

Cyclin-dependent kinase 5 (Cdk5) activity is important for the migration and invasion of cancer cells. Our results indicate that in the lung one of the mechanisms that hASH1 regulates—migration—takes place through induction of Cdk5 activity. Our data suggest that Cdk5 and its activator p35 promote lung cancer cell migration through hASH1-mediated signaling.
Abstract
Our previous data suggested that the human basic helix–loop–helix transcription factor achaete-scute homologue-1 (hASH1) may stimulate both proliferation and migration in the lung. In the CNS, cyclin-dependent kinase 5 (Cdk5) and its activator p35 are important for neuronal migration that is regulated by basic helix–loop–helix transcription factors. Cdk5/p35 may also play a role in carcinogenesis. In this study, we found that the neuronal activator p35 was commonly expressed in primary human lung cancers. Cdk5 and p35 were also expressed by several human lung cancer cell lines and coupled with migration and invasion. When the kinase activity was inhibited by the Cdk5 inhibitor roscovitine or dominant-negative (dn) Cdk5, the migration of lung cancer cells was reduced. In neuroendocrine cells expressing hASH1, such as a pulmonary carcinoid cell line, knocking down the gene expression by short hairpin RNA reduced the levels of Cdk5/p35, nuclear p35 protein, and migration. Furthermore, expression of hASH1 in lung adenocarcinoma cells normally lacking hASH1 increased p35/Cdk5 activity and enhanced cellular migration. We were also able to show that p35 was a direct target for hASH1. In conclusion, induction of Cdk5 activity is a novel mechanism through which hASH1 may regulate migration in lung carcinogenesis.
INTRODUCTION
Cyclin-dependent kinases (Cdks) belong to a large family of protein kinases (Dhavan and Tsai, 2001). Members of the family are essential for multiple cellular processes, including cell growth and differentiation (Xie and Tsai, 2004). Active Cdk5 is important for neural cell (NC) migration during development (Xie and Tsai, 2004). Unlike other Cdks, Cdk5 activity is mainly regulated by the association with p35, a protein often but not exclusively associated with neural tissues, and to lesser degree by p39 (Tsai et al., 1994). Cdk5 is able to perform substrate phosphorylation in different cellular compartments, including the cytoplasm and nucleus (Fu et al., 2006; Zhang et al., 2008).
Recently Cdk5 has been recognized as playing an important role outside the CNS as well. Specifically, overexpression of Cdk5 was related to an increased rate of cell migration in mouse corneal epithelial cells (Zelenka, 2004; Tripathi and Zelenka, 2009, 2010) and in the regulation of pancreatic insulin production (Lilja et al., 2001; Ubeda et al., 2004; Zheng et al., 2010). In addition, Cdk5 regulates cell motility and metastasis in prostate (Lin et al., 2004; Strock et al., 2006), pancreas (Feldmann et al., 2010), and thyroid cancer cells (Lin et al., 2007). Importantly, inhibition of Cdk5 activity reportedly decreased cancer cell migration and invasion (Lin et al., 2004; Goodyear and Sharma, 2007). However, the mechanism of Cdk5 involvement in tumorigenesis remains unknown. The activity of Cdk5 and its activator p35 have yet to be studied in depth in human lung cancer.
Achaete-scute complex homologue-1 (ASH1) was first identified as a basic helix–loop– helix (bHLH) transcription factor essential for neurogenesis in fetal nervous system (Guillemot et al., 1993). In normal and neoplastic lung, the expression of human ASH1 (hASH1) is restricted to pulmonary neuroendocrine (NE) cells or lung cancer cells with NE features, such as small cell lung cancers (SCLCs) and pulmonary carcinoids (Borges et al., 1997; Sriuranpong et al., 2002; Miki et al., 2012). It has been previously shown that hASH1 regulates tumor initiation capacity in SCLC cells (Jiang et al., 2009), but the underlying mechanism remains unclear. In a mouse model, constitutive hASH1 expression in the airway epithelium under Clara cell 10-kDa protein (CC10) promoter results in outgrowth of cells that extend into proximal alveolar spaces, which develop a histologically distinct lesion. The process involves suppression of apoptosis as well as promotion of growth, cellular proliferation, and migration (Wang et al., 2007, 2009). In vitro, overexpression of human hASH1 in lung cancer cells induces expression of the neuronal stem cell marker nestin (Jensen-Taubman et al., 2010). It also mediates down-regulation of cell adhesion molecules, such as E-cadherin, and activates the Wnt/β-catenin signaling pathway (Osada et al., 2008). Taken together, these data reflect the possible involvement of hASH1 in cancer cell migration, which may contribute to metastatic capabilities.
In the present study, we examine the expression and activity of Cdk5 and p35 in human lung cancer and explore the possibility that they regulate lung cancer cell migration and invasion. We hypothesize that human hASH1 regulates lung tumor cell migration and invasion by altering Cdk5/p35 expression. We show for the first time that human lung cancer cells with elevated expression of Cdk5 and p35 also demonstrate Cdk5 activity associated with their migration and invasion. Furthermore, we show that hASH1 acts directly on the p35 gene, which was found to be expressed in SCLCs. Suppression of endogenous hASH1 by short hairpin RNA (shRNA) in lung NE carcinomas or transduction of hASH1 in non-NE pulmonary adenocarcinomas also altered the expression patterns of Cdk5/p35. These data support the hypothesis that expression of hASH1 promotes human lung cancer cell migration and invasion through activation of Cdk5.
RESULTS
Cdk5 and its activator p35 are expressed in human lung cancer
Recent studies have shown that outside the nervous system, common cancers may express Cdk5 and p35 (Strock et al. 2006; Feldmann et al., 2010; Liu et al., 2011). Western blot analysis showed p35 and Cdk5 proteins in five out of five well-characterized human lung cancer cell lines, as well as in the immortalized human bronchial epithelial cell line BEAS-2B (Figure 1A). The levels were comparable with those of NCs obtained from fetal mouse brain. Lower expression of p35, similar to that seen in the bronchial epithelial cells, was seen in two of the lung cancer cell lines. Expression of p35 and Cdk5 transcripts was confirmed at the mRNA level by quantitative reverse transcription PCR (qRT-PCR; Figure 1B). The highest expression was seen in all three SCLC cell lines (DMS53, H82, H889) when compared with the levels of the prostate cancer cell line PC3, which was previously shown to be positive for p35 and Cdk5 (Supplemental Figure S1, A–B; Strock et al., 2006). By immunohistochemistry, p35 and Cdk5 proteins in lung cancer cell lines, as well as primary tumors, were seen to be distributed in both nuclear and cytoplasmic compartments (Figure 1, C and D), as has been reported for NCs (Zhang et al., 2008). In some cases, the immunoreactivity was found in plasma membranes (Figure 1D). This was further confirmed by Western blots of nuclear and cytoplasmic fractions of the cells from H727 lung cancer cells (Figure S2). Densitometric analyses suggested that the cytoplasmic fractions contained greater quantities of Cdk5 and p35 than the nuclear ones. Moreover, Figure 1C also indicates that high p35 level in cytoplasmic extensions correlates with enhanced polarity in individual cells that are either preparing to or in the process of migration (Strock et al., 2006). These results show that Cdk5 and its activator p35 are expressed in human lung cancer cell lines of non–small cell lung cancer (NSCLCs) and SCLCs, at both the mRNA and protein levels. Moreover, p35 is also expressed in tumor samples of lung cancers, regardless of the histological type.
