

Neutrophils induce endothelial docking structures prior to crossing the blood vessel wall. The Rho guanine nucleotide exchange factor Trio regulates the formation of these structures through ICAM-1 clustering in a filamin-dependent fashion. We show that Trio is a crucial mediator of the signaling pathway that controls leukocyte extravasation through docking structure formation.
Abstract
Leukocyte transendothelial migration involves the active participation of the endothelium through the formation of apical membrane protrusions that embrace adherent leukocytes, termed docking structures. Using live-cell imaging, we find that prior to transmigration, endothelial docking structures form around 80% of all neutrophils. Previously we showed that endothelial RhoG and SGEF control leukocyte transmigration. In this study, our data reveal that both full-length Trio and the first DH-PH (TrioD1) domain of Trio, which can activate Rac1 and RhoG, interact with ICAM-1 and are recruited to leukocyte adhesion sites. Moreover, upon clustering of ICAM-1, the Rho-guanine nucleotide exchange factor Trio activates Rac1, prior to activating RhoG, in a filamin-dependent manner. We further show that docking structure formation is initiated by ICAM-1 clustering into ring-like structures, which is followed by apical membrane protrusion. Interestingly, we find that Rac1 is required for ICAM-1 clustering, whereas RhoG controls membrane protrusion formation. Finally, silencing endothelial Trio expression or reducing TrioD1 activity without affecting SGEF impairs both docking structure formation and leukocyte transmigration. We conclude that Trio promotes leukocyte transendothelial migration by inducing endothelial docking structure formation in a filamin-dependent manner through the activation of Rac1 and RhoG.
INTRODUCTION
Under inflammatory conditions and in diseases such as rheumatoid arthritis and atherosclerosis (Ross, 1999; Libby, 2002), leukocytes from the circulation are activated to adhere to and transmigrate across the blood vessel wall. Initial adhesion is mediated by the selectin family of adhesion receptors, which mediate leukocyte rolling and tethering to the endothelium. Chemokine-triggered activation of leukocytic β2 and β1 integrins then allows firm adhesion through binding to endothelial adhesion molecules ICAM-1 (CD54) and VCAM-1 (CD106), respectively (Ley et al., 2007; Vestweber, 2007; Nourshargh et al., 2010). Binding and subsequent clustering of ICAM-1 induces endothelial signaling, including a rise in cytosolic calcium, activation of the tyrosine kinase Src, activation of the small GTPase RhoA, formation of F-actin stress fibers, and phosphorylation of vascular endothelial (VE)-cadherin (Huang et al., 1993; Durieu-Trautmann et al., 1994; Etienne et al., 1998; Adamson et al., 1999; Wojciak-Stothard et al., 1999; Etienne-Manneville et al., 2000; Sans et al., 2001; Allingham et al., 2007; Alcaide et al., 2008; Turowski et al., 2008). Engagement of ICAM-1 leads to the redistribution of ICAM-1 and F-actin into a ring-like structure around the adherent leukocyte (Carman et al., 2003; Shaw et al., 2004). Anchorage of ICAM-1 to the F-actin cytoskeleton via adaptor proteins, such as filamin and cortactin, is essential for formation of ICAM-1 rings and efficient leukocyte transendothelial migration (TEM; Tilghman and Hoover, 2002; Yang et al., 2006; Kanters et al., 2008; van Buul et al., 2010; Schnoor et al., 2011). Detailed analyses of ICAM-1 rings also revealed large microvilli-like membrane protrusions that surround and finally embrace adherent leukocytes, forming cup-like structures known as endothelial docking structures or transmigratory cups (Barreiro et al., 2002; Carman et al., 2003; Carman and Springer, 2004). Endothelial docking structures may protect transmigrating leukocytes from hemodynamic shear forces and promote transmigration (Carman and Springer, 2004). We have shown that the small GTPase RhoG is activated after ICAM-1 clustering and is involved in the assembly of these docking structures (van Buul et al., 2007). Next to RhoA and RhoG, the Rho family GTPase Rac1 is also activated after clustering of ICAM-1 (van Buul et al., 2007), but its precise contribution to ICAM-1 signaling and docking structure formation is not yet understood.
The F-actin cross-linker protein filamin interacts with Rac1 and its guanine nucleotide exchange factors (GEFs) Trio (Bellanger et al., 2000) and Vav2 (Del Valle-Perez et al., 2010), and functions as a scaffold protein for Rac1 signaling (Jeon et al., 2008; Del Valle-Perez et al., 2010). We have previously shown that upon clustering of ICAM-1, filamin B interacts with the intracellular domain of ICAM-1 and is required for ICAM-1 ring formation and leukocyte TEM (Kanters et al., 2008). In this study, we investigated the involvement of the filamin-binding GEF Trio and the relative contributions of its substrates Rac1 and RhoG to endothelial docking structure formation and leukocyte TEM.
RESULTS
ICAM-1 clustering activates the GEF Trio
Endothelial docking structures are rich in F-actin and actin-binding proteins (Barreiro et al., 2002; Carman et al., 2003). For studying the dynamics of endothelial actin during leukocyte transmigration, green fluorescent protein (GFP)-LifeAct was expressed in endothelial cells. Three-dimensional live-cell imaging using confocal microscopy demonstrated that neutrophils induced F-actin–rich protrusions prior to transmigration (Figure 1A). The adhesion receptor ICAM-1 is anchored to the F-actin cytoskeleton via actin-binding adaptor proteins (van Buul and Hordijk, 2009). We have previously shown that the actin-binding protein filamin B is an important regulator of ICAM-1–mediated leukocyte TEM (Kanters et al., 2008). In addition to filamin B, endothelial cells also express its close homologue, filamin A. On small interfering RNA (siRNA)-mediated knockdown of filamin A or B in fibrosarcoma cells, the muscle-specific filamin isoform, filamin C, is upregulated (Baldassarre et al., 2009). We therefore analyzed the expression of filamin C protein in human umbilical vein endothelial cells (HUVEC). Filamin C protein expression was clearly detected (Figure 1B), in line with its previously reported mRNA expression in HUVEC (Xie et al., 1998). To assess whether filamin C is also able to interact with the intracellular domain of ICAM-1, we used a biotinylated peptide comprising the C-terminal 28 amino acids of ICAM-1 coupled to streptavidin agarose (Kanters et al., 2008), and pulldown assays were performed on HUVEC lysates. In both control and filamin A/B knockdown conditions, filamin C bound equally well to the ICAM-1 C-terminal peptide (Figure 1B). Moreover, our data showed that reduction of filamin B increases the binding of filamin A to the intracellular tail of ICAM-1 and vice versa, indicating that the filamins may compete for binding to the intracellular tail to ICAM-1, thereby affecting downstream signaling (Supplemental Figure S1). To examine the role of filamin in the activation of Rac1 upon ICAM-1 clustering, we therefore decided to reduce the expression all three filamin isoforms (A, B, and C) in HUVEC using siRNA (Figure 1C). Leukocyte binding to ICAM-1 was mimicked by clustering of ICAM-1 induced by polystyrene beads coated with anti–ICAM-1 antibodies (α-ICAM-1–antibody beads; van Buul et al., 2007, 2010). In siCtrl-transfected cells, ICAM-1 clustering induced Rac1 activation after 10 and 30 min (Figure 1D). However, in cells transfected with siRNA to all three filamin isoforms simultaneously, ICAM-1–induced Rac1 activation was severely impaired. In addition, analysis of RhoG activity following ICAM-1 clustering demonstrated that filamin is also required for the activation of RhoG (Figure 1E).
FIGURE 1:

Rac1 and RhoG activation upon ICAM-1 clustering requires filamin. (A) Formation of F-actin–positive docking structures around transmigrating neutrophils was visualized by GFP-LifeAct in TNF-α–stimulated HUVEC. Top, left, GFP-LifeAct recruitment at different Z-stack levels, as indicated above. Bottom, left, a docking structure along the x–z axis. The leukocyte is marked with a asterisk. Middle, a differential interference contrast (DIC) image of a transmigrating neutrophils (XY, asterisk). Right, a three-dimensional Z-stack projection (3D). (B) For assessment of filamin C interaction with ICAM-1, HUVEC were transfected with control or filamin A and B siRNA, and pulldown assays were performed with the ICAM-1 C-terminal peptide. Streptavidin agarose without peptide was used as a control. (C) siRNA-mediated knockdown of filamin A/B/C expression in HUVEC was confirmed by Western blotting. Knockdown of filamin does not affect ICAM-1 expression induced by 20-h TNF-α treatment. Actin was used as a control for equal sample loading. ICAM-1 was clustered (CL) with α-ICAM-1–antibody beads on siCtrl and siFilamin A/B/C-transfected HUVEC for 10 and 30 min, and Rac1-GTP (D) and RhoG-GTP (E) were precipitated with biotinylated Pak1-CRIB peptide coupled to streptavidin agarose and GST-ELMO coupled to glutathione Sepharose pulldown (PD) assays, respectively. Rac1.GTP and RhoG.GTP levels were quantified and corrected for Rac1 and RhoG total lysate levels with ImageJ. Quantification of Western blots represents an average of at least three independent experiments ± SEM. *, p < 0.05.
