) in a fume hood.
When ready to immunostain, place slides at room temperature and carefully aspirate the media from each well.
Add 500 μL of 4% paraformaldehyde solution to each chamber and incubate for 15 min at room temperature in a fume hood (see Note 13).
Remove paraformaldehyde and discard it into a hazardous waste container (see Note 13). Repeat step 3.
Remove paraformaldehyde solution as in step 4. Wash wells three times with 1× PBS for 3 min each time at room temperature on a rocker.
Add 500 μL of ice-cold methanol per well and incubate for 2 min at room temperature on a rocker. Remove methanol. Wash wells three times with 1× PBS for 3 min each time at room temperature on a rocker.
3.6.2. Immunofluorescence
Block with blocking buffer for 1 h at room temperature on a rocker.
Remove blocking buffer. Add primary antibodies in blocking buffer.
Incubate overnight with both primary antibodies prepared in the same microtube. Place slides in a humid chamber (to avoid evaporation) on a rocker at 4°C.
Remove primary antibodies. Wash wells three times with 1× PBS for 3 min each time at room temperature on a rocker.
Prepare fresh secondary antibodies in dilution buffer in the same tube. Incubate for 1 h in the dark at room temperature.
Remove secondary antibodies. Wash wells three times with 1× PBS for 3 min each time at room temperature on a rocker.
Remove the wells, gasket, and biocompatible adhesive from the slide (a razor blade may be useful).
Add a few drops of mounting medium to the slide surface and quickly place the coverslip on top making sure to remove all bubbles.
The slide can be viewed immediately or can be stored in the dark at 4°C for several months.
View slides with a fluorescence microscope. Ubiquitin staining – green fluorescence; β-III tubulin – red fluorescence; DAPI – blue fluorescence. See Fig. 8.
4. Notes
Do not heat-inactivate the normal FBS.
The final DMSO concentration should be 0.5%.
Both cell cultures as well as other types of neuronal cultures can be used for all the assays described here.
Prepare fresh each time. Do not freeze.
Make sure that the EDTA is the acid form with no salts. Sodium and potassium inhibit proteasome activity. Use the acid EDTA (powder) to adjust the pH of the Tris solution.
While aspirating upon washing, tilt dishes slightly to make sure that all of the PBS is removed.
To increase protein concentration per treatment, transfer cell lysate to another dish with the same treatment and harvest cells the same way. Combine all of the cell lysates corresponding to the same treatment into one 1.5-mL chilled microtube and place on ice.
It is recommended to use four 100-mm dishes per treatment since a large amount (2 mg) of protein is needed.
It is recommended to use two 100-mm dishes per treatment.
Pellet may be difficult to see in heavier fractions.
Keep the glass tube in boiling water for at least 5 min to make sure that the buffer is hot.
To test if the setup is properly sealed, add 100 μL of any dye to the wells selected for sample loading. Remove the top part of the manifold. The dye should form round dots on the nitrocellulose membrane. Smeared, not perfectly round dye dots indicate that the setup is not properly sealed (see Fig. 7a). Redo the sealing and test again until the dye dots are perfectly round.
Paraformaldehyde is a hazardous chemical. Respiratory, skin, and eye protection should be used when working with it. Work within a fume hood or with local exhaust ventilation. Absorb incidental spills with damp absorbent pads. Collect and submit for waste disposal. Request hazardous waste pickup service for disposal.
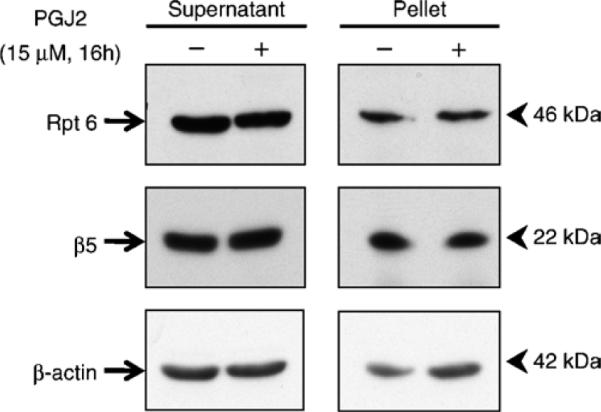
Fig. 1.

In-gel assay for proteasome activity and assembly in rat E18 primary cortical neuronal cultures. Crude extracts were prepared from control cultures (−) or cultures treated with 15 μM PGJ2 for 16 h (+). Cleared lysates (40 μg/sample) were subjected to nondenaturing gel electrophoresis. In (a) the proteasomal chymotrypsin-like activity was measured with Suc-LLVY-AMC by the in-gel assay. In (b and c) 26S and 20S proteasomes were detected by immunoblotting with an anti-β5 antibody (b), a subunit of the core proteasome particle (20S), and with the anti-Rpt6 antibody (c), an ATPase subunit of the 19S regulatory particle. Proteasomal 26S (two caps and one cap) and 20S forms are indicated by on the left.
Fig. 2.