FIGURE 1:
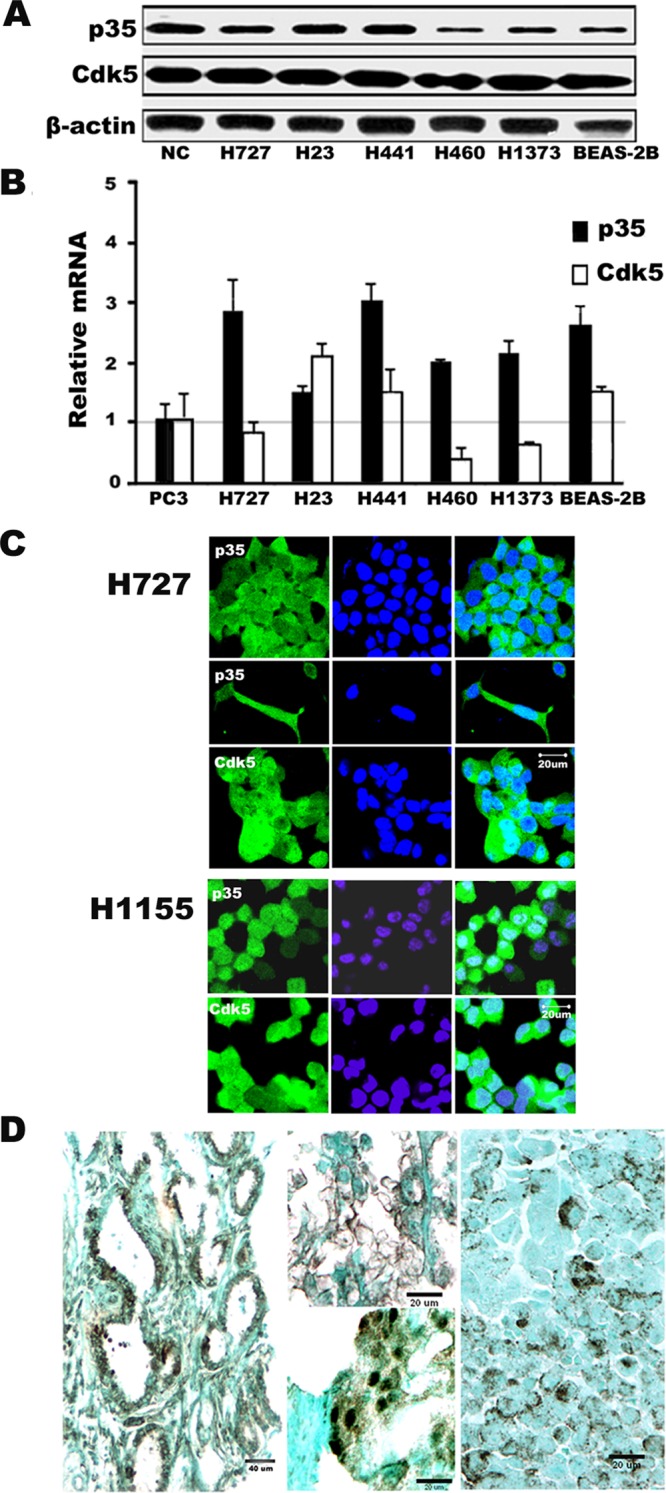
Expression of Cdk5 and the Cdk5 activator p35 in human lung cancer. (A) Western blotting of lung cancer (center lanes) and bronchial epithelial (BEAS-2B) cells shows expression of both p35 and Cdk5. NCs from mouse brain were used as a positive control (NC). (B) Relative mRNA expression by qRT- PCR shows high levels of p35 in all cell lines, while Cdk5 levels are variable. Expression is shown relative to the prostate cancer PC3 cells, which are designated as “1.” Mean ± SD from at least three independent experiments. (C) Photomicrographs of immunofluorescence (green) in two human lung cancer cell lines (H727 and H1155) reveal both cytoplasmic and nuclear distribution of p35 and Cdk5 protein. Nuclei were counterstained with DAPI (blue). Inset shows an elongated H727 cell with cytoplasmic projections positive for p35. (D) Photomicrographs of p35 expression in human lung cancers. Left, a combination of cytoplasmic and nuclear pattern (brown) in lung adenocarcinoma with glandular growth pattern. Top, center, membranous pattern on lung adenocarcinoma tumor cells that display a lepidic growth pattern. Bottom, center, predominantly nuclear pattern with some cytoplasmic immunoprecipitation and a cell at the bottom with membranous labeling in a poorly differentiated adenocarcinoma. Right, nonnuclear, granular pattern of immunoreactivity in SCLC. Because these cells typically have minimal cytoplasm, it is difficult to distinguish between membranous and cytoplasmic distribution. (Immunoperoxidase stain.)
Cdk5 is active in lung cancer cells
The kinase activity of Cdk5 has been extensively investigated in the CNS (Amin et al., 2002). Recently a number of studies have also provided evidence of Cdk5 activation beyond the neuronal system, including in prostate cancer cells (Strock et al., 2006), corneal epithelium (Gao et al., 2002), and pancreatic β cells (Ubeda et al., 2004). Because Cdk5 activity has not yet been studied in human lung cancer cells, we evaluated it here using an in vitro kinase assay. Cdk5 was immunoprecipitated from cultured human lung cancer cells, and the kinase activity was determined by using histone H1 (H1) as a substrate. In addition to elevated Cdk5 expression, Cdk5 activity was detected in human lung cancer H727 cells (Figure 2, A and B). The level of the activity was robust, albeit less than in NCs that were used as positive control. Roscovitine is known to block Cdk5 activity (Liebl et al., 2010; Zheng et al., 2010). We tested whether it inhibits Cdk5 activity in lung cancer cells. Confluent H727 cells were treated with different concentrations of roscovitine for 24 h; this was followed by a Cdk5 assay. As shown in (Figure 2C) 5–20 μM of roscovitine decreased Cdk5 activity in H727 cells.
FIGURE 2:

Cdk5 activity, migration, and invasion of H727 lung cancer cells is blocked by the Cdk5 inhibitor roscovitine. (A) Cdk5 assay of H727 human lung cancer cells with H1 phosphorylation. Autoradiographs (top) of phosphorylated H1 band and the corresponding Coomassie blue–stained gels of H1 (bottom). (B) Quantification of Cdk5 activity. NCs were used as a positive control. (C) Inhibition of Cdk5 activity by roscovitine. The bar graph indicates means ± SD from three independent experiments. (D) Phase-contrast photomicrographs of wound-healing assay using human H727 lung cancer cells following treatment with the Cdk5 inhibitor roscovitine. The ability of cells to migrate to the wound area was compared at 0 h and 24 h. Panels illustrate cellular migration without treatment and with treatment using the solvent DMSO and 20 μM of roscovitine (Ros). (E) Quantification (bar graph) of the wound areas is expressed as the percentage of the initially bare area at 0 h that is occupied by the migrating cells at 24 h. The occupied area decreased from 60 to 20% when cells were treated with roscovitine (p < 0.003) as compared with cells treated with DMSO. The solvent DMSO had no effect when compared with no treatment. (F) Quantification (bar graph) of cell invasion assay. H727 cells in serum-free media were plated onto Boyden chambers with Matrigel and allowed to invade toward serum in the absence (no treatment) or presence of roscovitine (Ros) for 24 h. The number of cells that invaded in the presence of 20 μM of roscovitine was significantly decreased compared with cells without treatment (p < 0.005). Minimal invasion was seen when the cells were plated in serum containing media (con, control without concentration gradient between the chambers). Values represent the mean ± SD of three wells from three experiments. (G) Quantification (bar graph) of wound repair assay using H727 cells transfected with dnCDK5. EV, empty virus.