Filamin can interact with two Rho-GEFs, Trio and Vav2 (Bellanger et al., 2000; Del Valle-Perez et al., 2010), which both mediate guanine nucleotide exchange on Rac1 and RhoG. Trio is an unusual multi-domain GEF protein, in that it encompasses two separate Dbl homology–pleckstrin homology (DH-PH) GEF units with different specificities. The N-terminal GEF unit (TrioD1) mediates nucleotide exchange on RhoG and Rac1, whereas the C-terminal GEF unit (TrioD2) can activate RhoA (Debant et al., 1996; Blangy et al., 2000). In contrast, Vav2 only has a single DH-PH catalytic subunit with very broad specificities for RhoG, Rac1, CDC42, and RhoA (Liu and Burridge, 2000; Wennerberg et al., 2002). Analysis of endogenous Trio and Vav2 showed expression in several endothelial cell types (Figure 2A). Interestingly, in HUVEC treated for 20 h with the inflammatory cytokine tumor necrosis factor α (TNF-α), Trio, but not Vav2, protein levels were increased (Figure 2A).
FIGURE 2:
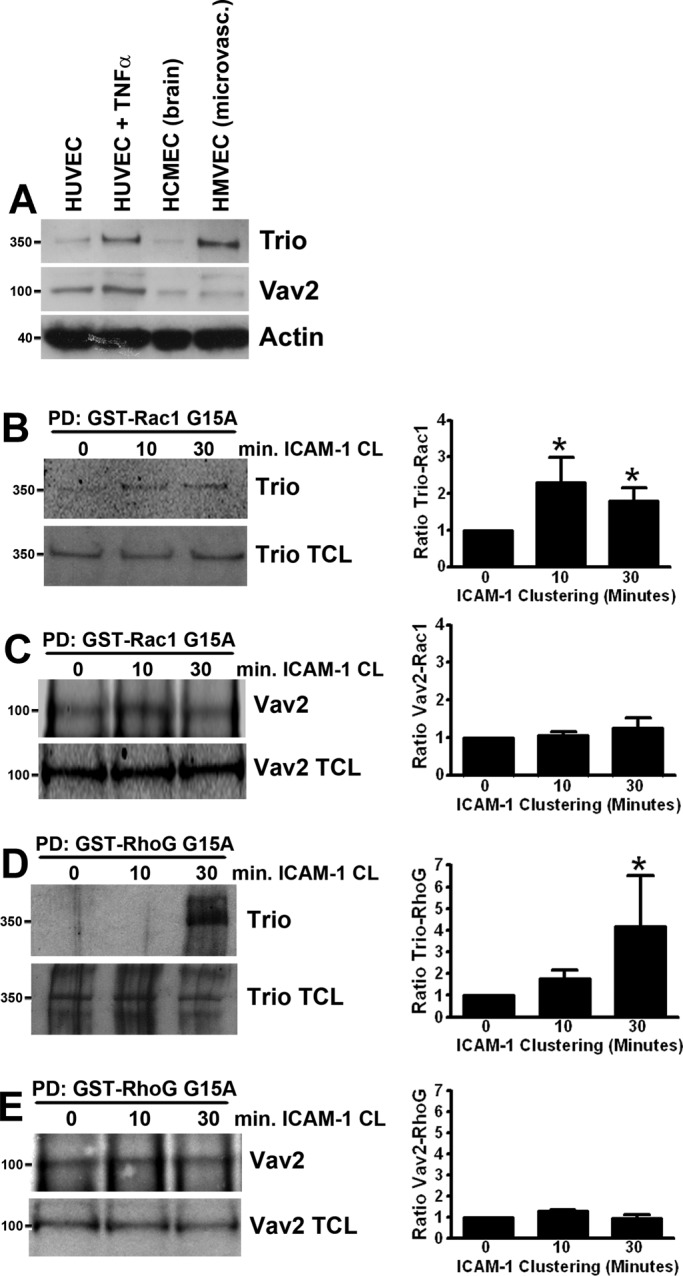
ICAM-1 clustering induces activation of the GEF Trio. (A) Trio (350 kDa) and Vav2 (100 kDa) protein expression in HUVEC (HUVEC, lane 1; HUVEC treated for 20 h with TNF-α, lane 2), human brain endothelial cells (hCMEC/D3, lane 3), and HMVEC (lane 4). Trio (B and D) and Vav2 (C and E) activation was assessed by their affinity in pulldown (PD) assays for GST-fusion protein of nucleotide-free mutants of Rac1 G15A (B and C) and RhoG G15A (D and E) coupled to glutathione Sepharose. Trio and Vav2 binding were quantified and corrected for Trio and Vav2 total lysate levels with ImageJ. Quantification of Western blots represents an average of at least three independent experiments ± SEM. *, p < 0.05.
To study the role of Trio and Vav2 in Rac1 and RhoG activation by ICAM-1 clustering in endothelial cells, we examined the binding of Trio and Vav2 to glutathione S-transferase (GST)-fusion proteins of nucleotide-free mutants of Rac1 (GST-Rac1 G15A) and RhoG (GST-RhoG G15A). Because these mutants are not able to bind GDP or GTP, they have a high affinity for GEFs and can therefore be used for purification of activated GEFs (Garcia-Mata et al., 2006). After clustering of ICAM-1 on TNF-α–stimulated HUVEC with α-ICAM-1–antibody beads, pulldown assays with the nucleotide-free mutants of RhoG and Rac1 were performed. Following 10 or 30 min of ICAM-1 clustering, increased binding of Trio (Figure 2B), but not Vav2 (Figure 2C) to nucleotide-free Rac1 was detected. Despite the fact that the purified RhoG G15A mutant protein is very unstable (Garcia-Mata et al., 2006), we were able to detect Trio binding to nucleotide-free RhoG after 30 min of ICAM-1 clustering (Figure 2D), whereas Vav2 interaction was not altered (Figure 2E). These data indicate that the GEF Trio, but not Vav2, is activated upon ICAM-1 clustering to mediate nucleotide exchange on Rac1 prior to RhoG.
Trio is localized at sites of ICAM-1 clustering
To study Trio localization during ICAM-1 clustering, we generated a GFP-fusion protein of full-length Trio (GFP-Trio FL). Expression of GFP-Trio FL together with mCherry-tagged ICAM-1 (ICAM-1-mCherry) in HeLa cells induced partial colocalization of the two proteins (Supplemental Video S1). However, when ICAM-1 was clustered, both ICAM-1 and Trio were recruited and colocalized to ring-like structures that formed around α-ICAM-1–antibody beads (Figure S2A and Video S1) and neutrophils (Figure S2). ICAM-1 clustering in TNF-α–stimulated primary HUVEC induced the recruitment of GFP-Trio FL and endogenous ICAM-1 around α-ICAM-1–antibody beads (Figure 3A) and adherent neutrophils (Figure 3B).
FIGURE 3:
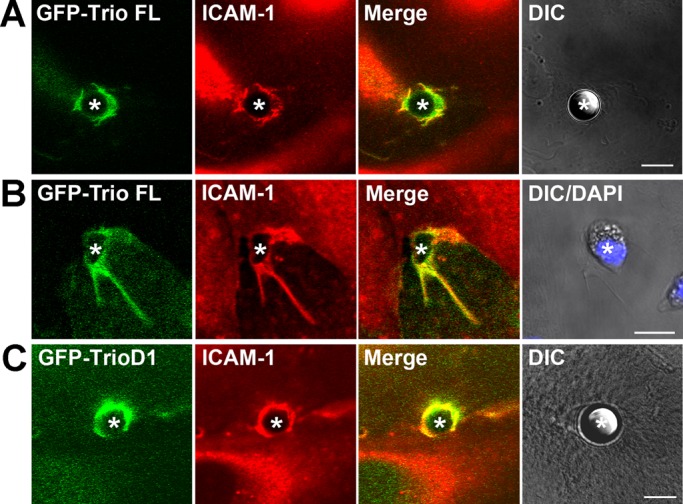
Trio and TrioD1 colocalize with ICAM-1 upon clustering. Endogenous ICAM-1 (red) in TNF-α–stimulated endothelial cells colocalizes with GFP-Trio full length to 10-μm α-ICAM-1–antibody beads (A, green) or to adherent neutrophils (B) and with TrioD1 (C, green) to these beads (asterisks). In (B), nuclei of neutrophils are visualized with DAPI staining and are included in the DIC images. Scale bars: 10 μm.
In addition to full-length Trio, we also generated a GFP-fusion protein of the N-terminal GEF domain of Trio (GFP-TrioD1). When expressed in HeLa cells or in HUVEC with an adenoviral approach, GFP-TrioD1 was recruited around α-ICAM-1–antibody beads together with endogenous ICAM-1 in HUVEC (Figure 3C) and with ICAM-1-mCherry in HeLa cells (Figure S2C). In addition, in HUVEC coexpressing GFP-TrioD1 and red fluorescent protein (RFP)-LifeAct (to visualize F-actin), both TrioD1 and F-actin formed a ring-like structure around transmigrating neutrophils (Video S2). To investigate whether TrioD1 recruitment is dependent on the intracellular C-terminal domain of ICAM-1, we coexpressed GFP-TrioD1 with a C-terminal deletion mutant of ICAM-1-mCherry (ICAM-1ΔC-mCherry) in HeLa cells and clustered ICAM-1 with α-ICAM-1–antibody beads. ICAM-1ΔC-mCherry was recruited around α-ICAM-1–antibody beads, albeit to a lesser extent than ICAM-1-mCherry, but no colocalization with GFP-TrioD1 was observed (Figure S2D). These results show that Trio is recruited to sites of ICAM-1–mediated leukocyte adhesion, which requires the intracellular domain of ICAM-1.