Setup for making a glycerol gradient. For explanation, see Subheading 3.2, steps 1–8.
Fig. 3.
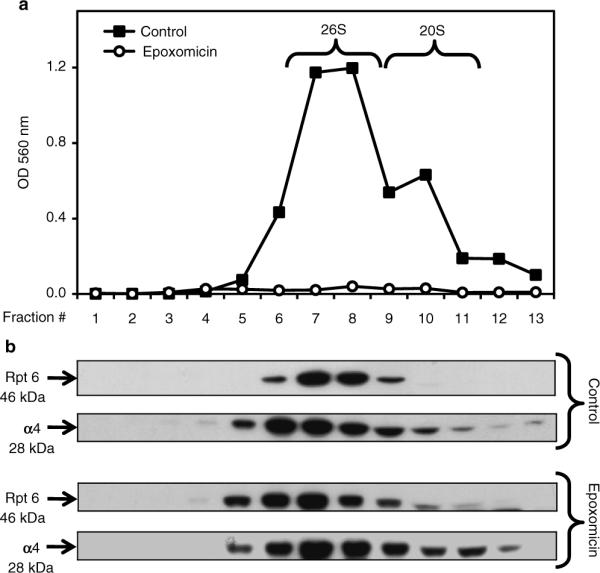
Sedimentation velocity of proteasomes in SK-N-SH cells. Cells were treated for 24 h with DMSO (control, vehicle) or epoxomicin (25 nM). Total lysates (2 mg protein/sample) were fractionated by glycerol density gradient centrifugation (10–40% glycerol corresponding to fractions 13 to 1). (a) Aliquots (50 μl) of each fraction obtained from control (black squares)- and epoxomicin (white circles)-treated cells were assayed for chymotrypsin-like activity with Suc-LLVY-AMC. (b) Immunoblot analyses of each fraction probed with antibodies that react with the proteasome (α4, core particle; Rpt6, 19S regulatory particle). Proteins were precipitated with acetone from 450 μl of each fraction. The fractions were obtained from control- and epoxomicin-treated cells.
Fig. 4.

Proteasome activities in SK-N-SH cells. Cells were treated for 24 h with DMSO (control, vehicle) or epoxomicin (25 nM). Proteasome activities were measured in cleared supernatants obtained from total cell homogenates (50 μg of protein/sample). Peptidase activities were assayed colorimetrically after a 24-h incubation at 37°C. The chymotrypsin-like activity was measured with Suc-LLVY-AMC, the trypsin-like activity with Z-GGR-βNA, and the caspase-like activity with Z-LLE-βNA.
Fig. 5.
Proteasome subunit levels in rat E18 primary cortical neuronal cultures. Aliquots of the supernatant (cleared lysate) and pellet fractions obtained from samples prepared for the “in-gel” assay (see Fig. 1) were run on SDS-PAGE (10% gel) followed by immunobloting with the same antibodies listed in Fig. 1 as well as with anti-β-actin. 40 μg of protein were loaded per lane.
Fig. 6.

Accumulation of ubiquitinated proteins in rat E18 primary cortical neuronal cultures. Cells were treated for 24 h with DMSO [control, vehicle (−)] or with 20 μM PGJ2 (+). Total cell extracts were subjected to western blot analysis (8% gel) to detect ubiquitinated proteins (40 μg of protein/lane). Equal protein loading was demonstrated by probing the immunoblots with the anti-β-actin antibody.
Fig. 7.

Filter trap assay to measure ubiquitin-protein aggregates in rat E18 primary cortical neuronal cultures. (a) In a preliminary test, waterproof drawing ink (100 μl per well) was applied to the nitrocellulose membrane to assess flow through each well. If vacuum is properly equilibrated, dye dots corresponding to each well should appear round and neat on the nitrocellulose membrane. (b) Influence of membrane composition and porosity on detection of ubiquitin-protein aggregates. Cells were treated for 16 h with DMSO [control, vehicle (0)] or with 15 μM or 20 μM PGJ2. Cell extracts were subjected to the filter trap assay to detect ubiquitin-protein aggregates (100 μg of protein/sample). Nitrocellulose-yielded optimal assay sensitivity relative to polyvinylidene fluoride (PVDF) or cellulose acetate.
Fig. 8.