Cdk5 activity required for migration and invasion of lung cancer cells
We next asked whether active Cdk5 in human lung cancer has a functional effect. Cell migration and invasion are critical biological processes during carcinogenesis (Chambers et al., 2002). Previous studies have shown that Cdk5 plays an important role in cell migration in NCs (Zheng et al., 2010), as well as in nonneuronal prostate cancer cells (Strock et al., 2006). To test whether Cdk5 activity detected in H727 lung cancer cells was relevant to cell migration, we performed a scratch wound assay in the presence or absence of the Cdk5 inhibitor roscovitine. In control conditions with untreated or dimethyl sulfoxide–treated (DMSO is a roscovitine solvent) cells, 60% of the wound gap was filled by migrating H727 cells in 24 h (Figure 2, D and E). The incubation of scratch-wounded culture with roscovitine arrested cell migration, as evidenced by only 20% of the gap being filled with cells at 24 h (Figure 2, D and E) compared with DMSO-treated cells. The difference between roscovitine-treated and control cells was statistically significant (p < 0.003). These data suggest that Cdk5 plays an important role in regulating the migration of H727 lung cancer cells. The ability of cells to penetrate through a basement membrane and invade in to adjacent tissues is also critical for the formation of metastases by cancer cells. As Cdk5 has been shown to be involved in cell invasion (Chambers et al., 2002), we examined its potential invasive activity in H727 lung cancer cells. The Boyden chamber assay with Matrigel was done in the presence or absence of roscovitine. H727 cells were placed in Matrigel on the filter of the upper chambers and 10% fetal bovine serum (FBS) was added as a chemoattractant to the lower chambers of 24-well dishes. As seen in Figure 2F, serum-starved H727 cells (DMSO only) had invaded toward serum at 24 h compared with control cells without serum (con). Following treatment with 20 μM roscovitine, the invasion of cells was decreased after 24 h when compared with cells that received only DMSO. The inhibitory effect of roscovitine was statistically significant (p < 0.005). Unstarved cells were used as a negative control. Finally, when cells were transfected with dominant-negative CDK5 (dnCDK5), there was a statistically significant decrease in their ability to close the wound (p <0.001; Figure 2G). The differences in cell migration and invasion seen in incubated cells at 0 and 24 h were not due to cell proliferation, as all experiments were performed in the presence of mitomycin C to block cell proliferation (unpublished data). Wound closure and Boyden chamber assay with Matrigel results showed that Cdk5 plays an important role in the regulation of lung cancer cell migration and invasion.
Expression of Cdk5/p35 and Cdk5 activity is regulated by hASH1 in human lung cancer cells
The Cdk5/p35 pathway is important for neuronal migration when it is coupled with proneural bHLH transcription factors in embryonic brain (Ge et al. 2006). The proneural bHLH transcription factor hASH1 is critical for NE differentiation in the lung (Borges et al., 1997). Because we found some of the highest levels of p35 expression, the neural activator of Cdk5 (Figure S1), in SCLC cells, which are characterized by NE and neural features, we examined the potential involvement of hASH1 in the activation of the Cdk5/p35 pathway. In fact, as already shown in Figures 1 and 2, Cdk5/p35 is active in the human lung cancer cell line H727 derived from a pulmonary carcinoid that represents another type of NE carcinoma typically expressing hASH1 (Linnoila, 1996; Miki et al., 2012). Figures 3A and S3 illustrate high expression of hASH1 mRNA in the pulmonary carcinoid H727 and the classic SCLC DMS53 and H889 cell lines, which are prototypes of human lung NE carcinomas. High levels of nuclear hASH1 are also found in SCLC tumor samples (Figure S3C). As expected, human adenocarcinoma cell lines and the immortalized human bronchial epithelial cells were negative. We used shRNA to suppress the endogenous hASH1 in H727 cells. As shown in Figure 3B, hASH1 level was significantly reduced in H727 cells (Figure 3, B and C) by shRNA (p < 0.002), indicating that it can efficiently target hASH1 mRNA. To determine whether the down-regulation of hASH1 affects the expression of Cdk5 and p35, we subjected protein lysates to Western blotting. The amount of Cdk5 and p35 in hASH1-shRNA transfected cells was much lower than in control H727 cells transfected with scrambled RNA (Figure 3, D and E). We also performed nuclear and cytoplasmic fractionation followed by Western blot assays to confirm the effect of hASH1 silencing on Cdk5 and p35 expression. Interestingly, we found that the nuclear p35 was greatly reduced when hASH1 was silenced by shRNA compared with that in the control H727 cells transfected by the scrambled RNA (Figure S4A). The reduction of p35 protein was statistically significant (Figure S4B). A decrease in Cdk5 was also observed (Figure S4). The results indicate that hASH1 regulates Cdk5/p35. It is thought that the complexing of p35 with Cdk5 occurs predominantly in the nucleus, which may explain the variance in subcellular responses to the hASH1 shRNA.
FIGURE 3:
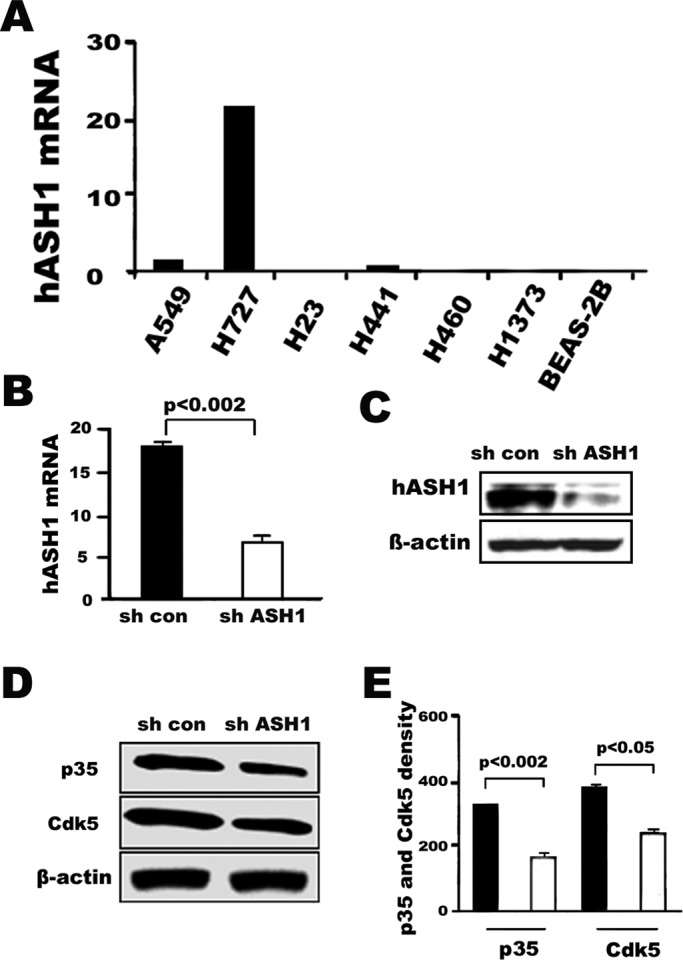
Down-regulation of hASH1 reduces the expression of p35 and Cdk5 in H727 lung cancer cells. (A) Relative expression of hASH1 mRNA by qRT-PCR in human lung cancer cell lines and bronchial epithelial BEAS-2B cells. The H727 lung cancer cell line was positive, which is consistent with the fact that it is derived from a pulmonary carcinoid. Carcinoids are well-differentiated NE cancers that express hASH1. The mRNA levels were normalized with that of the human lung adenocarcinoma A549 cell line designated as “1.” (B) Silencing hASH1 mRNA expression by shRNA in H727. By qRT-PCR, hASH1 expression was seen to be reduced more than 2.5-fold when compared with the expression by H727 cells transfected by scrambled RNA (con; p < 0.002). (C) Protein lysates from hASH1 shRNA transfected or scrambled vector–transfected cells were subjected to Western blotting. (D–E) Quantification of p35 and Cdk5 Western blot in (D) by densitometry (E; bar graph). shRNA-transfected H727 cells showed a significant reduction in p35 and Cdk5 expression compared with scrambled vector transfected cells (p < 0.002 and p < 0.05, respectively).
Consistent with its functional coupling, hASH1 immunoreactivity is colocalized with p35 in H727 cells, as demonstrated in Figure 4A. To further study the effects of hASH1 on tumor cell migration and invasion, we performed wound-healing and Boyden chamber with Matrigel assays with hASH1 shRNA-transfected H727 cells. We found that knockdown of hASH1 in H727 cells decreased migration by three-quarters compared with the ability of control cells (Figure 4, B and C). The invasive activity of H727 cells was also significantly blocked by hASH1 shRNA compared with that of control cells transfected with scrambled RNA (p < 0.02; Figure 4D). The data suggest that hASH1 may modulate the function of Cdk5/p35 pathway.