Trio interacts with ICAM-1
Because a cytoplasmic domain deletion mutant of ICAM-1 was unable to recruit TrioD1 to sites of ICAM-1 clustering, we tested whether Trio physically interacted with the C-terminal domain of ICAM-1. Pulldown assays were performed on lysates of cells that expressed Myc-TrioD1 with the ICAM-1 C-terminal peptide. Western blot analysis showed that Myc-TrioD1 (Figure 4A) interacted specifically with the ICAM-1 C-terminal peptide, but not with the peptide encoding the intracellular domain of VCAM-1 or control beads. The adaptor protein filamin binds to the intracellular domain of ICAM-1 (Figure 4B; Kanters et al., 2008) and has been shown to interact with Trio and to be required for Trio-mediated remodeling of the F-actin cytoskeleton (Bellanger et al., 2000). Therefore we tested whether filamin could mediate the interaction of Trio with ICAM-1 by using siRNA-mediated protein silencing of filamin. Surprisingly, silencing of filamin A or filamin B () did not reduce the binding of Trio to the ICAM-1 C-terminal peptide. Filamin A and B share ∼70% sequence homology, so they may be functionally redundant. However, in cells with simultaneous filamin A and B silencing, endogenous Trio or Myc-Trio FL interaction with the ICAM-1 C-terminal peptide also was not significantly impaired (Figures 4B and S3). We showed that a third isoform, filamin C, is also able to interact with ICAM-1 (Figure 1A). To investigate whether filamin C mediated the interaction between Trio and ICAM-1, we silenced filamin C expression using siRNA. Both knockdown of filamin C and knockdown of all three filamin A/B/C isoforms did not reduce the binding of Trio or Myc-Trio to the ICAM-1 C-terminal peptide (Figures 4B and S3). These data indicate that filamin plays a minor role in the interaction of Trio with ICAM-1.
FIGURE 4:
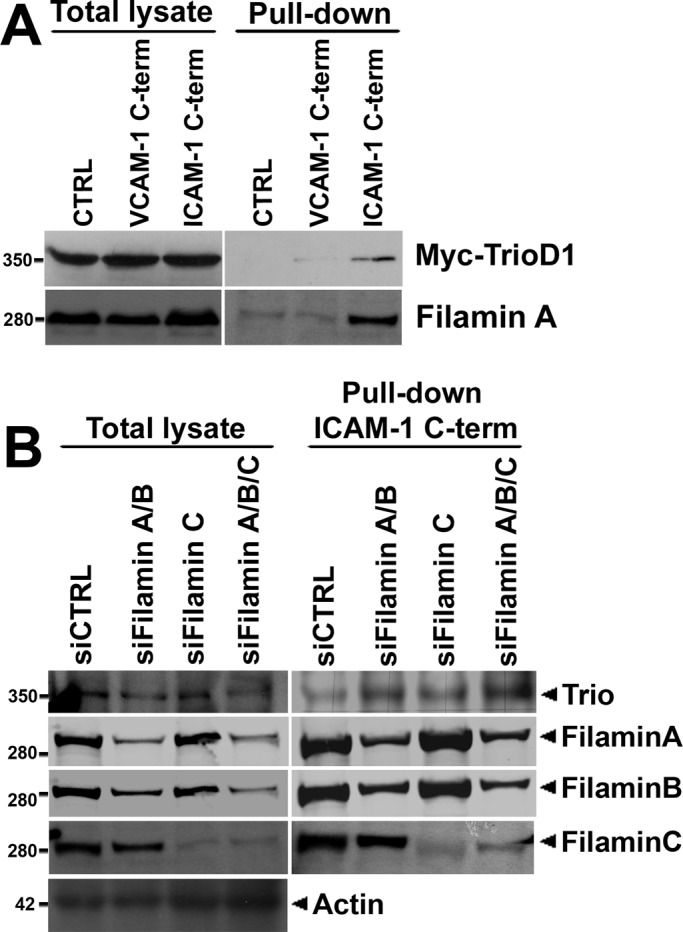
Trio interacts with ICAM-1 independently of filamin. (A) Cos7 cells were transiently transfected with Myc-TrioD1 (B), and pulldown assays were performed with the ICAM-1 C-terminal peptide, streptavidin-agarose without peptide (CTRL), or a peptide of the VCAM-1 C-terminal intracellular tail, as described in Materials and Methods. Interaction was assessed by immunoblotting with anti-Myc antibody. Filamin A binding to the ICAM-1 C-terminal peptide was used as a positive control. (B) HUVEC were transfected with siRNA targeting filamin A/B, filamin C, or filamin A/B/C together, and pulldown assays were performed with the ICAM-1 C-terminal peptide for endogenous Trio. No difference in binding of Trio to the ICAM-1 C-terminal peptide was observed. Actin was used as protein loading control. A representative result of three independent experiments is shown. Quantification is shown in Figure S3B.
Filamin is required for Trio activation upon ICAM-1 clustering
Filamin has been demonstrated to be a scaffold protein for Vav2-mediated Rac1 activation and Rac1 downstream signaling (Jeon et al., 2008; Del Valle-Perez et al., 2010). Bellanger and colleagues have shown that although the addition of filamin did not improve the in vitro guanine nucleotide exchange activity of TrioD1, filamin is required for TrioD1-induced membrane dynamics (Bellanger et al., 2000). Because we found filamin to be required for Rac1 activation upon ICAM-1 clustering (Figure 1, B and C), we examined whether filamin regulates the interaction of Trio with nucleotide-free Rac1 upon ICAM-1 clustering. Whereas ICAM-1 clustering with α-ICAM-1–antibody beads induced the interaction of Trio with nucleotide-free Rac1 G15A mutant under control conditions, knockdown of filamin A/B/C prevented this induction (Figure 5A). In addition, silencing of filamin A/B/C also prevented the ICAM-1–induced interaction of Trio with nucleotide-free RhoG G15A (Figure 5B). As a control, Vav2 was stained, but no interaction was found between Vav2 and RhoG G15A (Figure S4). These results therefore show that although Trio interacts independently of filamin with the ICAM-1 cytoplasmic domain, filamin is required for Trio activation downstream of ICAM-1 clustering. This suggests that filamin is a scaffold for Trio-mediated Rac1 and RhoG activation upon ICAM-1 clustering.
FIGURE 5:

Trio activation upon ICAM-1 clustering requires filamin. ICAM-1 was clustered (CL) for 10 and 30 min on TNF-α–stimulated HUVEC transfected with siRNA targeting filamin A/B/C. Trio activation was assessed by its affinity in pulldown (PD) assays with a GST-fusion protein of nucleotide-free mutant of Rac1 G15A (A) or RhoG G15A (B) coupled to glutathione Sepharose. Trio binding was quantified and corrected for Trio total lysate levels with ImageJ. Quantification of Trio binding to GST-Rac1 G15A Western blots represents an average of three independent experiments ± SEM.
Trio knockdown reduces neutrophil adhesion and TEM under flow
To test whether Trio is required for efficient leukocyte transendothelial migration, we initially reduced Trio expression with various siRNAs. However, knockdown efficiencies of only 5–10% were reached with this method. Also, use of different transfection reagents could not improve these results without strongly decreasing the viability of the endothelial cells. Therefore lentiviral expression of short hairpin RNA (shRNA) was used to reduce Trio expression. With this method, we effectively and reproducibly silenced Trio protein expression for 80–90% (Figure 6A).
FIGURE 6:

Trio knockdown or inhibition reduces neutrophil adhesion and TEM. HUVEC were transduced with shTrio-expressing lentivirus to silence Trio expression. (A) Knockdown of Trio expression was confirmed by Western blot. β-Catenin and actin were used as a control for equal loading. Neutrophil adhesion to and TEM across (B) shTrio-transduced and TNF-α–stimulated HUVEC was measured under physiological flow conditions as described in Materials and Methods. Data are means of three independent experiments ± SEM. (C) ICAM-1 expression in shTrio-transduced and TNF-α–stimulated HUVEC was analyzed by Western blotting. Actin was used as a control for equal loading. (D) TrioN was expressed using adenovirus into endothelial cells that were or were not silenced for Trio (shTrio). TEM assay under flow conditions was performed and showed that TrioN rescued impaired neutrophil TEM across Trio-deficient endothelial cells. The experiment was done twice in triplicate. Data are means ± SEM. (E and F) TNF-α–stimulated HUVEC were grown to confluency, treated with 100 μM ITX3 for 16 h or DMSO as a control. The resistance or confluency was not affected by the treatments. Neutrophil adhesion (E) and TEM (F) were measured under physiological flow conditions as described in Materials and Methods. Data are means of three independent experiments ± SEM. (G) HUVEC were microporated with GFP-LifeAct to visualize F-actin during live-cell imaging. TNF-α–stimulated HUVEC were treated with 100 μM ITX3 (lower panels) for 16 h or dimethyl sulfoxide (DMSO) as a control (top panels) and neutrophils were allowed to transmigrate through endothelial monolayers. The focal plane was set at the surface of the endothelium in ITX3-treated cells, since in contrast to controls cells (DMSO), no actin-positive rings are observed in the upper focal plane. Neutrophils are visualized in DIC panels. “XZ” panels show Z-stack reconstructions of endothelial docking structures. Asterisk indicates transmigrating neutrophils. (H) Quantification of GFP-LifeAct–positive docking structures around transmigrating neutrophils. Data are means of three independent experiments ± SEM. *, p < 0.05; ***, p < 0.001. Scale bar: 10 μm.
