

Abstract
Background
Analysis of elementary modes (EMs) is proven to be a powerful constraint-based method in the study of metabolic networks. However, enumeration of EMs is a hard computational task. Additionally, due to their large number, EMs cannot be simply used as an input for subsequent analysis. One possibility is to limit the analysis to a subset of interesting reactions. However, analysing an isolated subnetwork can result in finding incorrect EMs which are not part of any steady-state flux distribution of the original network. The ideal set to describe the reaction activity in a subnetwork would be the set of all EMs projected to the reactions of interest. Recently, the concept of "elementary flux patterns" (EFPs) has been proposed. Each EFP is a subset of the support (i.e., non-zero elements) of at least one EM.
Results
We introduce the concept of ProCEMs (Projected Cone Elementary Modes). The ProCEM set can be computed by projecting the flux cone onto a lower-dimensional subspace and enumerating the extreme rays of the projected cone. In contrast to EFPs, ProCEMs are not merely a set of reactions, but projected EMs. We additionally prove that the set of EFPs is included in the set of ProCEM supports. Finally, ProCEMs and EFPs are compared for studying substructures of biological networks.
Conclusions
We introduce the concept of ProCEMs and recommend its use for the analysis of substructures of metabolic networks for which the set of EMs cannot be computed.
Background
Metabolic pathway analysis is the study of meaningful minimal pathways or routes of connected reactions in metabolic network models [1,2]. Two closely related concepts are often used for explaining such pathways: elementary modes (EMs) [3,4] and extreme pathways (EXPAs) [5]. Mathematically speaking, EMs and EXPAs are generating sets of the flux cone [1,6]. Several approaches have been proposed for the computation of such pathways [7-14].
EM and EXPA analysis are promising approaches for studying metabolic networks [15,16]. However, due to the combinatorial explosion of the number of such pathways [17,18], this kind of analysis cannot be performed for "large" networks. Recent advances in the computation of EMs and extreme rays of polyhedral cones [12,13] have made it possible to compute tens of millions of EMs, but computing all EMs for large genome-scale networks may still be impossible. Additionally, one is often interested only in a subset of reactions, and not all of them. Therefore, even if the EMs are computable, possibly many of them are not relevant because they are not related to the reactions of interest.
The goal of the present paper is to introduce the new concept of Projected Cone Elementary Modes (ProCEMs) for the analysis of substructures of metabolic networks. The organisation is as follows. Firstly, the mathematical concepts used in the text are formally defined. Secondly, we review the studies which have tried to investigate (some of) the EMs or EXPAs of large-scale networks. In the next step, we present the concept of ProCEMs and propose a method to compute them. Finally, we compare ProCEMs with elementary flux patterns (EFPs) from the mathematical and computational point of view, and analyse some concrete biological networks.
Formal Definitions
We consider a metabolic network N with m internal metabolites and n reactions. Formally, we describe N by its stoichiometric matrix S ∈ ℝm × n and the set of irreversible reactions Irr ⊆ {1, . . ., n}. If steady-state conditions hold, i.e., there is no net production or consumption of internal metabolites, the set of all feasible flux distributions defines a polyhedral cone
| (1) |
which is called the (steady-state) flux cone [1,2].
A polyhedral cone in canonical form is any set of the form P = {x ∈ ℝn | Ax ≤ 0}, for some matrix A ∈ ℝk × n. To bring (1) in canonical form, we can replace the equalities Sv = 0 by the two sets of inequalities S · v ≤ 0 and -S · v ≤ 0. Furthermore, the inequalities vi ≥ 0, i ∈ Irr are multiplied by -1. Any non-zero element x ∈ P is called a ray of P. Two rays r and r' are equivalent, written r ≅ r', if r = λr', for some λ > 0. A ray r in P is extreme if there do not exist rays r', r"∈ P, r' ≇ r" such that r = r' + r".
For every v ∈ ℝn, the set supp(v) = {i ∈ {1, . . ., n} | vi ≠ 0 } is called the support of v.
A flux vector e ∈ C is called an elementary mode (EM) [3,4] if there is no vector v ∈ C \ {0} such that supp(v) ⊊ supp(e). Thus, each EM represents a minimal set of reactions that can work together in steady-state.
The set of all pairwise non-equivalent EMs, E = {e1, e2, . . ., es}, generates the cone C [3]. This means that every flux vector in C can be written as a non-negative linear combination of the vectors in E.
Given a set Q ⊆  ×
×  , where resp. are subspaces of ℝn of dimension p resp. q with p + q = n, the projection of Q onto is defined as
, where resp. are subspaces of ℝn of dimension p resp. q with p + q = n, the projection of Q onto is defined as
| (2) |
In the special case Q = {v}, we simply write instead of .