FIGURE 4:
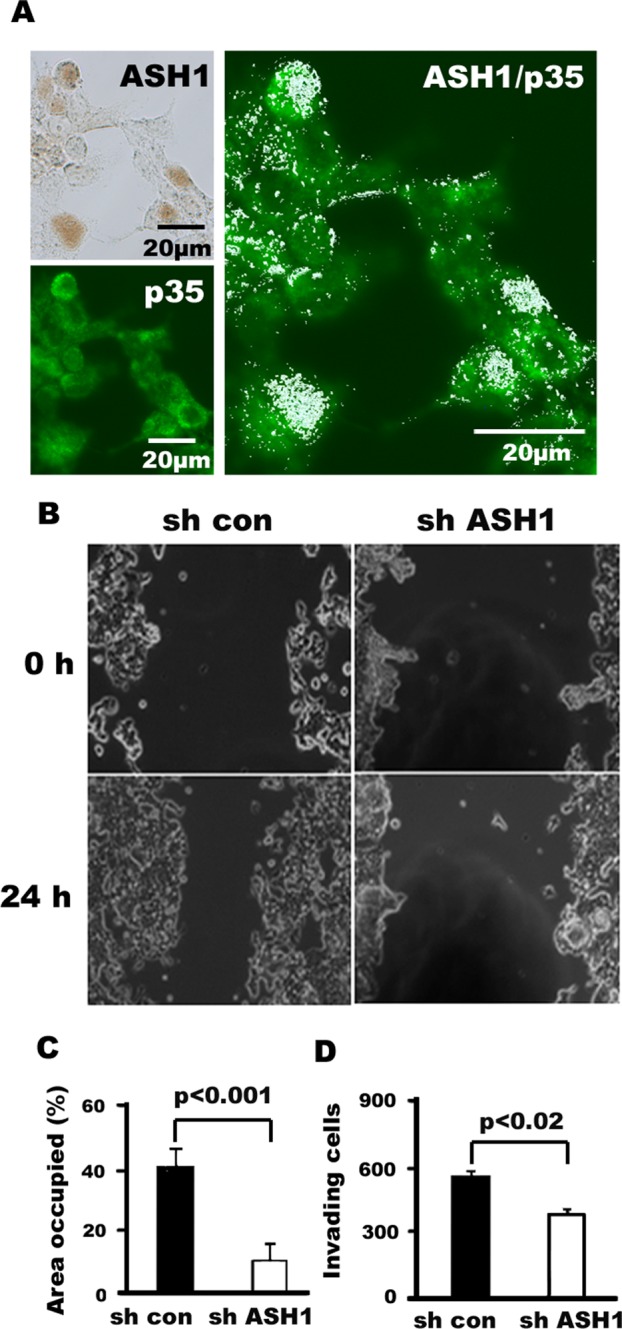
Silencing hASH1 in pulmonary carcinoid H727 cells leads to reduced migration and invasion. (A) Photomicrographs of immunohistochemical and immunofluorescence staining of hASH1 and p35 expression in H727 cells. Left, nuclear pattern of hASH1 immunoreactivity (top: brown, immunoperoxidase stain,) and cytoplasmic and nuclear distribution of p35 (bottom: green, immunofluorescence, bottom). Right, colocalization of hASH1 (nuclear, granular precipitate in pseudowhite) and p35 (green) in several cells (double staining with immunoperoxidase and immunofluorescence). (B) Phase-contrast photomicrographs of human carcinoid H727 cell migration assay. Down-regulation of hASH1 is associated with reduced cellular migration toward the wound at 24 h. (C) Quantification (bar graph) of the wound areas is expressed as the percentage of the initially bare area at 0 h that is occupied by the migrating cells at 24 h. The reduction in migration associated with shRNA was statistically significant (p < 0.002). (D) Silencing of hASH1 by shRNA significantly blocks invasion in H727 cells (p < 0. 05). The bars represent the means ± SD from three independent experiments.
To begin to explore the molecular mechanisms by which hASH1 regulates the migration through Cdk5/p35 pathway, we examined the SCLC cell line DMS53, which expresses high levels of hASH1 mRNA (Figure S3A). DMS53 typically forms colonies composed of roundish/polyclonal cells that adhere to the culture dish (Figure 5A). At any given time, a small number of cells can be seen to leave the colony and start migration (Figure 5A). Such polarized cells that are in the process of migration at edge of colonies revealed more intense p35 immunoreactivity. Figure 5C demonstrates that in most of the migrating cells hASH1 is expressed together with p35. The expression of hASH1 is nuclear. As with H727 cells, cellular migration in DMS53 cells was inhibited with the Cdk5 inhibitor roscovitine (Figure 5B). Within the promoter region of p35, there are multiple E-box elements, some of which are canonical to hASH1. Therefore we performed a chromatin immunoprecipitation (ChIP) assay in order to find out whether p35 is a direct target for hASH1. As illustrated in Figure 5D, the results suggest that p35 is downstream from hASH1. Moreover, in human SCLC tumor samples, most cells coexpressed hASH1 and p35 (Figure 5E).
FIGURE 5:

p35 is a direct target of hASH1, regulates migration in human SCLC cell line, and is coexpressed with hASH1 in primary SCLC tumor. (A) Photomicrograph of increased expression of p35 in migrating DMS53 SCLC cells. The most intense immunoreactivity is seen in groups of three to five cells (blue asterisk) that are migrating away from the main colony (circle), which is composed of cells with rounder shapes that appear minimally positive. (immunoperoxidase stain). (B) Quantification of migration (bar graph) of DMS53 SCLC cancer cells in scratch assay. Migration was significantly reduced by the CDK5 inhibitor roscovitine (p < 0.001), even at 6 h. (C) Photomicrographs of nuclear hASH1 (top, left: brown, immunoperoxidase) and p35 (top, right: green) immunofluorescence in migrating DMS53 cells. Colocalization (bottom) of nuclear hASH1 (pseudowhite) in several polarized, migrating DMS53 cells that are also positive for p35 (green; immunoperoxidase and immunofluorescence double labeling). (D) ChIP assay indicates direct binding of hASH1 (ASH1) to the CDK5 activator p35. (E) Photomicrograph of coexpression of nuclear hASH1 (ASH1, red) with p35 (green) in a human SCLC tumor sample. Most cells express both hASH1 and p35 (double immunofluorescence).
On the basis of the results in the NE lung cancer cell lines, we wanted to see whether overexpression of hASH1would affect the level of Cdk5/p35, Cdk5 activity, or Cdk5-dependent cell migration and invasion. The human adenocarcinoma cell line H23, which lacks endogenous hASH1 (Figure 3A), was transfected with hASH1. As shown in Figure 6A, a 10-fold increase in hASH1 mRNA expression was observed in the H23-hASH1 cells when compared with those transfected with vector alone (H23-V). By immunohistochemistry, hASH1 expression was seen to be exclusively nuclear (Figure 6B). This was associated with increased levels of Cdk5 mRNAs and proteins as analyzed by qRT-PCR and p35 mRNAs and proteins as analyzed by Western blotting (Figure 6, A–E). The total amount of p35 protein in H23-hASH1 cells was significantly increased when compared with that in H23-V cells (p < 0.001; Figure 6C). The p35 protein levels were comparable to those seen in NCs, which were used as a positive control (Figure 6, C and D). This was confirmed by immunofluorescence. As shown in Figure 6F, H23-hASH1 cells revealed strong p35 immunoreactivity that was both cytoplasmic and nuclear by location. We also observed strong p35 immunoreactivity localized to the cytoplasmic projections of polarized, potentially migrating H23-hASH1 cells (Figure 6G). Also shown in Figure 6G is the colocalization of nuclear hASH1 and cytoplasmic p35 in the same H23-hASH1 cells. The subcellular fractionation demonstrated that overexpression of hASH1 was associated with a significant increase in nuclear p35 (FigureS5). Evidence that linked overexpression of hASH1 with p35 in vivo came from a transgenic mouse model. In this model, hASH1 is constitutively expressed in the airway lining cells under a CC10 promoter, which results in histologically distinct metaplastic lesions—bronchiolization of the alveoli (BOA; Figure S6A). The airway lining and the lesions in the transgenic mice that express hASH1 were also positive for p35 (Figure S6B), while the p35 level was much lower in surrounding normal lung. Moreover, cellular colocalization (nuclear hASH1, cytoplasmic p35 in the same cell) was also seen in the tumor samples from mouse SCLC model (Meuwissen et al., 2003; Figure S6C).