Using a perfusion-based TEM flow model, we quantified neutrophil adhesion and TEM under physiological flow conditions. Under these circumstances, we observed a large reduction in neutrophil adhesion to endothelial cells, in which Trio expression was reduced (Figure 6B). Also neutrophil TEM across Trio-depleted cells was strongly reduced (Figure 6B). Verification of ICAM-1 protein expression in Trio-silenced endothelial cells revealed reduced ICAM-1 levels compared with control cells (Figure 6C). Nevertheless, we partially rescued neutrophil TEM under flow conditions by expressing the N-terminal part of Trio (TrioN), which included the spectrin repeats and the GEF-D1 domain, in endothelial cells (Figure 6D). This construct still activated Rac1 and RhoG, but was insensitive for the shRNA against endogenous Trio. Preliminary evidence from our lab suggested that Trio is involved in the signaling induced by TNF-α that leads to ICAM-1 up-regulation. This work is currently under investigation. Although Trio is evidently required for efficient neutrophil adhesion and TEM, we cannot exclude that reduced ICAM-1 expression attributed to the decrease observed in neutrophil adhesion and transmigration.
TrioD1 inhibition reduces leukocyte TEM
Because our previous results showed that ICAM-1 clustering induced Trio activation, as evidenced by increased binding to nucleotide-free Rac1 and RhoG, the involvement of the N-terminal TrioD1 GEF domain is of particular interest. Recently Bouquier and colleagues identified a novel, specific Trio inhibitor, ITX3, which targets the N-terminal RhoG- and Rac1-specific TrioD1 GEF domain of Trio (Bouquier et al., 2009) but does not inhibit the C-terminal RhoA-specific TrioD2 GEF domain of Trio or the GEFs Vav2, TIAM1, and GEF337/ARHGEF17. Analysis of Rac1 activity induced by GFP-TrioD1 in HEK293T or melanoma A7 cells indeed showed reduction to background levels when cells were pretreated with ITX3 (Figure S5A). In contrast, SGEF activity, known to activate RhoG (Ellerbroek et al., 2004), is not affected by ITX3, as was judged by dorsal ruffle formation (Figure S5B). We used ITX3 pretreatment of the endothelial cells to study neutrophil TEM under physiological flow conditions. In this assay, no difference in adhesion of neutrophils to ITX3-treated endothelial monolayers compared with carrier-treated cells was observed (Figure 6E). However, TEM of neutrophils across ITX3-treated endothelial cells was significantly reduced compared with carrier-treated cells (Figure 6F). Analysis of expression of ICAM-1 showed that, unlike knockdown of the complete Trio protein, ITX3 treatment did not interfere with ICAM-1 up-regulation (Figure S5C). Moreover, the inhibitor did not affect the electrical resistance of TNF-α–treated endothelial monolayers (unpublished data). These results show that functional TrioD1 GEF activity is required for efficient neutrophil TEM. Because we have shown that TrioD1 is recruited to neutrophil adhesion sites, we assessed the effect of TrioD1 inhibition on endothelial docking structure formation during neutrophil TEM. GFP-LifeAct was transfected into endothelial cells to study F-actin dynamics. Real-time imaging showed that 80% of the neutrophils induced F-actin–rich docking structures prior to transmigration. TrioD1 inhibition by ITX3 significantly impaired F-actin–positive docking structure formation around transmigrating neutrophils (Figure 6, G and H). These results demonstrate that TrioD1 GEF activity is required for both endothelial docking structure formation and neutrophil TEM.
Trio inhibition impairs ICAM-1 ring and docking structure formation
To study ICAM-1–dependent endothelial docking structure formation in more detail, we used α-ICAM-1–antibody beads to stimulate docking structure formation. Analysis of ICAM-1 recruitment around α-ICAM-1–antibody beads by confocal laser-scanning microscopy showed that Trio inhibition with ITX3 resulted in small ICAM-1 rings consisting of discontinuous ICAM-1 microclusters (Figure 7A). Quantification of ICAM-1 rings showed that ITX3 significantly reduced the formation of ring structures (Figure 7B). Instead, in ITX3-treated cells, significantly more discontinuous ICAM-1 rings that also lacked cup-like membrane protrusions were observed. Analysis of the F-actin cytoskeleton revealed that in ITX3-treated cells significantly fewer F-actin rings formed around α-ICAM-1–antibody beads (Figure 7, A and C). Rather, ITX3-treated cells were often characterized by large and thick bundles of F-actin just underneath adherent beads, sometimes colocalized with discontinuous ICAM-1 microclusters. Previously several actin-binding proteins, such as α-actinin, cortactin, and filamin (Carpen et al., 1992; Tilghman and Hoover, 2002; Celli et al., 2006; Yang et al., 2006; Kanters et al., 2008; van Buul et al., 2010), have been identified as interacting with ICAM-1 and anchoring ICAM-1 to the F-actin cytoskeleton. Analysis of the localization of these actin-binding proteins showed that, whereas filamin A and B recruitment around α-ICAM-1–antibody beads was not affected by Trio inhibition with ITX3, recruitment of cortactin and α-actinin-4 was reduced to a similar extent as F-actin (Figure S6, A–C), suggesting that Trio may regulate the recruitment of cortactin and α-actinin-4 upon ICAM-1 clustering.
FIGURE 7:

Trio inhibition with ITX3 impairs endothelial docking structure formation. (A) ICAM-1 was clustered with 10-μm α-ICAM-1–antibody beads on TNF-α–stimulated HUVEC were treated with 100 μM ITX3 for 16 h or DMSO as a control. ICAM-1 and F-actin localization were analyzed by immunofluorescence. Beads are visualized in DIC panels and indicated by arrowheads. (B) TNF-α–stimulated HUVEC transduced with ICAM-1-GFP adenovirus and treated with ITX3 or DMSO as a control were imaged live, and continuous and discontinuous rings around α-ICAM-1–antibody beads were quantified after 30 min of ICAM-1 clustering. Rings were quantified around > 50 beads per experiment. Data are means of eight experiments ± SEM. (C) F-actin ring quantification. F-actin rings were quantified around > 50 beads per experiment. Data are means of four independent experiments ± SEM. (D) ICAM-1 ring diameter in TNF-α–stimulated HUVEC transduced with ICAM-1-GFP adenovirus and treated with ITX3 or DMSO as a control. ICAM-1 is shown in green and the bead in red. (E) ICAM-1 ring diameter quantification. ICAM-1 ring diameter was measured for ∼30 beads per experiment. Data are means of eight experiments ± SEM. (F) Reconstruction of Z-stack imaging in TNF-α–stimulated HUVEC transduced with ICAM-1-GFP adenovirus and treated with ITX3 or DMSO as a control shows ICAM-1–positive docking structure around a α-ICAM-1–antibody bead. Average length of ICAM-1–positive membrane protrusions around ∼30 α-ICAM-1–antibody beads per experiment was measured in TNF-α–stimulated HUVEC treated with ITX3 (G) or with Rac1 inhibitor NSC23766 (H). Data are means of three independent experiments ± SEM. (I). Schematic representation of the effect of Trio inhibition by ITX3 on endothelial docking structure formation. *, p < 0.05; **, p < 0.01. Scale bars: 10 μm.
Quantification of the size of ICAM-1–positive rings showed that the average diameter of the rings in control cells is 9.5 μm, which is close to the actual 10-μm diameter of the beads (Figure 7, D and E). In contrast, ITX3-treated cells significantly decreased the average diameter to 6.5 μm (Figure 7, D and E). Measuring the length of the apical membrane protrusions formed around α-ICAM-1–antibody beads demonstrated that the membrane projections in ITX3-treated cells were also significantly decreased, reaching 2.6 μm, compared with 5.2 μm in control cells (Figure 7, F and G). In addition, in ITX3-treated cells, membranes did not fuse to form a cup-like docking structure as in control cells, but rather consisted of single microvilli that differed in length (Figure 7F and Videos S3 and S4). Thus these data demonstrate that TrioD1 inhibition with ITX3 severely impaired both the coalescence of ICAM-1 microclusters into mature ICAM-1 rings and the growth and fusion of ICAM-1–positive microvilli into cup-like docking structures (Figure 7I).
The Rac1 inhibitor NSC23766 has been shown to bind Rac1 and to block the interaction of Rac1 with its Rho-GEFs Trio and TIAM1 (Gao et al., 2004). Previously we have shown that Rac1 inhibition with NSC23766 also impaired ICAM-1 ring formation around α-ICAM-1–antibody beads (van Buul et al., 2010), but docking structure formation was not investigated. Therefore we also measured the length of apical membrane protrusions around α-ICAM-1–antibody beads in NSC23766-treated endothelial cells. The average length of the membrane projections was also significantly reduced in NSC23766-treated cells, although to a lesser extent (Figure 7H). These results suggest that Trio and Rac1 are important mediators of ICAM-1–induced endothelial docking structure formation.
Trio mediates docking structure formation through Rac1 and RhoG
We previously showed that RhoG is implicated in ICAM-1–induced docking structure formation (van Buul et al., 2007). Because Trio mediates nucleotide exchange on RhoG and Rac1 with its N-terminal GEF domain, we first analyzed the kinetics of GTPase activation downstream from ICAM-1 clustering. Interestingly, Rac1 activation was measured after 5–10 min of ICAM-1 clustering, after which it declined (Figure 8A). RhoG activation, on the other hand, was measured after 30 min, declining at 60 min (Figure 8A). Next we analyzed Rac1 and RhoG activation after ICAM-1 clustering in ITX3-treated endothelial cells. Trio inhibition with ITX3 prevented Rac1 activation after 10 min of ICAM-1 clustering (Figures 8B and S7A). However, after 30 min of ICAM-1 clustering, Rac1 activity was detected, suggesting that Trio inhibition with ITX3 delays ICAM-1–induced Rac1 activation. In addition, ICAM-1–induced RhoG activation was also decreased by ITX3 treatment (Figures 8B and S7B).