Now consider a metabolic network 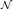 with p + q reactions and a subnetwork given by a subset of p "interesting" reactions. For the flux cone C of we assume , where the reactions of correspond to the subspace . The projection of the cone C on the subspace is again a polyhedral cone, called the projected cone on . Any elementary mode of the projected cone will be called a projected cone elementary mode (ProCEM). The projection of an elementary mode e ∈ C to the subspace will be called a projected elementary mode (PEM). As we will see in the sequel, the two concepts of PEM and ProCEM are closely related but different.
with p + q reactions and a subnetwork given by a subset of p "interesting" reactions. For the flux cone C of we assume , where the reactions of correspond to the subspace . The projection of the cone C on the subspace is again a polyhedral cone, called the projected cone on . Any elementary mode of the projected cone will be called a projected cone elementary mode (ProCEM). The projection of an elementary mode e ∈ C to the subspace will be called a projected elementary mode (PEM). As we will see in the sequel, the two concepts of PEM and ProCEM are closely related but different.
If the subnetwork has to be analysed, PEMs might be more relevant than EMs, as they are in lower dimension and easier to study. However, the only method currently known to compute PEMs is to enumerate the complete set of EMs and then to project these onto the subspace of interest. As we will see, ProCEMs provide an interesting alternative in this situation.
The State of the Art
As mentioned above, the set of EMs of a genome-scale network may be large, and in general, cannot be computed with the available tools. Even if this is possible, one cannot simply extract interesting information from it. Therefore, a subset of EMs (or in case that we are interested in a subset of reactions, the set of PEMs) should be computed to reduce the running time and/or output size of the algorithm. Several approaches to this problem have been proposed in the literature. These strategies can be classified into four main categories:
Computation of a Subset of EMs
The first strategy is to constrain the complete set of EMs (or EXPAs) to a subset describing a phenotype space or a set of phenotypic data. For example, Covert and Palsson [19] showed that consideration of regulatory constraints in the analysis of a small "core metabolism" model can reduce the set of 80 EXPAs to a set of 2 to 26 EXPAs, depending on the applied regulatory constraints. On the other hand, Urbanczik [20] suggested to compute "constrained" elementary modes which satisfy certain optimality criteria. As a result, instead of a full enumeration of EMs, only a subset of them should be computed, which results in a big computational gain. The idea of reducing the set of EMs has been used recently in an approach called yield analysis [21]. In this approach, the yield space (or solution space) is defined as a bounded convex hull. Then, the minimal generating set spanning the yield space is recalculated, and therefore, all EMs with negligible contribution to the yield space can be excluded. The authors show that their method results in 91% reduction of the EM set for glucose/xylose-fermenting yeast.
Computation of EMs in Isolated Subsystems
A second strategy to focus on the EMs (or EXPAs) of interest is to select a (possibly disconnected) subsystem, rather than the complete metabolic model, by assuming all other reactions and metabolites to be "external", and computing the EMs (or EXPAs) of this selected subsystem. This idea, i.e., cutting out subsystems or splitting big networks into several subsystems, is broadly used in the literature (e.g., see [22-34]). In some of these studies, not only the network boundary is redrawn, but also some reactions may be removed for further simplifying the network.
Although this strategy is useful, it can result in serious errors in the computational analysis of network properties [35]. For example, dependencies and coupling relationships between reactions can be influenced by redrawing the system boundaries [36]. Burgard et al. [37] showed that subsystem-based flux coupling analysis of the H. pylori network [25] results in an incomplete detection of coupled reactions. Kaleta et al. [35] suggest that neglecting such a coupling can lead to fluxes which are not part of any feasible EM in the original complete network. Existence of such infeasible "pathway fragments" [38] can result in incorrect conclusions.
To better understand this problem, we consider Figure 1A as an example. Let us assume that we are interested in a subnetwork composed of reactions 1, . . ., 9. This subnetwork is called SuN. If we simply assume the "uninteresting" reactions and metabolites to be the external reactions and metabolites, we will obtain the subsystem shown in Figure 1B. This subnetwork has only four EMs, two of which are not part of any feasible steady-state flux vector in the complete network. For example, the EM composed of reactions 5 and 7 in Figure 1B cannot appear in steady-state in the original complete network, because the coupling between reaction 1 and reaction 5 is broken. Therefore, analyzing this subnetwork instead of the original network can result in false conclusions.
Figure 1.

An example metabolic subnetwork. (A): A small metabolic network with 17 reactions. Metabolites are shown as nodes, while reactions are shown by arrows. Reactions 1, 8, 9, 15 and 16 are boundary reactions, while all other reactions are internal reactions. We might be interested only in a subnetwork containing nine reactions: 1, . . ., 9, which are shown by thick arrows. This subnetwork will be called SuN. (B): The reduced subsystem comprising only the nine interesting reactions.
Computation of Elementary Flux Patterns
We observed that some errors may appear in the analysis of isolated subsystems. One possible solution to this problem is to compute a "large" subset of PEMs, or alternatively, as suggested by Kaleta et al. [35], to compute the support of a subset of PEMs. These authors proposed a procedure to compute the elementary flux patterns (EFPs) of a subnetwork within a genome-scale network. A flux pattern is defined as a set of reactions in a subnetwork that is included in the support of some steady-state flux vector of the entire network [35]. A flux pattern is called an elementary flux pattern if it cannot be generated by combination of two or more different flux patterns. Each EFP is the support of (at least) one PEM. It is suggested that in many applications, the set of EFPs can be used instead of EMs [35].