FIGURE 6:
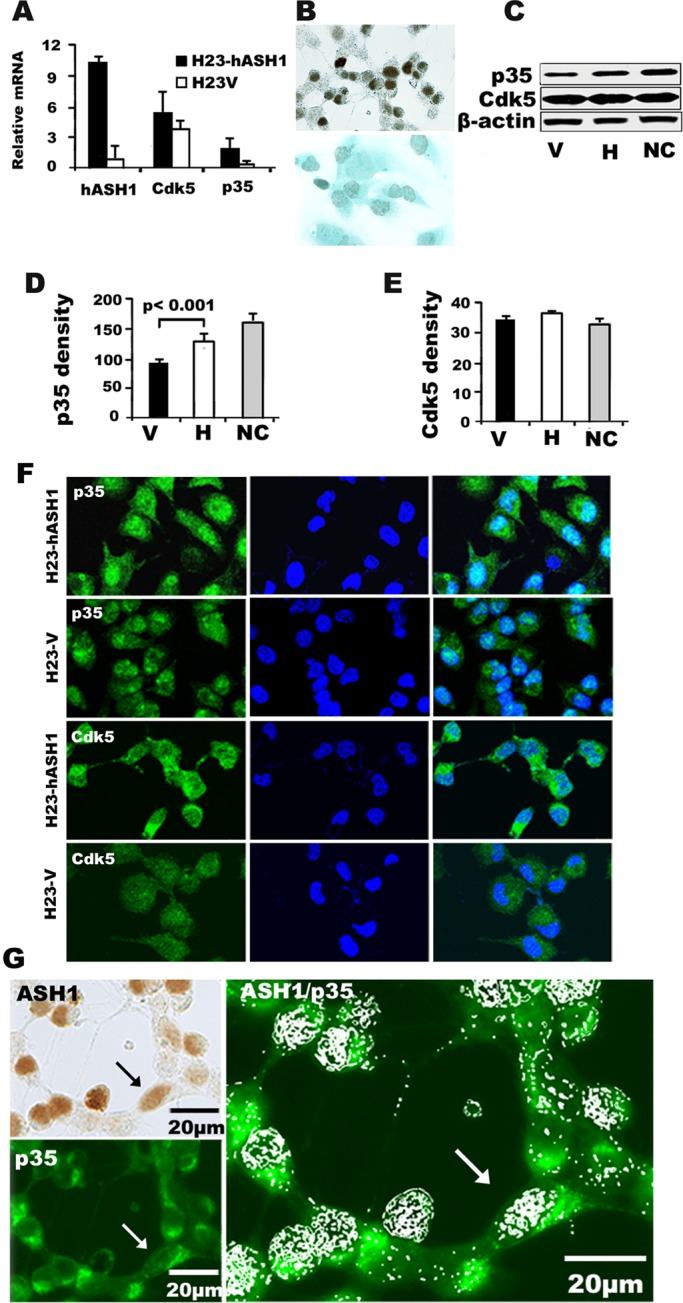
Overexpression of hASH1 in human lung adenocarcinoma cells leads to an increase in p35 expression in vitro. (A) By qRT-PCR, expression of hASH1 and p35 mRNAs was seen to increase in H23 cells transfected with hASH1 (H23-hASH1). Total RNA was extracted from stably expressing H23-hASH1 or control H23 cells (H23-V) that were transfected by vector only. hASH1 expression was increased 10-fold in transfected cells compared with empty vector. hASH1 overexpression significantly increased p35 (p < 0.01) mRNA expression. (B) Intense nuclear hASH1 immunoreactivity in H23-hASH1 cells (top panel). H23-V cells were negative (bottom panel) (immunoperoxidase staining). (C) Total protein lysates from H23- hASH1 (H) or control H23V (V) cells were analyzed by Western blot analysis The levels of p35 and Cdk5 were compared with those seen in NCs from fetal mouse brain, which served as a positive control. (D and E) Quantification of the results in (C) by densitometry showed a significant increase (p < 0.001) in p35 in H compared with V cells. NC, neural cells from embryonic brain. The bars represent means ± SD from three independent experiments. (F) Photomicrographs p35 (green) and Cdk5 (green) expression in H23-hASH1 and H23-V cells by immunofluorescence (left). Nuclei (blue) were counterstained with DAPI (middle). Light blue nuclear staining indicates coexpression (overlay in right). The overall intensity of immunofluorescence was higher in H23-hASH1 cells. (G) Photomicrographs of nuclear expression (brown) of hASH1 in H23-hASH1 cells (top, left: immunoperoxidase staining) and the same view showing p35 (green) expression (bottom, left: immunofluorescence). Colocalization of nuclear hASH1 (pseudowhite grains) and p35 (green) by immunoperoxidase and immunofluorescence, respectively, in H23-hASH1 cells. Black and white arrows point to a typical polarized double-positive cell with cytoplasmic extensions.
To assess the implications of hASH1 overexpression, we examined the Cdk5 activity. Endogenous Cdk5 was immunoprecipitated with Cdk5 antibody from human H23 lung cancer cell lysates. There was elevated kinase activity in H23-hASH1 cells, as compared with that in the H23-V control cells. However, the activity of the lung cancer cells was lower than that of NCs obtained from embryonic mouse brain. To establish a functional relationship between hASH1 and Cdk5 in H23 cancer cells, we studied migration. The results of the in vitro scratch wound assay showed that the H23-hASH1 cells migrated more efficiently than the H23-V control cells (Figure 7, C and D). The difference was statistically significant (p < 0.001; Figure 7D), suggesting that hASH1 is important for the motility of lung cancer cells. Moreover, the migrative activity of hASH1-H23 cells was blocked by the Cdk5 inhibitor roscovitine (Figure 7, C and D). The effect of roscovitine was minimal in H23-V cells. No impact was detected by the roscovitine solvent DMSO (unpublished data). To confirm the finding, we also transfected hASH1-H23 cells with dnCdk5 and tested their migrational capacity using the in vitro scratch wound assay. As shown in Figure 7E, the migration of hASH1-H23 lung cancer cells was significantly reduced (p < 0.013). The results indicate that exogenous hASH1 has a strong effect on Cdk5 activity and Cdk5-mediated cell migration of human lung cancer cells. Taken together, the data provide evidence that Cdk5/p35 is intimately coupled with the proneural bHLH transcription factor hASH1 in lung carcinogenesis.
FIGURE 7:

Overexpression of hASH1 in H23 lung adenocarcinoma cells leads to increased Cdk5 activity and migration that is blocked by dnCdk5. (A) Analysis of Cdk5 activity in vitro using H1 as a substrate. hASH1-transfected H23 (H23-hASH1) cells showed increased Cdk5 activity compared with H23-V control cells. NCs of embryonic mouse brain were used as a positive control (NC). Top, autoradiograph of phosphorylated H1 ([32P]H1); bottom, corresponding Coomassie blue–stained gel of H1. (B) Bar graph of the radioactivity measured in a liquid scintillation counter. Data represent mean ± SD of three experiments. (C) Phase-contrast micrographs and (D) quantitation of in vitro wound assay. The cells expressing hASH1 (H23-hASH1) display accelerated cell migration (a and b) compared with vector-transfected (H23-V) control cells (c and d; p < 0.001). Treatment with 20 μM of roscovitine reduces the hASH1-mediated cell migration (e and f; p < 0.03) over a 24-h time period. Cell migration was expressed as percentage of the wound area occupied by migrating cells. Data represent mean ± SD of three independent experiments. (E) Migration in the in vitro wound-healing assay was reduced by the expression of dnCdk5 in the hASH1-containing H23 cells. The difference between empty virus (EV) and dnCdk5-transfected H23-hASH1 cells was statistically significant (p < 0.013). Data represent mean ± SD of three experiments. (F) Western blot of dnCdk5 and endogenous Cdk5 in H23-hASH1 cells. trans, transfected with dnCDK5; con, control cells transfected with empty virus.