FIGURE 8:

Sequential Rac1 and RhoG activation upon ICAM-1 clustering. (A) ICAM-1 was clustered (CL) with α-ICAM-1–antibody beads for indicated time points, and Rac1-GTP and RhoG-GTP were precipitated as described in Materials and Methods. ICAM-1 is shown as loading control. Graph on the right shows quantification of the blots by ImageJ. Dotted line represents active Rac1 levels (square) and filled line represents active RhoG levels (circle). Data are mean of three independent experiments ± SEM. (B) Graphs show quantification of active Rac1 levels (left) or active RhoG levels (right) upon ICAM-1 clustering. ITX3 inhibits ICAM-1–induced Rac1 and RhoG activation. Data are an average of three independent experiments ± SEM.
To study the individual contributions of Rac1 and RhoG to endothelial docking structure formation, we silenced Rac1 and RhoG expression using lentivirally delivered shRNA constructs (Figure 9A). Both knockdown of Rac1 and RhoG expression significantly reduced the average length of endothelial docking structures (Figures 9B and S7C). However, whereas hardly any ICAM-1 rings were observed and ICAM-1 remained in separate microclusters in Rac1-silenced cells, RhoG-silenced cells were still able to form ICAM-1 rings (Figure 9C), suggesting differential roles of Rac1 and RhoG in endothelial docking structure formation. Similar results were obtained when using primary isolated neutrophils (Figure 9D). Moreover, reduced Rac1 or RhoG expression by RNA interference in endothelial cells significantly impaired neutrophil transmigration (Figures 9E and S7D). These results therefore show that upon ICAM-1 clustering, Trio mediates activation of Rac1 and RhoG, which differentially regulate ICAM-1 ring formation, docking structure formation, and consequently, TEM.
FIGURE 9:

Trio mediates endothelial docking structure formation through Rac1 and RhoG. (A) To silence Rac1 and RhoG expression, we used lentivirally delivered constructs targeting Rac1 and RhoG mRNA in TNF-α–stimulated HUVEC. Knockdown of Rac1 and RhoG expression was confirmed by Western blotting and did not affect ICAM-1 expression. Tubulin or actin was used as loading control. (B) Average length of ICAM-1–positive membrane protrusions around ∼30 α-ICAM-1–antibody beads per experiment was quantified. Data are means of three independent experiments ± SEM. (C) ICAM-1 was clustered with 10-μm α-ICAM-1–antibody beads on TNF-α–stimulated HUVEC with silenced Rac1 or RhoG expression. ICAM-1 and F-actin localization were analyzed by immunofluorescence. Beads are visualized in DIC panels and indicated by arrowheads. Scale bars: 10 μm. (D) Neutrophils were allowed to adhere to TNF-α–stimulated HUVEC silenced for Rac1 or RhoG. x–z projections show the absence of ICAM-1–positive protrusions upon Rac1 or RhoG silencing. Scale bars: 20 μm. (E) Neutrophil TEM were significantly impaired upon Rac1 or RhoG silencing in TNF-α–stimulated HUVEC. Data are means of three independent experiments ± SEM. *, p < 0.01.
DISCUSSION
The endothelium extends large membrane projections that embrace the adherent leukocyte and promote efficient diapedesis (Barreiro et al., 2002; Carman and Springer, 2004). Rho-GTPases and their GEFs are known to be the central orchestrators of membrane dynamics through remodeling of the F-actin cytoskeleton. In this study, we show that the multi-domain GEF Trio is recruited upon ICAM-1 engagement to sites of leukocyte adhesion and activates the GTPases Rac1 and RhoG through its N-terminal TrioD1 GEF domain. By controlling the activity of Rac1 and RhoG, Trio regulates both local F-actin polymerization and ICAM-1 clustering leading to the assembly of endothelial docking structures (Figure 10). Furthermore, Trio-dependent assembly of endothelial docking structures is required for efficient neutrophil diapedesis under physiological flow conditions.
FIGURE 10:

Model for ICAM-1–induced endothelial docking structure formation. Clustering of ICAM-1 induces the recruitment of the F-actin cross-linker protein filamin. Filamin functions as a scaffold for subsequent activation of the GEF Trio. In turn, Trio activates the GTPase Rac1 through its N-terminal GEF domain, leading to the recruitment of other adapter proteins, such as cortactin and α-actinin-1/4. ICAM-1 clustering also induces the activation of the GTPase RhoG through Trio and SGEF. On their activation, Rac1 and RhoG induce remodeling of the F-actin cytoskeleton, leading to endothelial docking structure formation. Formation of docking structures serves to guide adherent leukocytes during diapedesis. Image is not drawn to scale.
The intracellular domain of ICAM-1 is essential for mediating endothelial docking structure formation and leukocyte extravasation (Greenwood et al., 2003; Lyck et al., 2003; Oh et al., 2007; van Buul et al., 2007). Because this domain comprises only 28 amino acids and does not contain apparent signaling motifs, signaling likely occurs via interacting adaptor proteins. We have previously shown that the F-actin cross-linker protein filamin B interacts directly with the intracellular domain of ICAM-1 and is required for ICAM-1 ring formation and leukocyte TEM (Kanters et al., 2008). Filamin has been demonstrated to interact with the GTPase Rac1 (Ohta et al., 1999) and to function as a scaffold for Rac1 signaling (Jeon et al., 2008; Del Valle-Perez et al., 2010). Bellanger and colleagues have shown that filamin also binds to the PH domain of the Rac1- and RhoG-specific TrioD1 GEF unit of Trio and is required for TrioD1-induced membrane dynamics (Bellanger et al., 2000). Our results show that although Trio interacts with ICAM-1 independently of filamin, filamin is required for ICAM-1–induced Trio activation, suggesting that filamin may function as a scaffold that connects Trio to upstream regulatory proteins. Nevertheless, the potential mechanism of Trio activation is unclear. We found that inhibition of TrioD1-GEF activity by ITX3 does not affect the recruitment of filamin to clustered ICAM-1, confirming that filamin functions upstream of Trio activation. Interestingly, TrioD1 inhibition did impair the recruitment of the actin-binding proteins cortactin and α-actinin-4, which are known to be involved in ICAM-1 clustering (Tilghman and Hoover, 2002; Celli et al., 2006; Yang et al., 2006), suggesting the presence of different complexes with ICAM-1 that may be differentially regulated in time. Indeed, several reports have shown that the association of cortactin to the cortical actin cytoskeleton is regulated by Rac1 (Head et al., 2003; Weed et al., 1998), suggesting that TrioD1-induced Rac1 activation may also be involved in cortactin recruitment during ICAM-1–induced docking structure formation (Figure 10).
ICAM-1–mediated docking structure formation around adherent leukocytes and α-ICAM-1–antibody beads resembles the process of phagocytic cup formation by professional phagocytes. We found that inhibiting the TrioD1-GEF domain or silencing of Rac1 and RhoG expression largely prevents the formation of endothelial docking structures. Indeed, Trio has also been implicated upstream of RhoG and Rac1 during phagocytosis of apoptotic cells in Caenorhabditis elegans and in phagocytic LR73 cells (deBakker et al., 2004), suggesting the use of analogous signaling pathways. However, whereas phagocytic cells completely ingest particles or cells, we hardly ever observed complete engulfment of α-ICAM-1–antibody beads. In general, ICAM-1–induced docking structures do not extend much above the 5-μm midline of adherent beads. Nevertheless, we observed that overexpression of the TrioD1 GEF domain in endothelial cells does result in total engulfment of a large portion of the α-ICAM-1–antibody beads (van Rijssel, unpublished observations), suggesting that endogenous Trio signaling leading to docking structure formation is probably terminated by negative feedback loops.
During phagocytic cup formation, Rho-GTPases such as Rac1, Rac2, and CDC42 exhibit different spatial activation patterns and have distinct contributions to this process (Hoppe and Swanson, 2004). In this study, we observed that Rac1 controlled ICAM-1 ring formation, whereas RhoG primarily regulated the formation of cup-like membrane protrusions, suggesting that Rac1-mediated ICAM-1 ring formation could be a prerequisite for subsequent RhoG-dependent endothelial docking structure formation. In support of this idea, we observed a more rapid activation of Rac1, which was followed by RhoG activation. These observations are in line with the faster kinetics of Trio binding to nucleotide-free Rac1 than to RhoG, suggesting that Trio mediates nucleotide exchange on Rac1 prior to RhoG. As both can be activated by the TrioD1 GEF domain, there might be competition between Rac1 and RhoG for binding to TrioD1. Recent data from our group already indicated that TrioD1-induced Rac1 activation increased when RhoG expression was silenced (van Rijssel et al., 2012). Given that Rac1 has been shown to interact with filamin (Ohta et al., 1999; Jeon et al., 2008), this may favor initial Rac1 activation by Trio, whereas RhoG may be activated at a later stage. Alternatively, Trio may also activate Rac1 and RhoG within different spatially restricted signaling complexes. Interestingly, TrioD1 inhibition with ITX3 did not completely abolish Rac1 activation, but rather caused a delay, suggesting the presence of alternative Rac1 activation pathways. As for RhoG, we previously identified SGEF as being involved in RhoG activation upon ICAM-1 clustering (van Buul et al., 2007). Because SGEF interacts with the cytoplasmic domain of ICAM-1 in a direct manner, it is difficult to envision both Trio and SGEF as part of the same ICAM-1–containing complex. Instead, Trio and SGEF may form different complexes with ICAM-1 that may connect RhoG to different effectors, and in this way control local RhoG signaling within endothelial docking structures. Nevertheless, since Trio-induced Rac1 activation is required for the initial ICAM-1 ring formation, Trio-Rac1 signaling seems to be dominant over RhoG signaling in the process of TEM.