Although EFPs are promising tools for the analysis of metabolic pathways, they also have their own shortcomings. The first important drawback of EFPs is that they cannot be used in place of EMs in certain applications [9], where the precise flux values are required. For example, in the identification of all pathways with optimal yield [23,39] and in the analysis of control-effective fluxes [27,28,40], the flux values of the respective reactions in the EMs should be taken into account.
Another important shortcoming of EFP analysis is that it is possible to have very different EMs represented by the same EFP, since flux values are ignored in EFPs. For example, consider the case that two reactions i and j are partially coupled [37]. This means that there exist at least two EMs, say e and f, such that ei/ej ≠ fi/fj [41]. However, if we consider a subnetwork composed of these two reactions, then we will only have one EFP, namely {i, j}. From the theoretical point of view, finding all EMs that correspond to a certain EFP is computationally hard (see Theorem 2.7 in [42]).
Every EFP is related to at least one EM in the original metabolic network. However, one of the limitations of EFP analysis is that EFPs are activity patterns of some EMs, not necessarily all of them. We will show this by an example. In Figure 1A, the flux cone is a subset of ℝ17, while the subnetwork SuN induces a 9-dimensional subspace . If G is the set of EMs in Figure 1A, then the set of PEMs can be computed as . The set of the 10 PEMs of SuN in Figure 1A is shown in Table 1. For the same network and subnetwork, we used EFPTools [43] to compute the set of the EFPs. The resulting 7 EFPs are also presented in Table 1. If we compare the PEMs and EFPs, we find out that the support of each of the first 7 PEMs is equal to one of the EFPs. However, for the last three PEMs no corresponding EFP can be found in Table 1. This is due to the fact that supp(p8) = E4 ∪ E5, supp(p9) = E3 ∪ E5, and supp(p10) = E1 ∪ E2. Hence, the flux patterns corresponding to these PEMs are not elementary. Therefore, some EMs may exist in the network which have no corresponding EFP on a certain subnetwork. This means that by EFP analysis possibly many EMs of the original network cannot be recovered. Informally speaking, we ask whether the set of EFPs is the largest set of PEM supports which can be computed without enumerating all EMs.
Table 1.
List of elementary flux patterns, projected cone elementary modes and projected elementary modes of SuN.
| EFPs | EFP set | ProCEM | PEM | vector |
|---|---|---|---|---|
| E1 | {9} | u1 | p1 | (0, 0, 0, 0, 0, 0, 0, 0, 1) |
| E2 | {8} | u2 | p2 | (0, 0, 0, 0, 0, 0, 0, 1, 0) |
| E3 | {1, 4} | u3 | p3 | (1, 0, 0, 1, 0, 0, 0, 0, 0) |
| E4 | {1, 2, 3} | u4 | p4 | (1, 1, 1, 0, 0, 0, 0, 0, 0) |
| E5 | {1, 5, 7} | u5 | p5 | (1, 0, 0, 0, 1, 0, 1, 0, 0) |
| E6 | {1, 4, 6, 7} | u6 | p6 | (1, 0, 0, 1, 0, 1, 1, 0, 0) |
| E7 | {1, 2, 3, 6, 7} | u7 | p7 | (1, 1, 1, 0, 0, 1, 1, 0, 0) |
| - | - | u8 | p8 | (1, 1, 1, 0, 1, 0, 1, 0, 0) |
| - | - | u9 | p9 | (1, 0, 0, 1, 1, 0, 1, 0, 0) |
| - | - | - | p10 | (0, 0, 0, 0, 0, 0, 0, 1, 1) |
Flux through reactions 1, . . ., 9, respectively, are the elements of the shown vectors. Zero vector and also the empty set are excluded.
Projection Methods
A possible strategy to simplify the network analysis is to project the flux cone down to a lower-dimensional space of interest. In other words, if we are interested in a subnetwork, we may project the flux cone onto the lower-dimensional subspace defined by the "interesting" reactions. Note that projecting the flux cone is in general different from removing reactions from the network. Consider the simple network shown in Figure 2A and a graphical representation of its corresponding flux space in Figure 2B (here, the axes x1, x2, x3 correspond to reactions 1, 2, 3, thus the flux cone is the open triangle shown in light gray). This network has two EMs, which are the generating vectors of the flux cone, g1 and g2. Now, if we are interested in a subnetwork composed of reactions 1 and 2, then we can project the flux cone to the 2D subspace produced by these two reactions. This is comparable to light projection on a 3D object to make 2D shadows. The projected cone is shown in dark gray. When the flux cone is projected onto the lower-dimensional space, new generating vectors may appear. In this example, g1 and g3 (in 2D space) are the generating vectors of the projected cone. Intuitively, one can think about g3 as the projected flux vector through reaction 1 and 3. This projected flux cone is certainly different from the flux cone of a network made by deleting reaction 3 (Figure 2C). Such a network has only one EM, and its corresponding flux cone can be generated by only one vector, namely, g1.
Figure 2.