DISCUSSION
Cdk5 is a multifunctional protein kinase in the CNS that has been implicated in cell survival, death, and migration (Lagace et al., 2008; Huang et al., 2009; Kuo et al., 2009). Cdk5 activity has also been reported to promote cancer cell invasion and migration (Strock et al., 2006; Feldmann et al., 2010). However, the presence of Cdk5 activity and its functional role in lung cancer cells have not been explained. In the present study, we show for the first time that lung cancer cells have Cdk5 activity that mediates lung cancer cell migration and invasion. We also show that p35, the best-characterized activator of Cdk5 (Lew et al., 1994; Tsai et al., 1994) is commonly expressed both in primary lung cancers and in human lung cancer cell lines. Although the mRNA or protein levels of p35 and Cdk5 in lung cancer cells may be lower than those of NCs derived from embryonic mouse brain, there was enough p35 and Cdk5 activity in lung cancer cells to have functional impact. The finding that Cdk5 activity leads to lung cancer cell migration is analogous to a previous report on the promotion of prostate cancer cell migration by Cdk5 (Strock et al., 2006).
For clinical and pathological reasons, lung cancers are divided into SCLCs, which represent ∼15% of all lung cancers, and NSCLCs. SCLC is the most common and most virulent of NE cancers. Up to one-third of all lung cancers may have NE features. It was previously shown by Liu et al. (2011) that Cdk5 and/or p35 are commonly expressed in two-thirds of NSCLCs and may indicate poor prognosis. In the current study, we have extended these observations to lung NE carcinomas, including SCLC and carcinoid. About one-half of SCLC tumors expressed p35. We were able to show that lung cancer cell lines with high levels of Cdk5/p35 also showed Cdk5 activity. While we only studied a few cell lines, we were able to provide evidence that the lung NE lineage-specific oncogene ASH1 may be important for the regulation of Cdk5 and p35 when they are involved in cellular migration.
The ability of roscovitine to inhibit Cdk5 activity is well known (Goodyear and Sharma, 2007). As in prostate cancer, roscovitine treatment of lung cancer cells resulted in the reduction of cell migration and invasion by lung cancer cells in vitro (Strock et al. 2006). The specificity was confirmed by transfecting lung cancer cells with dnCdk5, which led to decreased Cdk5 activity and impaired migration. At the morphological level, both Cdk5 and p35 proteins were likely to accumulate in the polarizing lung cancer cells, as is typical for a migrating phenotype seen at the edge of a closing wound or an expanding colony of growing cells. The proteins most likely associate with the plasma membrane at the cytoplasmic projections to assist cell motility through phosphorylation of a variety targets (Huang et al., 2009). Taken together, our data show a critical requirement for Cdk5 activity and its main activator p35 in lung cancer cell migration and invasion.
For the purpose of the present study, we focused on the impact of hASH1 on the expression and activation of Cdk5/p35 in relation to lung cancer cell migration and invasion. In the developing brain, bHLH factors such as ASH1 are essential both for neuronal differentiation and the migration of maturing neurons to their proper locations, which is mediated by Cdk5/p35 pathway (Ge et al., 2006). In the lung, ASH1 is also pivotal for NE cell differentiation (Borges et al., 1997). In this paper, we show using ChIP assay that the neural Cdk5 activator p35 is a direct downstream target for hASH1. While the expression of both Cdk5 and p35 is common in lung cancer, the highest levels of the neuronal activator p35 were found in the NE SCLC and carcinoid cell lines that also express hASH1.
p35 was detected by immunohistochemistry in nuclear, cytoplasmic, and membranous locations in primary lung cancers. This was supported by fractionation assays in lung cancer cell lines and is analogous with neuronal data suggesting that nuclear location is important for the activity of p35, which is associated with migration in postmitotic neurons (Fu et al., 2006). Moreover, nuclear expression is necessary for the formation of Cdk5/p35 complex. It is thought that loss of nuclear Cdk5 leads to cell cycle entry (Zhang et al., 2008), suggesting an additional role of Cdk5 in cell cycle regulation and proliferation. When shRNA was used to suppress endogenous hASH1 expression in the human pulmonary carcinoid cell line H727, there was reduced Cdk5 and p35 expression. Interestingly, we found that most of the p35 protein in the nuclear extract was lost. So hASH1 seems to play a critical role in regulating the expression of p35 and its ability to trigger Cdk5 activation. We also found that knockdown of hASH1 caused inhibition of cell migration and invasion. These effects resemble the roscovitine-treated H727 cell response to cell migration and invasion. Conversely, when hASH1 was overexpressed in the lung adenocarcinoma cell line H23, which normally is negative for hASH1, there was increased expression of Cdk5 and p35 in conjunction with increased cell migration and invasion. Whether endogenous or overexpressed, the localization of hASH1 was always nuclear, consistent with it being a transcription factor. On the other hand, the presence of Cdk5 in the cytoplasmic compartment is significant, as its main substrates are cytoskeletal proteins (Zhang et al., 2008). Taken together, the results suggest that, as in the nervous system, basic HLH factors in the lung, such as hASH1, may regulate migration and invasion.
Like Cdk5, hASH1 is also multifunctional as a member of a large basic HLH family (Castro et al., 2011). In the lung, hASH1 is critical for the NE differentiation (Borges et al., 1997; Miki et al. 2012) and most, but not all, of its functions are lineage-specific (Nishikawa et al., 2011). Overexpression of hASH1 in the human adenocarcinoma cell line A549 leads to reduced expression of E-cadherin (Osada et al., 2008), which is an important member of the cadherin family. Down-regulation of E-cadherin gene expression is a crucial step in tumor cell migration and invasion through surrounding tissues (Bremnes et al., 2002; Moersig et al., 2002; Liu et al., 2009). We have shown that constitutive expression of hASH1 in vivo in non-NE cells along the airway epithelium in transgenic mice leads to BOA, a model for putative premalignant lesion in humans (Linnoila et al., 2000; Wang et al., 2007; Jensen-Taubman et al., 1998). While multiple pathways commonly activated in carcinogenesis may be involved in bronchiolization, we focused on migration by MMP7, the expression of which is regulated by hASH1 (Wang et al., 2009). In this study, we found that the Cdk5 main activator p35 is a direct target of hASH1 and is coexpressed in the BOA lesions of the mouse model, while the expression in the lung otherwise is low. In the light of the current in vitro data, it is possible that one of the mechanisms by which ASH1 contributes to lung carcinogenesis is through Cdk5/p35-mediated migration, similar to its role in the embryonic brain. On the other hand, both Cdk5 and p35 are widely expressed in lung cancers (Liu et al., 2011), regardless of the cell type, which suggests they are also linked to other pathways. One such pathway may be ras-related, as indicated by a study on pancreatic cancer (Feldmann et al., 2011; Eggers et al., 2011), as K-ras mutations are common also in NSCLCs.
MATERIALS AND METHODS
Cell lines and tissues
The human SCLC cell line DMS53, a gift from Douglas Ball (Johns Hopkins University, Baltimore, MD), was cultured in Waymouth's medium supplemented with 16% FBS. The other human cell lines were obtained from the American Type Culture Collection (Manassas, VA) and cultured at 37ºC in RPMI 1640 supplemented with 10% heat-inactivated FBS, 100 U penicillin, and 100 μg/ml streptomycin. An shRNAmir GIPZ lentiviral vector (Open Biosystems, Huntsville, AL) targeting the hASH1 mRNA was used for knocking down hASH1 expression in H727 cells as previously described (Wang et al., 2010). The target sequence of hASH1 shRNA is 5′-CGCTCAGAACAGTATCTTT-3′ (Clone ID # V2HS_15334; Open Biosystems).