Since the first detailed description of the docking structures in 2002 by Barreiro and coworkers, numerous studies have observed both in vitro and in vivo the presence of these cup-like membrane protrusions that surround adherent leukocytes during their transmigration through the endothelium (Barreiro et al., 2002; Carman et al., 2003; Carman and Springer, 2004; Wolburg et al., 2005; Doulet et al., 2006; van Buul et al., 2007; Cayrol et al., 2008; Phillipson et al., 2008; Riethmuller et al., 2008; Petri et al., 2011). The docking structures were initially proposed as structures that captured leukocytes, promoting firm adhesion (Barreiro et al., 2002). However, our results show that although Trio inhibition severely impairs docking structure formation, adhesion of neutrophils under physiological flow conditions is not affected, suggesting that the formation of docking structures is not involved in adhesion strengthening. In line with this, Carman and Springer have shown that preventing docking structure formation through inhibition of F-actin or microtubule polymerization did not affect leukocyte adhesion to ICAM-1 under increasing shear flow conditions (Carman et al., 2003). Instead, our data show that Trio inhibition impairs neutrophil diapedesis under physiological flow conditions, showing that Trio-mediated docking structure formation facilitates leukocyte TEM rather than adhesion.
Carman and Springer demonstrated that endothelial docking structures are strongly correlated with transmigrating leukocytes and proposed that endothelial docking structures facilitate leukocyte diapedesis by providing increased endothelial membrane for the migrating leukocyte in order to direct leukocyte TEM (Carman and Springer, 2004). In addition, in vivo studies have shown that these apical endothelial membrane protrusions also function to “seal” the transmigrating leukocyte, in this way preventing plasma leakage during leukocyte diapedesis (Phillipson et al., 2008; Petri et al., 2011). Interestingly, in addition to its role in endothelial docking structure formation, we found that Trio is also required for endothelial monolayer integrity. It is therefore tempting to speculate that Trio functions as a gatekeeper during leukocyte extravasation by allowing leukocyte diapedesis through endothelial docking structure formation and at the same time maintaining endothelial barrier function by sealing off the transmigration sites and regulating the integrity of the adherens junctions.
In conclusion, in this study we have demonstrated that ICAM-1–induced docking structures are required for leukocyte TEM and are controlled by Rac1 and RhoG through the Rho-GEF Trio, in a filamin-dependent manner (Figure 10). This work shows new mechanistic insights in the regulation and function of docking structures in the process of leukocyte extravasation.
MATERIALS AND METHODS
Antibodies and reagents
Trio (clone D-20), Vav2 (clone H-200), ICAM-1 (clone H-108), and VE-cadherin (F8) antibodies were from Tebu-Bio (Heerhugowaard, Netherlands). ICAM-1 mAb (clone BBIG-I1/11C81) was purchased from R&D Systems (Abingdon, United Kingdom). Actin (clone AC-40) mAb antibody was purchased from Sigma-Aldrich (Zwijndrecht, Netherlands). Filamin A mAb (clone PM6/317) antibody was purchased from Serotec (Oxford, United Kingdom). Polyclonal filamin B (A301-726A) antibody was from Bethyl Laboratories (Montgomery, AL). Mouse polyclonal Trio antibody was from Abnova (Heidelberg, Germany). Polyclonal filamin C antibody was from Kinasource (Dundee, United Kingdom). Polyclonal α-actinin-4 antibody was purchased from Enzo Life Sciences BVBA (Zandhoven, Belgium). Cortactin (4F11) mAb was from Millipore (Leiden, Netherlands). c-Myc (clone 9E10) antibody was purchased from Invitrogen (Breda, Netherlands). Rac1 (clone 102) mAb antibody was purchased from BD Biosciences (Breda, Netherlands). RhoG monoclonal antibody was a kind gift of J. Meller and M. A. Schwartz (University of Virginia, Charlottesville, VA). Secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse, swine anti-rabbit, and rabbit anti-goat antibodies were purchased from Dako (Heverlee, Belgium). Monoclonal GFP (JL-8) antibody, secondary goat anti-rabbit IR 680, goat anti-mouse IR 800, and donkey anti-goat IR 800 antibodies were purchased from Licor Westburg (Leusden, Netherlands). Texas Red- and Alexa Fluor 633–conjugated phalloidin were from Invitrogen (Breda, Netherlands). TNF-α was purchased from Peprotech EC (London, United Kingdom). NSC23766 was purchased from Merck (Nottingham, United Kingdom). ITX3 was from Chembridge (San Diego, CA).
Cell culture and transfection
HEK-293, COS-7, and HeLa were maintained in Iscove's modified Dulbecco's medium (IMDM; BioWhittaker, Verviers, Belgium) containing 10% (vol/vol) heat-inactivated fetal calf serum (Invitrogen, Breda, Netherlands), 300 μg/ml glutamine, 100 U/ml penicillin and streptomycin, at 37°C and 5% CO2. Cells were transiently transfected with the expression vectors indicated in each experiment according to the manufacturer's protocol with TransIT-LT1 reagent (Mirus, Madison, WI). Primary HUVEC were purchased from Lonza (Baltimore, MD) and cultured during regular passaging following fibronectin (10 μg/ml) coating of the tissue culture flasks (TPP, Switzerland) or glass slides in EGM2-containing SingleQuots (Lonza). Endothelial cells were cultured until passage 9. Human microvascular endothelial cells (HMVEC) were purchased from Lonza. HCMEC/D3 human brain endothelial cells were a kind gift from P. O. Couraud, CNRS, Paris, France.
Knockdown with siRNA
siRNA duplexes against filamin A and B have been described previously (Kanters et al., 2008). As control siRNA, the following sequence targeting the firefly luciferase GL2 gene (Elbashir et al., 2001) was used: CGUACGCGGAAUACUUCGA. Oligos were purchased from Eurogentec (Liege, Belgium). Filamin C siRNA SMARTpool was purchased from Dharmacon (Lafayette, CO). Cells were transfected with 1 nM siRNA by means of interferin transfection reagent (Polyplus/Westburg, Leusden, Netherlands) according to the manufacturer's instructions. After 72 h, cells were assayed as described below. Note that the oligos efficiently reduced protein expression in human (HUVEC) as well as monkey (Cos7) cells.
Cloning of GFP-Trio FL
Trio fragment (1091–8593) was digested out of Myc-Trio FL (McPherson et al., 2005) into peGFP(C1)-TrioN with NruI and KpnI, resulting in peGFP(C1)-Trio FL pt1. The C-terminal part (8594–9117) was obtained by PCR amplification with Myc-Trio as template and JR43F (GAGATCGGTACCTGCACAACTGCAGGA) and JR44R (GAGATC GGTACCTCAAACTCTAGGCAGAAGC) as 5′ and 3′ primers, respectively. The PCR product was cloned as a KpnI-digested fragment into peGFP(C1)-Trio FL pt1, resulting in peGFP(C1)-Trio FL.
Western blotting
SDS–PAGE samples were analyzed on 7.5, 10, or 15% (wt/vol) polyacrylamide gels, depending on the size of the proteins of interest, and transferred onto nitrocellulose (Whatman, Piscataway, NJ) or PVDF membrane (Bio-Rad, Hercules, CA). Following blocking in 5% (wt/vol) low-fat milk in TBST Tris-buffered saline–Tween-20 (TBST), the blots were incubated with the primary antibody for 1 h at room temperature, washed 3 times for 20 min in TBST, and subsequently incubated with HRP-coupled secondary antibodies (dilution: 1:7000) in TBST for 1 h at room temperature; this was followed by three washes with TBST for 20 min each and development of the blot by enhanced chemiluminescence (ECL) or SuperSignal West nano-ECL (ThermoScientific, Etten-Leur, The Netherlands). Alternatively, blots were incubated with IR 680 or IR 800 dye-conjugated secondary antibodies (dilution 1:5000) in TBST for 1 h at room temperature; this was followed by three washes with TBST for 20 min each. Infrared signal was detected and analyzed with the Odyssey infrared detection system (LI-COR Biosciences, Lincoln, NE). For Trio protein expression, 3–8% (wt/vol) Tris-acetate precast gels (Invitrogen) were used according to the manufacturer's instructions, and samples were transferred onto nitrocellulose membrane by blotting for 18 h at 20 mA.
Confocal laser-scanning microscopy
For immunofluorescence, cells were grown on fibronectin-coated 14-mm coverslips. After treatment, cells were washed in cold PBSA (phosphate-buffered saline [PBS], 0.5% [wt/vol] bovine serum albumin [BSA], 1 mM CaCl2, 0.5 mM MgCl2) and fixed in 4% (vol/vol) formaldehyde in PBS (+1 mM CaCl2, 0.5 mM MgCl2) for 10 min. After fixation, cells were permeabilized in PBS-T (PBS + 1% Triton X-100 and 10% glycerol) for 4 min; this was followed by a blocking step in PBS supplemented with 2% (wt/vol) BSA. Cells were incubated with primary and secondary antibodies, and after each step were washed three times in PBSA. Coverslips were mounted with Vectashield with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Peterborough, UK) on microscope slides. For live-cell imaging, cells were seeded on 30-mm coverslips and transfected or transduced, as indicated. After 24–72 h, cells were placed in a heating chamber at 37°C and 5% CO2 and imaged with a confocal microscope (LSM510 META; Carl Zeiss MicroImaging, Jena, Germany).