Flux cone projection. (A): A small metabolic network. The reactions in the interesting subnetwork are shown as thick arrows. (B): The flux cone of this network, shown in light gray, can be generated by vectors g1 and g2. The projected cone is shown in dark gray. The projected cone can be generated by g1 and g3 in a 2D plane. (C): the same metabolic network as in A, but with reaction 3 removed. The flux cone of this network is generated by only one vector, namely g1.
Historically, the idea of flux cone projection has already been used in some papers. Wiback and Palsson [44] suggested that the space of cofactor production of red blood cell can be studied by projecting the cell-scale metabolic network onto a 2D subspace corresponding to ATP and NADPH production. A similar approach was used by Covert et al. [19] and also by Wagner and Urbanczik [45] to analyze the relationship between carbon uptake, oxygen uptake and biomass production. All the above studies considered very small networks. Therefore, the authors computed the extreme rays of the flux cone and then projected them onto the subspace of interest, without really projecting the flux cone. Urbanczik and Wagner [46] later introduced the concept of elementary conversion modes (ECMs), which are in principle the extreme rays of the cone obtained by projecting the original flux cone onto the subspace of boundary reactions. They suggest that the extreme rays of this "conversion" cone, i.e., the ECMs, can be computed even for large networks [47].
Following this idea, we introduce the ProCEM set ("Projected Cone Elementary Mode" set), which is the set of EMs of the projected flux cone. In contrast to [46], we formulate the problem in a way that any subnetwork can be chosen, not only the boundary reactions. Additionally, we compare the closely related concepts of ProCEMs, PEMs and EFPs.
Method and Implementation
Computational Procedure
Our algorithm needs three input objects: the stoichiometric matrix S ∈ ℝm×n of the network is , the set of irreversible reactions Irr ⊆ {1, . . ., n}, and the set of reactions ∑ ⊆ {1, . . ., n} in the subnetwork of interest, while as an output it will return the complete set of ProCEMs. The computation of ProCEMs is achieved in three main consecutive steps.
Step 1 - Preprocessing: The aim of this step is to remove inconsistencies from the metabolic network and to transform it into a form suitable for the projection in Step 2. First, based on ∑ we sort the columns of S in the form:
| (3) |
where the reaction corresponding to the i-th column belongs to ∑ iff the i-th column is in Ā. Next, the blocked reactions [37] are removed. Finally, each of the reversible reactions is split into two irreversible "forward" and "backward" reactions. The final stoichiometric matrix will be in the form:
| (4) |
where the columns of A represent the "interesting" reactions after splitting reversible reactions and removing the blocked reactions. In the following, we assume that A (resp. B) has p (resp. q) columns. Given S', the steady-state flux cone in canonical form will look as follows
| (5) |
where matrix G (resp. H) represent the columns to be kept (resp. eliminated):
| (6) |
Here Ip denotes the p × p identity matrix, and 0p,q the p × q zero matrix.
Step 2 - Cone Projection: In this step, the flux cone is projected, eliminating the reactions corresponding to columns in H. Several methods have been proposed in the literature for the projection of polyhedra [48]. For our purpose we chose the block elimination method [49]. This method allows us to find an inequality description of the projected cone by enumerating the extreme rays of an intermediary cone called the projection cone. In our case, the projection cone is defined as
| (7) |
where HT denotes the transpose of H.
We enumerate the extreme rays {r1, r2, . . ., rk} of W using the double description method [50]. The projected cone is given by
| (8) |
where
| (9) |
This representation of the projected cone contains as many inequalities as there are extreme rays in W, thus a large number of them might be redundant [48]. These redundant inequalities are removed next (see below).
Step 3 - Finding ProCEMs: In the final step, the extreme rays of the projected cone, i.e., the ProCEMs, are enumerated. Similarly as in Step 2, the double description method is employed to enumerate the extreme rays of .
With the block elimination algorithm, it is also possible to perform the projection in an iterative manner. This means that rather than eliminating all the "uninteresting" reactions in one step, we can partition these in t subsets and then iteratively execute Step 2, eliminating every subset of reactions one by one. By proceeding in this fashion, the intermediary projection cones, W1, W2, . . ., Wt get typically smaller, thus enumerating their extreme rays requires less memory. On the other side, the more sets we partition into, the slower the projection algorithm usually gets.
Implementation and Computational Experiments
The ProCEM enumeration algorithm has been implemented in MATLAB v7.5. In our implementation, polco tool v4.7.1 [12,13] is used for the enumeration of extreme rays (both in Step 2 and 3). For removing redundant inequalities in Step 2, the redund method from the lrslib package v4.2 is used [51]. All computations are performed on a 64-bit Debian Linux system with Intel Core 2 Duo 3.0 GHz processor. A prototype implementation is available on request from the authors.
Dataset
The metabolic network model of red blood cell (RBC) [44] is used in this study. The network is taken from the example metabolic networks associated with CellNetAnalyzer [52] and differs slightly from the original model. Additionally, we studied the plastid metabolic network of Arabidopsis thaliana [53] (see Additional file 1). Then, the subnetwork of "sugar and starch metabolism" is selected as the interesting subnetwork of the plastid metabolic network.