To establish active hASH1 expressing NCI-H23 (H23) human lung cancer stable cell lines, we inserted the hASH1 gene into pcDNA 3.1/V5-His-TOPO TA plasmid (Invitrogen, Carlsbad, CA) and transfected it into TOP 10 Escherichia coli cells. Transfection of H23 cells was performed as described previously (Wang et al., 2007). The transfected cells were subsequently selected in the presence of G418 (Invitrogen, Carlsbad, CA) to establish stable clones and designated “H23-hASH1.” For dnCdk5 transfection (Nikolic et al., 1996), H727, H23-hASH1, or HEK-293 cells were grown to 70% confluency in six-well plates at a density of 3 × 105 cells/well. The cells were transiently transfected with 2 μg dnCdk5 cDNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Nontransfected cells were used to detect endogenous Cdk5 level. Transfected HEK-293 cells were used as a positive control.
Archival Formalin-fixed, paraffin-embedded human lung cancer specimens were obtained from the National Disease Research Interchange (Philadelphia, PA) and the National Cancer Institute (NCI)–Navy Medical Branch, National Naval Medical Center (Bethesda, MD). A multitumor human lung cancer tissue array of NSCLCs was provided by Stephen M. Hewitt at NCI–Tissue Array Research Program Laboratory, Advanced Technology Center (Gaithersburg, MD), and a tissue array of SCLCs was received from Pierre Massion, Vanderbilt-Ingram Cancer Center (Nashville, TN). All material used in the current study was derived from investigations performed after approval by local institutional review boards. Formalin-fixed, paraffin-embedded specimens of mouse tissues came from our previously published studies (Linnoila et al. 2000; Meuwissen et al., 2003). All animals were housed disease-free and handled in a humane manner at a facility accredited by and following the guidelines of a protocol approved by the NCI Animal Care and Use Committee.
Western blot analysis
For each cell line, 5 × 105 cells were plated in 100-mm dishes. Whole-cell protein lysates were extracted using M-PER lysis buffer (Thermo Fisher Scientific, Rockford, IL) with protease inhibitor cocktail (Roche, Somerville, MA), and the assay was carried out as previously described (Zheng et al., 2005). The blots were probed with the following primary antibodies: anti-Cdk5 (C8, 1:500), anti-p35 (C19, 1:500; Santa Cruz biotechnology, Santa Cruz, CA), and anti-β actin (1:1000; Abcam, Cambridge, MA). For quantification of the results, densitometry was performed by digitizing original films using professional options in an Epson 4490 Photo scanner and by measuring the bands using ImageJ software (http://rsb.info.nih.gov/ij).
Preparation of the nuclear and cytoplasmic extracts
Nuclear and cytoplasmic extracts were obtained using NE-PER extraction reagent kit (Thermo Fisher Scientific) according to the manufacturer's protocol. In brief, H23 and H727 (5 × 105) cells were harvested by being scraped from dishes and lysed in ice-cold NE-PER lysis buffer. After centrifugation for 5 min at 16, 000 × g at 4º C, the supernatant (cytoplasmic fraction) was collected in a prechilled tube. The pellet, which contained nuclear proteins, was resuspended in ice-cold NER buffer and incubated on ice for 40 min by overtaxing 15 s every 10 min. After that, the samples were centrifuged (16,000 × g for 10 min at 4º C), and the supernatant's nuclear fraction was transferred to a prechilled tube. Cytoplasmic and nuclear fractions were stored at −80ºC until use. Equal amounts (50 μg) of each cytoplasmic and nuclear protein sample were resolved on SDS–PAGE gel and transferred to a polyvinylidene difluoride membrane. The blot was probed with anti-Cdk5, anti-p35, anti-actin, or anti-H1. Anti-mouse horseradish peroxidase (HRP) or anti-rabbit HRP at a dilution of 1:10,000 was used as the secondary antibody with Super Signal West Pico Chemiluminescent substrate for detection of HRP. Anti–β-actin and anti- H1 antibodies were used as loading and quality controls for cytoplasmic and nuclear extracts, respectively.
In vitro Cdk5 assay
For the Cdk5 assay, H727 cells or stably hASH1 transfected H23 cells were treated with 5–20 μM of the Cdk5 inhibitor roscovitine or DMSO or left untreated. After 24 h, the cells were lysed on ice with lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM ethylene glycol tetraacetic acid, 1% SDS, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, and 2.5 mM NaVo4 supplemented with a mixture of protease inhibitors and 1 mM phenylmethylsulfonyl fluoride. NCs obtained from embryonic mouse brains were included as a positive control. Total protein samples (500 μg) were precleared with protein A PLUS-agarose beads (Sigma-Aldrich, St. Louis, MO), rotating at 4ºC for 1 h. After centrifugation for 3 min at 10, 000 × g at 4ºC, the precleared samples were mixed with anti-Cdk5 (C8) antibody and incubated overnight at 4ºC. The complexes were then incubated for 4 h at 4ºC with Protein A-agarose beads. The immunoprecipitant was washed four times with lysis buffer and once with 1X kinase buffer (25 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 5 mM β-glycerophosphate, 0.1 mM NaVO4, 2 mM dithiothreitol, 50 μM ATP). Immmunopreciptated Cdk5 was subjected to kinase assays in the presence of 10 μCi [γ32P]ATP and 1 μg H1 protein as a substrate at 30ºC for 30 min. The reaction was stopped by adding a tricloroacetic acid. Samples (25 μl) were spotted in duplicate onto p18 phosphocellulose paper. After air drying, the phosphocellulose paper was washed four times for 15 min each time in 75 mM phosphoric acid, air dried, and transferred to vials containing Bio-Safe II Scintillation solution (Research Products International, Mount Prospect, IL). The radioactivity was measured in a liquid scintillation counter. The Cdk5 activity was expressed in picomoles of ATP/min/μg protein. For detection of H1, sample aliquots were electrophorized on polyacrylamide gel and stained with Coomassie blue.
ChIP assay
ChIP assay was performed following Jiang et al. (2009), using a Magna-ChIP A kit (Millipore, Billerica, MA) on DMS53 cells. Cross-linking was performed via 10-min incubation with 1% formaldehyde. After lysis, sonication yielded an average fragment size of 200–1000 base pairs. Solubilized chromatin was immunoprecipitated with mouse immunoglobulin G (negative control) or the monoclonal antibody against ASH1 (BD Biosciences, San Jose, CA) overnight at 4ºC. Magnetic protein A beads were added, and specimens were incubated at 4ºC for 2 h. PCR was performed using primers directed at conserved regions containing the ASH1-binding E-box motif identified in the proximal promoter regions of the p35 gene. Input represents a 10-fold dilution of unprecipitated genomic DNA.
Immunohistochemistry and immunofluorescence
Immunohistochemistry was done using the avidin-biotinylated peroxidase method and Vectastain ABC kits (Vector, Burlingame, CA). Primary antibodies were incubated overnight at 4°C, and signals were visualized using 3,3′-diaminobenzidine tetrahydrochloride, as previously described (Wang et al., 2009). Slides were counterstained with Mayer's hematoxylin or Light Green and viewed using a Nikon E400 microscope. Immunofluorescence was performed as described previously (Zheng et al., 2010). For cell preparations, approximately 4 × 104 H727, DMS53, stable hASH1 expressing H23, or vector control cells were plated on glass coverslips (Fisher Scientific, Waltham, MA) in six-well plates for 2 d. The cells were washed twice with phosphate-buffered saline (PBS) and then fixed with 4% paraformaldehyde in PBS for 30 min. They were permeabilized and blocked in 5% FBS in PBS containing 0.1% Triton X-100 for 1 h. Cell and tissue slides were incubated with primary antibodies (prepared in blocking buffer) against Cdk5 (1:100), p35 (1:100), and ASH1 (1:20–50) (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4ºC. Following five washes, the cells were incubated with Alexa Fluor 594 conjugated anti-mouse secondary antibody or Alexa Fluor 388 conjugated anti-rabbit antibody (1:500 or 1:1000) for 45 min to 2 h at room temperature, washed three times with PBS, optionally stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) for 10 min, and mounted onto slides in Fluoromount-G (Southern Biotech, Birmingham, AL). The fluorescence images were observed under a laser-scanning confocal microscope (Zeiss LSM510) using 40× non-oil or 63× oil immersion objective.