Adenovirus production
The ICAM-1-GFP fragment was obtained by PCR amplification with peICAM-1-GFP (Barreiro et al., 2002) as template and JR11F (GAGATCGTCGACATG GCTCCCAGCAGCCC) and JR18R (GAGATCGCGGCCGCTTTACTTGTAC) as 5′ and 3′ primers, respectively. The PCR product was cloned as a SalI/NotI fragment into pENTR1A (Invitrogen). GFP-TrioD1 was obtained by PCR amplification with peGFP-TrioD1 as template and primer pairs JR22F (GAGATCGTCGACCTG GTTTAGTGAACCGTCAGA) and JR4R (GAGATCGAATTCCTAGGCGTCATTG CTGGAGAC). The PCR product was cloned as a SalI/EcoRI fragment into pENTR1A (Invitrogen). pENTR1A-constructs were recombined into the pAd/CMV/V5-DEST vector (Invitrogen) with Clonase II enzyme mix according to the manufacturer's instructions (Invitrogen). Adenovirus expressing ICAM-1-GFP or GFP-TrioD1 was produced by transfecting PacI-digested (Westburg, Leusden) constructs into HEK293T cells.
shRNA lentivirus production
shRNA constructs (Sigma Mission library) targeting Trio (TRC_10561), Rac1 (TRC_4873), and RhoG (TRC_48019), or nontargeting shCtrl construct were packaged into lentivirus in HEK293T cells with third-generation lentiviral packaging plasmids (Dull et al., 1998). Lentivirus-containing supernatant was harvested on days 2 and 3 after transfection. Lentivirus was concentrated by centrifugation at 20,000 × g for 2 h.
Fusion proteins
The fusion proteins GST-ELMO, GST-Rac1 G15A, and GST-RhoG G15A were purified from BL21 Escherichia coli cells (Agilent Technologies, Amstelveen, Netherlands) with glutathione–Sepharose 4B, as previously described (Ellerbroek et al., 2004). GST-fusion proteins were stored in 30% (vol/vol) glycerol at −80°C.
Pulldown assay
A synthetic, biotinylated peptide encoding the intracellular domains of human ICAM-1 or VCAM-1, or empty beads, was used in pulldown assays, as previously described (Kanters et al., 2008). In short, a confluent monolayer of cells in a 100-mm Petri dish was washed with cold PBS (+ 1 mM CaCl2; 0.5 mM MgCl2) and lysed in cold NP-40 lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 10% [vol/vol] glycerol, and 1% [vol/vol] Nonidet P40) supplemented with protease inhibitors (complete mini EDTA, Roche, Almere, The Netherlands) for 5 min. After lysis, cell debris was removed by centrifugation (10,000 × g, 5 min at 4°C). Supernatant was incubated with 30 μg of biotinylated peptide comprising the intracellular tail of ICAM-1 or indicated controls and 30 μl (concentration 2.4 mg/ml packed gel) of streptavidin agarose (Sigma-Aldrich, Zwijndrecht, Netherlands) for 2–3 h under constant head-over-head rotation at 4°C. Beads were subsequently washed five times with NP40 lysis buffer and resuspended in SDS–PAGE sample buffer.
RhoG and Rac1 activation assay
A confluent monolayer of HUVEC in a 100-mm Petri dish was washed with cold PBS (+ 1 mM CaCl2; 0.5 mM MgCl2) and lysed in 50 mM Tris, pH 7.4, 0.5 mM MgCl2, 500 mM NaCl, 1% (vol/vol) Triton X-100, 0.5% (wt/vol) deoxycholic acid, and 0.1% (wt/vol) SDS supplemented with protease inhibitors. Lysates were cleared at 14,000 × g for 5 min. GTP-bound RhoG was isolated by rotating supernatants for 30 min with 60–90 μg of GST-ELMO (GST-fusion protein containing the full-length RhoG effector ELMO) conjugated to glutathione–Sepharose beads (GE Healthcare, Zeist, Netherlands; van Buul et al., 2007; Wittchen and Burridge, 2008). GTP-bound Rac1 was isolated with biotinylated Pak1-Crib peptide coupled to streptavidin agarose (ten Klooster et al., 2006). Beads were washed four times in 50 mM Tris, pH 7.4, 0.5 mM MgCl2, 150 mM NaCl, 1% (vol/vol) Triton X-100, and protease inhibitors. Pulldowns and lysates were immunoblotted with monoclonal RhoG and Rac1 antibodies.
Antibody-coated beads
Polystyrene beads (3-μm beads for biochemical assays and 3- or 10-μm beads for immunofluorescence; Polysciences, Eppelheim, Germany) were pretreated with 8% (vol/vol) glutaraldehyde overnight, washed five times with PBS, and incubated with 300 μg/ml ICAM-1 mAb according to the manufacturer's protocol. Free glutaraldehyde groups were blocked with 0.5 M ethanolamine and 2 mg/ml BSA.
Electric cell-substrate impedance sensing
Monolayer permeability was determined by measuring the electrical resistance with electric cell–substrate impedance sensing (ECIS). Electrode arrays (10WE; Applied BioPhysics) were precoated with 10 mM l-cysteine (Sigma-Aldrich, Steinheim, Germany) for 15 min at 37°C, after which they were washed twice with 0.9% (wt/vol) NaCl and subsequently coated with fibronectin (Sigma-Aldrich) in 0.9% (wt/vol) NaCl for 1 h at 37°C. A measure of 1 × 105 cells per well (0.8 cm2) were seeded, grown to confluency, and treated with TNF-α for 20 h. Electrical resistance was continuously measured at 37°C and 5% CO2 with ECIS-Model-9600 Controller (Applied BioPhysics, Troy, NY).
Neutrophil isolation
Polymorphonuclear neutrophils were isolated from whole blood derived from healthy donors by means of a Ficoll-Paque plus (Pharmacia Biotech, Uppsala, Sweden) density gradient. After erythrocyte lysis in a ice-cold isotonic lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA pH 7.4), neutrophils were washed once with lysis buffer, once with 10% (vol/vol) trisodium citrate/PBS, resuspended in HEPES medium (20 mM HEPES, 132 mM NaCl, 6 mM KCl, 1 mM CaCl2, 1 mM MgSO4, 1.2 mM K2HPO4, 5 mM glucose [all from Sigma-Aldrich], and 0.4% [wt/vol] human serum albumin [Sanquin Reagents], pH 7.4), and kept at room temperature for not longer than 4 h until use.
Neutrophil TEM across Transwell
Transmigration of neutrophils over endothelial monolayers was analyzed with Falcon FluoroBlok Transwell inserts with 3.0-μm pore size (BD Biosciences, Breda, Netherlands). At 24 h after transduction with shCtrl or shTrio-producing lentivirus, 2 × 105 HUVEC per well were seeded on fibronectin-coated filters and grown for another 48 h. At 20 h prior to the experiment, HUVEC were stimulated with 10 ng/ml TNF-α. Freshly isolated, CellTrace calcein green–labeled (Invitrogen) neutrophils were added to the upper compartment, and cells were allowed to migrate toward 10 nM formyl-Met-Leu-Phe (fMLP) in the lower compartment for 30 min. During this time, fluorescence was measured with a Tecan GENios Plus microplate reader with a 530/30 band-pass filter. The percentage of TEM was calculated as (fluorescence of calcein-labeled neutrophils that transmigrated across a Transwell filter coated with HUVEC)/(fluorescence of calcein-labeled neutrophils input) × 100.
Neutrophil TEM under flow
HUVEC were cultured in a fibronectin-coated Ibidi μ-slide VI0.4 (Ibidi, München, Germany) for 2–3 d until confluency and were stimulated overnight with TNF-α (10 ng/ml). Freshly isolated neutrophils were resuspended at 0.4 × 106 cells/ml in HEPES medium and were incubated for 30 min at 37ºC. Neutrophils were perfused over HUVEC monolayers at 0.5 ml/min (shear stress of 1.0 dyne/cm2). After 3 min, the neutrophil-containing HEPES solution was replaced by HEPES medium for 30 min. During this time leukocyte–endothelial interactions were recorded in six random fields with a Zeiss Axiovert 200 microscope (10× objective) equipped with a motorized stage. Images were recorded with Zeiss Axiovision 4.7 software. Live imaging was performed at 37ºC and 5% CO2. Transmigrated neutrophils were distinguished from those adhering to the apical surface of the endothelium by their transition from bright to phase-dark morphology. HUVEC that were pretreated with ITX3 were carefully washed before being exposed to flow. Owing to the washing and flow conditions, no ITX3 was present in the medium at times of neutrophil TEM. ITX3 was incubated overnight but was also active after only 1 h of incubation on HUVEC. In addition, ITX3 did not need to be present in the medium to inhibit Trio activity, as wash-out experiments showed that ITX3 was able to inhibit Trio activity for at least 60 min after wash-out.
Statistical analysis
Statistical comparisons between experimental groups were performed by the Student's t test. A two-sided p value of ≤0.05 was considered significant. Unless otherwise stated, a representative experiment out of at least three independent experiments is shown.
Supplementary Material
Acknowledgments
Myc-TrioD1 and HA-TrioD2 were kind gifts of P. Fort, and GFP-Trio FL was a kind gift of A. Debant (both at Macromolecular Biochemistry Research Center, Montpellier, France). Myc-Trio FL was a kind gift of B. Eipper, University of Connecticut, Farmington, CT. Filamin B-GFP was a kind gift from A. Sonnenberg, NKI, Amsterdam, Netherlands. RhoG mAb was a kind gift from J. Meller and M. A. Schwartz, University of Virginia, Charlottesville, VA. ICAM-1-GFP was a kind gift from O. Barreiro and F. Sanchez-Madrid, Madrid, Spain. ICAM-1ΔC-GFP was a kind gift of C. D. Jun, Gwangju Institute of Science and Technology, Gwangju, Korea. We thank D. Roos for critically reading the manuscript. Erik Mul, Niels Heemskerk, and Hakima Belkasim are acknowledged for their technical assistance. J.D.v.B. is supported by the Dutch Heart Foundation (grant no. 2005T039) and a Netherlands Organization for Scientific Research (NWO) Veni grant (916.76.053). J.v.R. is supported by Academic Medical Center (AMC) Research B.V. M.H. was funded by the Landsteiner Foundation for Blood Transfusion Research (project no. 0903).