Results and Discussion
Mathematical Relationships among PEMs, EFPs and ProCEMs
From Table 1, one can observe that the set of ProCEMs in Figure 1A is included in the set of PEMs. Additionally, the set of EFPs is included in the set of ProCEM supports. Here, we prove that these two properties are true in general. This means that the analysis of ProCEMs has at least two advantages compared to the analysis of EFPs. Firstly, ProCEMs can tell us about the flux ratio of different reactions in an elementary mode, while EFPs can only tell us whether the reaction has a non-zero value in that mode. Secondly, enumeration of ProCEMs may result in modes which cannot be obtained by EFP analysis.
Theorem 1. In a metabolic network with irreversible reactions only, let J (resp. P) be the set of ProCEMs (resp. PEMs) for a given set of interesting reactions. Then J ⊆ P.
Proof. We have to show that for every u ∈ J there exists an elementary mode e ∈ C in such that . We know that for any u ∈ J there exists v ∈ C such that .
Any v ∈ C can be written in the form , where e1, . . ., er are elementary modes of 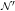 and c1, . . ., cr >0. It follows that .
and c1, . . ., cr >0. It follows that .
If all the vectors are pairwise equivalent, u is a PEM.
Otherwise, u is a linear combination of at least two non-equivalent PEMs, which are vectors in .
This implies that u is not an extreme ray of , in contradiction with Lemma 1 in [9] saying that in a metabolic network with irreversible reactions only, the EMs are exactly the extreme rays. □
Theorem 2. In a metabolic network with irreversible reactions only, let E (resp. J) be the set of EFPs (resp. ProCEMs) for a given set of interesting reactions. Then, E ⊆ {supp(u) | u ∈ J}.
Proof. Suppose that for some F ∈ E, there exists no v ∈ J such that F = supp(v). Since F is an EFP, there exists p ∈ P such that F = supp(p). It follows p ∉ J, but , where C is the flux cone. Therefore, there exist r ≥ 2 different ProCEMs, say u1,..., ur ∈ J, such that , with ck >0 for all k. Since uk ≥ 0, for all k, we have , with supp(uk) ≠ supp(p) for all k. Since supp(uk) is a flux pattern for all k, this is a contradiction with F being an EFP. □
Computing the Set of EFPs from the Set of ProCEMs
Here, we present a simple algorithm to show that it is possible to compute the set of EFPs when the set of ProCEMs is known. Table 2 summarizes this procedure.
Table 2.
Algorithm 1: Computing the set of EFPs based on the set of ProCEMs
| Input: |
| • J (the set of ProCEMs) |
| Output: |
| • E (the set of EFPs) |
| Initialization: |
| E := ∅; |
| Main procedure: |
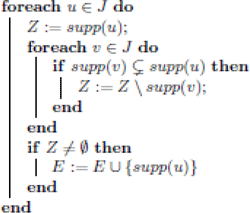 |
We know that the support of every ProCEM u is a flux pattern Z. In the main procedure, we check whether every such flux pattern is elementary or not. If Z is not elementary, then it is equal to the union of some other flux patterns. Therefore, if all other flux patterns which are subsets of supp(u) are subtracted from Z, this set becomes empty. This algorithm has the complexity , where q is the number of ProCEMs and n is the number of reactions.
Comparing EFPs and ProCEMs
Analysis of Subnetworks in the Metabolic Network of RBC
In order to compare our approach (computation of ProCEMs) with the enumeration of EFPs, we tested these methods for analysing subnetworks of the RBC model [44]. Again, we split every reversible reaction into one forward and one backward irreversible reaction. The resulting network contains 67 reactions, including 20 boundary reactions, and a total number of 811 EMs. For comparing the methods, the set of all boundary reactions was considered as the interesting subsystem, resulting in 502 PEMs.
When we computed the EFPs of this network by EFPTools [43], only 90 EFPs are determined. However, for the same subnetwork, we computed 252 ProCEMs. This means that the ProCEMs set covers more than half of the PEMs, while the EFPs set covers less than one fifth of the PEMs. These results confirm the relevance of using ProCEMs for the analysis of subnetworks.
In order to compare the computation of EFPs and ProCEMs, the following task was performed on the RBC model [44]. In each iteration, a random subnetwork containing r reactions was selected. Then, EFPs and ProCEMs were computed. The task was repeated for different subnetwork sizes. The computational results can be found in Figure 3.
Figure 3.

ProCEM vs. EFP computation. Left: Number of ProCEMs and EFPs computed for random subnetworks of different sizes. Right: The computation times (per second) required for computing the ProCEMs and EFPs in the left chart. ▲: ProCEMs; O: EFPs. Each experiment is repeated 100 times. Confidence intervals in this plot are based on one-sample t-test (95% c.i.). For large subnetworks (r > 40), we did not compute the EFPs because the program was very slow.
From Figure 3, it can be seen that EFP computation is faster than ProCEM computation for small subnetworks. However, when the subnetwork size r increases, computation of ProCEMs does not become slower, while computation of EFPs significantly slows down. This is an important observation, because the difference between the number of EFPs and ProCEMs also increases with r.
Analysis of Subnetworks in the Plastid Metabolic Network of A. thaliana
ProCEM analysis becomes important when PEMs cannot be computed. This may happen frequently in the analysis of large-scale metabolic networks, as memory consumption is a major challenge in computation of EMs [12]. In such cases, cone projection might still be feasible.