Immunofluorescence detection of ubiquitin-protein aggregates in rat E18 primary cortical neuronal cultures. Ubiquitinated proteins and βIII-tubulin immunofluorescence staining of cortical cultures treated with DMSO (top 4 panels) or 15 μM PGJ2 for 16 h (bottom 4 panels). Nuclei are stained with DAPI. Large arrows point to protein aggregates and small arrows to dystrophic neurites. Scale bar = 10 μm.
Table 1.
Native gel solutions
| Gel components | 3% (top) | 4% (middle) | 5% (bottom) |
|---|---|---|---|
| Layer height (cassette 8 × 8 cm) | 3 cm | 3 cm | 2 cm |
| 30% acrylamide/Bis solution, 29:1 | 1,260 μL | 1,680 μL | 2,100 μL |
| Rhinohide polyacrylamide gel strengthener | 240 μL | 320 μL | 400 μL |
| Ammonium persulfate (1.5%, w/v) | 600 μL | 600 μL | 600 μL |
| Gel buffer (complete with ATP and DTT) | 9.888 mL | 9.388 mL | 8.888 mL |
| TEMED | 12 μL | 12 μL | 12 μL |
| Total volume | 12 mL | 12 mL | 12 mL |
Table 2.
SDS-polyacrylamide minigel solutions
| Gel components | 8% | 10% | Stacking |
|---|---|---|---|
| 30% acrylamide/Bis solution, 29:1 | 2.7 mL | 3.4 mL | 1.03 mL |
| Resolving buffer | 1.3 mL | 1.3 mL | None |
| Stacking buffer | None | None | 2.07 mL |
| Ammonium persulfate (1.5%, w/v) | 515 μL | 515 μL | 0.41 mL |
| Water | 5.37 mL | 4.67 mL | 3.6 mL |
| SDS (10%, m/v) | 105 μL | 105 μL | 0.83 mL |
| TEMED | 10 μL | 10 μL | 6 μL |
| Total volume | 10 mL | 10 mL | 8 mL |
Acknowledgments
Supported by NIH: [NIA-AG028847 to M.F.-P.; NINDS-NS41073 (SNRP) to M.F.-P. (head of subproject); NCRR-RR03037 (infrastructure) to Hunter College, City University of New York].
References
- 1.Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998;21:516–520. doi: 10.1016/s0166-2236(98)01276-4. [DOI] [PubMed] [Google Scholar]
- 2.Huang Q, Figueiredo-Pereira ME. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis. 2010;15(11):1292–1311. doi: 10.1007/s10495-010-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Melandri F, Berdo I, Jansen M, Hunter L, Wright S, Valbrun D, Figueiredo-Pereira ME. delta12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochem Biophys Res Commun. 2004;319:1171–1180. doi: 10.1016/j.bbrc.2004.05.098. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Aris VM, Ogburn KD, Soteropoulos P, Figueiredo-Pereira ME. Prostaglandin J2 alters pro-survival and pro-death gene expression patterns and 26S proteasome assembly in human neuroblastoma cells. J Biol Chem. 2006;281:21377–21386. doi: 10.1074/jbc.M601201200. [DOI] [PubMed] [Google Scholar]
- 6.Biedler JL, Roffler-Tarlov S, Schachner M, Freedman LS. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978;38:3751–3757. [PubMed] [Google Scholar]
- 7.Li Z, Jansen M, Ogburn K, Salvatierra L, Hunter L, Mathew S, Figueiredo-Pereira ME. Neurotoxic prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal cells through the p38MAPK pathway: a death wish? J Neurosci Res. 2004;78:824–836. doi: 10.1002/jnr.20346. [DOI] [PubMed] [Google Scholar]
- 8.Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- 9.Elsasser S, Schmidt M, Finley D. Characterization of the proteasome using native gel electrophoresis. Methods Enzymol. 2005;398:353–363. doi: 10.1016/S0076-6879(05)98029-4. [DOI] [PubMed] [Google Scholar]
- 10.Eytan E, Ganoth D, Armon T, Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc Natl Acad Sci USA. 1989;86:7751–7755. doi: 10.1073/pnas.86.20.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirano Y, Murata S, Tanaka K. Large-and small-scale purification of mammalian 26S proteasomes. Methods Enzymol. 2005;399:227–240. doi: 10.1016/S0076-6879(05)99015-0. [DOI] [PubMed] [Google Scholar]
- 12.Wilk S, Orlowski M. Evidence that pituitary cation-sensitive neutral endopeptidase is a multicatalytic protease complex. J Neurochem. 1983;40:842–849. doi: 10.1111/j.1471-4159.1983.tb08056.x. [DOI] [PubMed] [Google Scholar]
- 13.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 14.Chang E, Kuret J. Detection and quantification of tau aggregation using a membrane filter assay. Anal Biochem. 2008;373:330–336. doi: 10.1016/j.ab.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]