Abbreviations used:
- BSA
bovine serum albumin
- DH-PH
Dbl homology–pleckstrin homology
- DIC
differential interference contrast
- DMSO
dimethyl sulfoxide
- ECIS
electric cell–substrate impedance sensing
- ECL
enhanced chemiluminescence
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HMVEC
human microvascular endothelial cells
- HRP
horseradish peroxidase
- HUVEC
human umbilical vein endothelial cells
- PBS
phosphate-buffered saline
- RFP
red fluorescent protein
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TBST
Tris-buffered saline–Tween-20
- TEM
transendothelial migration
- TNF-α
tumor necrosis factor α
- VE
vascular endothelial
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-11-0907) on June 13, 2012.
REFERENCES
- Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J Immunol. 1999;162:2964–2973. [PubMed] [Google Scholar]
- Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- Baldassarre M, Razinia Z, Burande CF, Lamsoul I, Lutz PG, Calderwood DA. Filamins regulate cell spreading and initiation of cell migration. PLoS One. 2009;4:e7830. doi: 10.1371/journal.pone.0007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol. 2000;2:888–892. doi: 10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- Blangy A, Vignal E, Schmidt S, Debant A, Gauthier-Rouviere C, Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J Cell Sci. 2000;113:729–739. doi: 10.1242/jcs.113.4.729. [DOI] [PubMed] [Google Scholar]
- Bouquier N, Vignal E, Charrasse S, Weill M, Schmidt S, Leonetti JP, Blangy A, Fort P. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem Biol. 2009;16:657–666. doi: 10.1016/j.chembiol.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. 2003;171:6135–6144. doi: 10.4049/jimmunol.171.11.6135. [DOI] [PubMed] [Google Scholar]
- Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpen O, Pallai P, Staunton DE, Springer TA. Association of intercellular adhesion molecule-1 (ICAM-1) with actin-containing cytoskeleton and α-actinin. J Cell Biol. 1992;118:1223–1234. doi: 10.1083/jcb.118.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol R, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. 2008;9:137–145. doi: 10.1038/ni1551. [DOI] [PubMed] [Google Scholar]
- Celli L, Ryckewaert JJ, Delachanal E, Duperray A. Evidence of a functional role for interaction between ICAM-1 and nonmuscle α-actinins in leukocyte diapedesis. J Immunol. 2006;177:4113–4121. doi: 10.4049/jimmunol.177.6.4113. [DOI] [PubMed] [Google Scholar]
- deBakker CD, et al. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol. 2004;14:2208–2216. doi: 10.1016/j.cub.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Debant A, Serra-Pages C, Seipel K, O'Brien S, Tang M, Park SH, Streuli M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA. 1996;93:5466–5471. doi: 10.1073/pnas.93.11.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle-Perez B, Martinez VG, Lacasa-Salavert C, Figueras A, Shapiro SS, Takafuta T, Casanovas O, Capella G, Ventura F, Vinals F. Filamin B plays a key role in vascular endothelial growth factor-induced endothelial cell motility through its interaction with Rac-1 and Vav-2. J Biol Chem. 2010;285:10748–10760. doi: 10.1074/jbc.M109.062984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulet N, Donnadieu E, Laran-Chich MP, Niedergang F, Nassif X, Couraud PO, Bourdoulous S. Neisseria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J Cell Biol. 2006;173:627–637. doi: 10.1083/jcb.200507128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieu-Trautmann O, Chaverot N, Cazaubon S, Strosberg AD, Couraud PO. Intercellular adhesion molecule 1 activation induces tyrosine phosphorylation of the cytoskeleton-associated protein cortactin in brain microvessel endothelial cells. J Biol Chem. 1994;269:12536–12540. [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, DeMali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15:3309–3319. doi: 10.1091/mbc.E04-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Amos CL, Walters CE, Couraud PO, Lyck R, Engelhardt B, Adamson P. Intracellular domain of brain endothelial intercellular adhesion molecule-1 is essential for T lymphocyte-mediated signaling and migration. J Immunol. 2003;171:2099–2108. doi: 10.4049/jimmunol.171.4.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head JA, Jiang D, Li M, Zorn LJ, Schaefer EM, Parsons JT, Weed SA. Cortactin tyrosine phosphorylation requires Rac1 activity and association with the cortical actin cytoskeleton. Mol Biol Cell. 2003;14:3216–3229. doi: 10.1091/mbc.E02-11-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon YJ, et al. Filamin B serves as a molecular scaffold for type I interferon-induced c-Jun NH2-terminal kinase signaling pathway. Mol Biol Cell. 2008;19:5116–5130. doi: 10.1091/mbc.E08-06-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanters E, van Rijssel J, Hensbergen PJ, Hondius D, Mul FP, Deelder AM, Sonnenberg A, van Buul JD, Hordijk PL. Filamin B mediates ICAM-1-driven leukocyte transendothelial migration. J Biol Chem. 2008;283:31830–31839. doi: 10.1074/jbc.M804888200. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not β1 integrins. Mol Cell Biol. 2000;20:7160–7169. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyck R, Reiss Y, Gerwin N, Greenwood J, Adamson P, Engelhardt B. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood. 2003;102:3675–3683. doi: 10.1182/blood-2003-02-0358. [DOI] [PubMed] [Google Scholar]
- McPherson CE, Eipper BA, Mains RE. Multiple novel isoforms of Trio are expressed in the developing rat brain. Gene. 2005;347:125–135. doi: 10.1016/j.gene.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11:366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- Oh HM, Lee S, Na BR, Wee H, Kim SH, Choi SC, Lee KM, Jun CD. RKIKK motif in the intracellular domain is critical for spatial and dynamic organization of ICAM-1: functional implication for the leukocyte adhesion and transmigration. Mol Biol Cell. 2007;18:2322–2335. doi: 10.1091/mbc.E06-08-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci USA. 1999;96:2122–2128. doi: 10.1073/pnas.96.5.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri B, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. 2011;117:942–952. doi: 10.1182/blood-2010-02-270561. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One. 2008;3:e1649. doi: 10.1371/journal.pone.0001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmuller C, Nasdala I, Vestweber D. Nano-surgery at the leukocyte-endothelial docking site. Pflugers Arch. 2008;456:71–81. doi: 10.1007/s00424-007-0412-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Sans E, Delachanal E, Duperray A. Analysis of the roles of ICAM-1 in neutrophil transmigration using a reconstituted mammalian cell expression model: implication of ICAM-1 cytoplasmic domain and Rho-dependent signaling pathway. J Immunol. 2001;166:544–551. doi: 10.4049/jimmunol.166.1.544. [DOI] [PubMed] [Google Scholar]
- Schnoor M, Lai FPL, Zarbock A, Kläver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SK, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor β-Pix. J Cell Biol. 2006;172:759–769. doi: 10.1083/jcb.200509096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman RW, Hoover RL. The Src-cortactin pathway is required for clustering of E-selectin and ICAM-1 in endothelial cells. FASEB J. 2002;16:1257–1259. doi: 10.1096/fj.01-0969fje. [DOI] [PubMed] [Google Scholar]
- Turowski P, et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121:29–37. doi: 10.1242/jcs.022681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–1293. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul JD, Hordijk PL. Endothelial adapter proteins in leukocyte transmigration. Thromb Haemost. 2009;101:649–655. [PubMed] [Google Scholar]
- van Buul JD, van Rijssel J, van Alphen FP, Hoogenboezem M, Tol S, Hoeben KA, van Marle MJ, Mul EP, Hordijk PL. Inside-out regulation of ICAM-1 dynamics in TNF-α-activated endothelium. PLoS One. 2010;5:e11336. doi: 10.1371/journal.pone.0011336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijssel J, Hoogenboezem M, Wester L, Hordijk PL, Van Buul JD. The N-Terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS One. 2012;7:e29912. doi: 10.1371/journal.pone.0029912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D. Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev. 2007;218:178–196. doi: 10.1111/j.1600-065X.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- Weed SA, Du Y, Parsons JT. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J Cell Sci. 1998;111:2433–2443. doi: 10.1242/jcs.111.16.2433. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Ellerbroek SM, Liu RY, Karnoub AE, Burridge K, Der CJ. RhoG signals in parallel with Rac1 and Cdc42. J Biol Chem. 2002;277:47810–47817. doi: 10.1074/jbc.M203816200. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, Burridge K. Analysis of low molecular weight GTPase activity in endothelial cell cultures. Methods Enzymol. 2008;443:285–298. doi: 10.1016/S0076-6879(08)02014-4. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005;109:181–190. doi: 10.1007/s00401-004-0928-x. [DOI] [PubMed] [Google Scholar]
- Xie Z, Xu W, Davie EW, Chung DW. Molecular cloning of human ABPL, an actin-binding protein homologue. Biochem Biophys Res Commun. 1998;251:914–919. doi: 10.1006/bbrc.1998.9506. [DOI] [PubMed] [Google Scholar]
- Yang L, Kowalski JR, Yacono P, Bajmoczi M, Shaw SK, Froio RM, Golan DE, Thomas SM, Luscinskas FW. Endothelial cell cortactin coordinates intercellular adhesion molecule-1 clustering and actin cytoskeleton remodeling during polymorphonuclear leukocyte adhesion and transmigration. J Immunol. 2006;177:6440–6449. doi: 10.4049/jimmunol.177.9.6440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.