As an example, the metabolic network of A. thaliana plastid was studied (Additional file 1). This network contains 102 metabolites and 123 reactions (205 reactions after splitting reversible reactions). Using efmtool (and also polco) [12], even after specifying 2 GB of memory, computation of EMs was not possible due to running out of memory. Therefore, for no subnetwork of the plastid network, PEMs could be computed. However, if the analysis is restricted to the 57 reactions involved in sugar and starch metabolism (see Additional file 1), one can compute the ProCEMs or EFPs of this subnetwork. We computed the ProCEMs as described in the Method and Implementation section, using a projection step size of 5 reactions. The complete set of 1310 ProCEMs was computed in approximately 15 minutes. However, when we tried to compute the set of EFPs using EFPTools [35,43], only 279 EFPs were computed after 4 days of running the program (270 EFPs were computed in the first two days). On the other hand, using a Matlab implementation of Algorithm 1, the complete set of 1054 EFPs was obtained in 30 seconds. In conclusion, in metabolic networks for which the set of EMs cannot be enumerated, ProCEMs prove to be a useful concept to get insight into reaction activities.
Conclusions
In this paper, we introduce the concept of projected cone elementary modes (ProCEMs). The set of ProCEMs covers more PEMs than EFPs. Therefore, ProCEMs contain more information than EFPs. The set of ProCEMs is computable without enumerating all EMs. Is there a bigger set of vectors that covers even more PEMs and does not require full enumeration of EMs? This question is yet to be answered. One possible extension to this work is to use a more efficient implementation of polyhedral projection. With such an implementation, analysis of different subnetworks in genome-scale network models using ProCEMs is an interesting possibility for further research. For example, the ProCEMs can be used in the identification of all pathways with optimal yield [23] and in the analysis of control-effective fluxes [27].
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The original idea was presented by SAM, LD and AB. The mathematical results are presented by SAM, and improved by all authors. Implementation of the ProCEM method and performing the computational experiments are done by LD. The manuscript was originally drafted by SAM, and improved by all authors. The final version of the manuscript was read and approved by all authors.
Supplementary Material
Plastid metabolic network. In the first tab of this Excel file, general information about the plastid network of A. thaliana is mentioned. In the second tab, stoichiometric matrix and the set of reversible reactions (as a binary vector) is included. In the third tab, the reactions involved in sugar and starch metabolism are listed.
Contributor Information
Sayed-Amir Marashi, Email: marashi@molgen.mpg.de.
Laszlo David, Email: Laszlo.David@fu-berlin.de.
Alexander Bockmayr, Email: Alexander.Bockmayr@fu-berlin.de.
References
- Klamt S, Stelling J. Two approaches for metabolic pathway analysis? Trends in Biotechnology. 2003;21:64–69. doi: 10.1016/S0167-7799(02)00034-3. [DOI] [PubMed] [Google Scholar]
- Terzer M, Maynard ND, Covert MW, Stelling J. Genome-scale metabolic networks. WIREs Systems Biology and Medicine. 2009;1:285–297. doi: 10.1002/wsbm.37. [DOI] [PubMed] [Google Scholar]
- Schuster S, Hilgetag C. On elementary flux modes in biochemical reaction systems at steady state. Journal of Biological Systems. 1994;2:165–182. doi: 10.1142/S0218339094000131. [DOI] [Google Scholar]
- Schuster S, Fell DA, Dandekar T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nature Biotechnology. 2000;18:326–332. doi: 10.1038/73786. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Letscher D, Palsson BO. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. Journal of Theoretical Biology. 2000;203:229–248. doi: 10.1006/jtbi.2000.1073. [DOI] [PubMed] [Google Scholar]
- Jevremovic D, Trinh CT, Srienc F, Boley D. On algebraic properties of extreme pathways in metabolic networks. Journal of Computational Biology. 2010;17:107–119. doi: 10.1089/cmb.2009.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer T, Sánchez-Valdenebro I, Nuño JC, Montero F, Schuster S. METATOOL: for studying metabolic networks. Bioinformatics. 1999;15:251–257. doi: 10.1093/bioinformatics/15.3.251. [DOI] [PubMed] [Google Scholar]
- Wagner C. Nullspace Approach to Determine the Elementary Modes of Chemical Reaction Systems. Journal of Physical Chemistry B. 2004;108:2425–2431. doi: 10.1021/jp034523f. [DOI] [Google Scholar]
- Gagneur J, Klamt S. Computation of elementary modes: a unifying framework and the new binary approach. BMC Bioinformatics. 2004;5:175. doi: 10.1186/1471-2105-5-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt S, Gagneur J, von Kamp A. Algorithmic approaches for computing elementary modes in large biochemical reaction networks. IEE Proceedings - Systems Biology. 2005;152:249–255. doi: 10.1049/ip-syb:20050035. [DOI] [PubMed] [Google Scholar]
- von Kamp A, Schuster S. Metatool 5.0: fast and flexible elementary modes analysis. Bioinformatics. 2006;22:1930–1931. doi: 10.1093/bioinformatics/btl267. [DOI] [PubMed] [Google Scholar]
- Terzer M, Stelling J. Large-scale computation of elementary flux modes with bit pattern trees. Bioinformatics. 2008;24:2229–2235. doi: 10.1093/bioinformatics/btn401. [DOI] [PubMed] [Google Scholar]
- Terzer M, Stelling J. Proceedings of the 8th International Conference on Parallel Processing and Applied Mathematics (PPAM 2009), Volume 6068 of Lecture Notes in Computer Science. Wroclaw, Poland; 2010. Parallel Extreme Ray and Pathway Computation; pp. 300–309. [DOI] [Google Scholar]
- Bell SL, Palsson BO. Expa: a program for calculating extreme pathways in biochemical reaction networks. Bioinformatics. 2005;21:1739–1740. doi: 10.1093/bioinformatics/bti228. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Schuster S, Palsson BO, Heinrich R. Metabolic pathway analysis: Basic concepts and scientific applications in the post-genomic era. Biotechnology Progress. 1999;15:296–303. doi: 10.1021/bp990048k. [DOI] [PubMed] [Google Scholar]
- Trinh CT, Wlaschin A, Srienc F. Elementary mode analysis: a useful metabolic pathway analysis tool for characterizing cellular metabolism. Applied Microbiology and Biotechnology. 2009;81:813–826. doi: 10.1007/s00253-008-1770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt S, Stelling J. Combinatorial complexity of pathway analysis in metabolic networks. Molecular Biology Reports. 2002;29:233–236. doi: 10.1023/A:1020390132244. [DOI] [PubMed] [Google Scholar]
- Yeung M, Thiele I, Palsson BO. Estimation of the number of extreme pathways for metabolic networks. BMC Bioinformatics. 2007;8:363. doi: 10.1186/1471-2105-8-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Palsson BO. Constraints-based models: regulation of gene expression reduces the steady-state solution space. Journal of Theoretical Biology. 2003;221:309–325. doi: 10.1006/jtbi.2003.3071. [DOI] [PubMed] [Google Scholar]
- Urbanczik R. Enumerating constrained elementary flux vectors of metabolic networks. IET Systems Biology. 2007;1(5):274–279. doi: 10.1049/iet-syb:20060073. [DOI] [PubMed] [Google Scholar]
- Song HS, Ramkrishna D. Reduction of a set of elementary modes using yield analysis. Biotechnology and Bioengineering. 2009;102:554–568. doi: 10.1002/bit.22062. [DOI] [PubMed] [Google Scholar]
- Nuño JC, Sánchez-Valdenebro I, Pérez-Iratxeta C, Meléndez-Hevia E, Montero F. Network organization of cell metabolism: monosaccharide interconversion. Biochemical Journal. 1997;324:103–111. doi: 10.1042/bj3240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S, Dandekar T, Fell DA. Detection of elementary flux modes in biochemical networks: A promising tool for pathway analysis and metabolic engineering. Trends in Biotechnology. 1999;17:53–60. doi: 10.1016/S0167-7799(98)01290-6. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Palsson BO. Assessment of the metabolic capabilities of Haemophilus influenzae Rd through a genome-scale pathway analysis. Journal of Theoretical Biology. 2000;203:249–283. doi: 10.1006/jtbi.2000.1088. [DOI] [PubMed] [Google Scholar]
- Schilling CH, Covert MW, Famili I, Church GM, Edwards JS, Palsson BO. Genome-scale metabolic model of Helicobacter pylori 26695. Journal of Bacteriology. 2002;184:4582–4593. doi: 10.1128/JB.184.16.4582-4593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S, Pfeiffer T, Moldenhauer F, Koch I, Dandekar T. Exploring the pathway structure of metabolism: decomposition into subnetworks and application to Mycoplasma pneumoniae. Bioinformatics. 2002;18:351–361. doi: 10.1093/bioinformatics/18.2.351. [DOI] [PubMed] [Google Scholar]
- Stelling J, Klamt S, Bettenbrock K, Schuster S, Gilles ED. Metabolic network structure determines key aspects of functionality and regulation. Nature. 2002;420:190–193. doi: 10.1038/nature01166. [DOI] [PubMed] [Google Scholar]
- Çakır T, Kırdar B, Ülgen KO. Metabolic Pathway Analysis of Yeast Strengthens the Bridge between Transcriptomics and Metabolic Networks. Biotechnology and Bioengineering. 2004;86:251–260. doi: 10.1002/bit.20020. [DOI] [PubMed] [Google Scholar]
- Schwarz R, Musch P, von Kamp A, Engels B, Schirmer H, Schuster S, Dandekar T. YANA - a software tool for analyzing flux modes, gene-expression and enzyme activities. BMC Bioinformatics. 2005;6:135. doi: 10.1186/1471-2105-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verwoerd W. MODSIM07. Christchurch, New Zealand; 2007. Identifying coherent subnetworks in genome scale metabolic networks; pp. 2013–2019. [Google Scholar]
- Verwoerd WS. A new computational method to split large biochemical networks into coherent subnets. BMC Systems Biology. 2011;5:25. doi: 10.1186/1752-0509-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Varner JD, Ramkrishna D. A hybrid model of anaerobic E. coli GJT001: combination of elementary flux modes and cybernetic variables. Biotechnology Progress. 2008;24:993–1006. doi: 10.1002/btpr.73. [DOI] [PubMed] [Google Scholar]
- Teusink B, Wiersma A, Jacobs L, Notebaart RA, Smid EJ. Understanding the adaptive growth strategy of Lactobacillus plantarum by in silico optimisation. PLoS Computational Biology. 2009;5:e1000410. doi: 10.1371/journal.pcbi.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenanov D, Kaleta C, Petzold A, Hoischen C, Diekmann S, Siddiqui RA, Schuster S. Theoretical study of lipid biosynthesis in wild-type Escherichia coli and in a protoplast-typeL-form using elementary flux mode analysis. FEBS Journal. 2010;277:1023–1034. doi: 10.1111/j.1742-4658.2009.07546.x. [DOI] [PubMed] [Google Scholar]
- Kaleta C, de Figueiredo LF, Schuster S. Can the whole be less than the sum of its parts? Pathway analysis in genome-scale metabolic networks using elementary flux patterns. Genome Research. 2009;19:1872–1883. doi: 10.1101/gr.090639.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marashi SA, David L, Bockmayr A. On flux coupling analysis of metabolic subsystems. Journal of Theoretical Biology. 2012;302:62–69. doi: 10.1016/j.jtbi.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Burgard AP, Nikolaev EV, Schilling CH, Maranas CD. Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Research. 2004;14:301–312. doi: 10.1101/gr.1926504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Belta C. Exploiting the pathway structure of metabolism to reveal high-order epistasis. BMC Systems Biology. 2008;2:40. doi: 10.1186/1752-0509-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S, Klamt S, Weckwerth W, Moldenhauer F, Pfeiffer T. Use of network analysis of metabolic systems in bioengineering. Bioprocess and Biosystems Engineering. 2001;24:363–372. doi: 10.1007/s004490100253. [DOI] [Google Scholar]
- Zhao Q, Kurata H. Genetic modification of flux for flux prediction of mutants. Bioinformatics. 2009;25:1702–1708. doi: 10.1093/bioinformatics/btp298. [DOI] [PubMed] [Google Scholar]
- Marashi SA, Bockmayr A. Flux coupling analysis of metabolic networks is sensitive to missing reactions. BioSystems. 2011;103:57–66. doi: 10.1016/j.biosystems.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Acuña V, Marchetti-Spaccamela A, Sagot M, Stougie L. A note on the complexity of finding and enumerating elementary modes. BioSystems. 2010;99:210–214. doi: 10.1016/j.biosystems.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Kaleta C. EFPTools, for computing elementary flux patterns (EFPs) 2009. http://users.minet.uni-jena.de/~m3kach/EFPA/
- Wiback SJ, Palsson BO. Extreme pathway analysis of human red blood cell metabolism. Biophysical Journal. 2002;83:808–818. doi: 10.1016/S0006-3495(02)75210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C, Urbanczik R. The geometry of the flux cone of a metabolic network. Biophysical Journal. 2005;89:3837–3845. doi: 10.1529/biophysj.104.055129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanczik R, Wagner C. Functional stoichiometric analysis of metabolic networks. Bioinformatics. 2005;21:4176–4180. doi: 10.1093/bioinformatics/bti674. [DOI] [PubMed] [Google Scholar]
- Urbanczik R. SNA-a toolbox for the stoichiometric analysis of metabolic networks. BMC Bioinformatics. 2006;7:129. doi: 10.1186/1471-2105-7-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CN, Kerrigan EC, Maciejowski JM. On polyhedral projection and parametric programming. Journal of Optimization Theory and Applications. 2008;138:207–220. doi: 10.1007/s10957-008-9384-4. [DOI] [Google Scholar]
- Balas E, Pulleyblank W. The perfectly matchable subgraph polytope of a bipartite graph. Networks. 1983;13:495–516. doi: 10.1002/net.3230130405. [DOI] [Google Scholar]
- Fukuda K, Prodon A. Double description method revisited. Combinatorics and Computer Science: 8th Franco-Japanese and 4th Franco-Chinese Conference. Brest, France, Volume 1120 of Lecture Notes in Computer Science. 1996. pp. 91–111. [DOI]
- Avis D. In: Polytopes - Combinatorics and Computation, Oberwolfach Seminars. Kalai G, Ziegler G, editor. Birkhäuser-Verlag; 2000. lrs: A revised implementation of the reverse search vertex enumeration algorithm; pp. 177–198. [Google Scholar]
- Klamt S, Saez-Rodriguez J, Gilles ED. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Systems Biology. 2007;1:2. doi: 10.1186/1752-0509-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Dal'Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiology. 2010;152:579–589. doi: 10.1104/pp.109.148817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plastid metabolic network. In the first tab of this Excel file, general information about the plastid network of A. thaliana is mentioned. In the second tab, stoichiometric matrix and the set of reversible reactions (as a binary vector) is included. In the third tab, the reactions involved in sugar and starch metabolism are listed.