Double labeling was performed by applying immunoperoxidase or immunofluorescence methods for ASH1, which was followed by immunofluorescence for p35. Results were viewed using a Nikon E400 microscope equipped with bright-field and epifluorescence illuminations, a camera, and MetaMorph software. Pseudowhite coloring was created in Photoshop 6.0 using the steps image→mode→grayscale→indexed color→color table and selecting spectrum mode.
qRT-PCR
Total RNA was extracted from several different lung cancer cell lines using the RNeasy Mini Kit (Qiagen, Valencia, CA). The human prostate cancer cell line PC3 was included as a positive control.
RNA (1 μg) was converted to cDNA using SuperScript III First-Strand kit (Invitrogen) according to the manufacturer's protocol. The relative levels of mRNA expression were quantified by using an iCycler iQ Multi-Color real-time PCR detection system (Bio-Rad, Hercules, CA) with a set of primers specific for Cdk5, p35, hASH1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Midland Certified Reagent Company, TX). GAPDH RNA levels were used as an endogenous control to adjust differences in the amount of template added to the reaction. Amplification was performed as previously described (Wang et al., 2009). Data are expressed as fold change versus controls (endogenous, scrambled vector–transfected cells or vector-transfected cells). The values represent the mean ± SD of three wells from three experiments.
Cell migration assay
Cell migration activity was determined by in vitro wounding (“wound-healing”) assay. Cells were cultured to confluence in six-well plates with RPMI 1640 containing 10% FBS. To prevent proliferation, we also used 10 μg/ml of mitomycin C (Calbiochem, San Diego, CA; Daniel and Groves, 2002). Linear wounds were created using a sterile pipette tip. The debris was washed away with PBS, and plates were incubated in fresh medium for 24 h. Cells were cultured in the presence or absence of 20 μM roscovitine. In these experiments, a DMSO vehicle was used as a control. For quantification, the wound gap closing process was digitally photographed using an inverted microscope with 10× objective at 0 h (time of wounding), 8 h, 18 h, or 24 h time points and measured using MetaMorph software. To serially measure the same fields the (x,y) coordinates of the plate position on the microscope stage were recorded, and the locations were also marked by reference points on the reverse sides of the plates.
To confirm that the mitomycin C was blocking the proliferation of lung cancer cells, cells were seeded at 10,000 cells/well into 96-well plates and incubated overnight. The H727 and H23 cells were pretreated with 10 μg/ml mitomycin C at 37°C for 40 min. After a 40-min incubation, they were washed with media and allowed to continue to grow for 24 h in fresh growth media. The growth curve was established using a sulforhodamine B–based assay. The results showed that mitomycin C treatment prevented the growth, as the curve was flattened (unpublished data).
Cell invasion assay
The ability of cells to invade through extracellular material was evaluated using the Boyden chamber with a Matrigel layer in the presence or absence of 20 μM roscovitine. DMSO (the solvent for roscovitine) treatment was used as control. Approximately 5 × 104 tumor cells were seeded into Matrigel-coated Boyden chambers (8-μm pore; BD Biosciences, San Jose, CA) in RPMI medium containing 0.1% FBS. Medium containing 10% FBS was used as a chemoattractant in the lower chamber. After incubation at 37ºC for 24h, the noninvading cells on the upper surface of the membrane were removed with cotton-tipped swabs, and cells on the lower surface of the membrane were stained with Diff-Quick solution (Diff-Quik Stain Set; Siemens Healthcare Diagnostics, Deerfield, IL). The degree of cellular invasion was determined by counting the number of cells that invaded through the Matrigel per high-power field.
Statistical analysis
Statistical data evaluation was performed using Student's t test. p values of <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Kathy Kelly for the discussions during the project. The authors are also grateful to Kathy Keefe and Timothy Healy for their help in proofreading the manuscript. This research was supported by the NCI Intramural Research Program of the National Institutes of Health. The authors are grateful to Steve Hewitt and Pierre Massion for providing the tissue microarrays of human lung cancers.
Abbreviations used:
- ASH1
achaete-scute homologue-1
- bHLH
basic helix-loop-helix
- BOA
bronchiolization of the alveoli; CC10, Clara cell 10-kDa protein
- Cdk
cyclin-dependent kinase
- ChIP
chromatin immunoprecipitation
- DMSO
dimethyl sulfoxide
- dnCdk5
dominant-negative Cdk5
- FBS
fetal bovine serum
- H1
histone H1
- hASH1
human achaete-scute homologue-1
- HRP
horseradish peroxidase
- NC
neural cell
- NCI
National Cancer Institute
- NE
neuroendocrine
- NSCLC
non–small cell lung cancer
- PBS
phosphate-buffered saline
- qRT-PCR
quantitative reverse transcription PCR
- SCLC
small cell lung cancer
- shRNA
short hairpin RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-12-1010) on June 13, 2012.
REFERENCES
- Amin ND, Albers W, Pant HC. Cyclin-dependent kinase 5 (cdk5) activation requires interaction with three domains of p35. J Neurosci Res. 2002;67:354–362. doi: 10.1002/jnr.10116. [DOI] [PubMed] [Google Scholar]
- Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, Ball DW. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature. 1997;386:852–855. doi: 10.1038/386852a0. [DOI] [PubMed] [Google Scholar]
- Bremnes RM, Veve R, Hirsch FR, Franklin WA. The E-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer. 2002;36:115–124. doi: 10.1016/s0169-5002(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Castro DS, et al. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Gene Dev. 2011;25:930–945. doi: 10.1101/gad.627811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Daniel RJ, Groves RW. Increased migration of murine keratinocytes under hypoxia is mediated by induction of urokinase plasminogen activator. J Invest Dermatol. 2002;119:1304–1309. doi: 10.1046/j.1523-1747.2002.19533.x. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Eggers JP, et al. Cyclin-dependent kinase 5 is amplified and over-expressed in pancreatic cancer and activated by mutant K-Ras. Clinical Cancer Res. 2011;17:6140–6150. doi: 10.1158/1078-0432.CCR-10-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, Goggins M, Maitra A, Nelkin BD. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460–4469. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Choi YK, Qu D, Yu Y, Cheung NS, Qi RZ. Identification of nuclear import mechanisms for the neuronal Cdk5 activator. J Biol Chem. 2006;281:39014–39021. doi: 10.1074/jbc.M512663200. [DOI] [PubMed] [Google Scholar]
- Gao C, Negash S, Guo HT, Ledee D, Wang HS, Zelenka P. CDK5 regulates cell adhesion and migration in corneal epithelial cells. Mol Cancer Res. 2002;1:12–24. [PubMed] [Google Scholar]
- Ge W, et al. Coupling of cell migration with neurogenesis by proneural bHLH factors. Proc Natl Acad Sci USA. 2006;103:1319–1324. doi: 10.1073/pnas.0510419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear S, Sharma MC. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp Mol Pathol. 2007;82:25–32. doi: 10.1016/j.yexmp.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell. 1993;75:463–476. doi: 10.1016/0092-8674(93)90381-y. [DOI] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Yousefi N, Chen Z, Jacobson K, Ginsberg MH. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat Cell Biol. 2009;11:624–630. doi: 10.1038/ncb1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Taubman S, Steinberg SM, Linnoila RI. Bronchiolization of the alveoli in lung cancer: pathology, patterns of differentiation and oncogene expression. Int J Cancer. 1998;75:489–496. doi: 10.1002/(sici)1097-0215(19980209)75:4<489::aid-ijc1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Jensen-Taubman S, Wang XY, Linnoila RI. Achaete-scute homologue-1 tapers neuroendocrine cell differentiation in lungs after exposure to naphthalene. Toxicol Sci. 2010;117:238–248. doi: 10.1093/toxsci/kfq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, Nelkin BD, Ball DW. Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res. 2009;69:845–854. doi: 10.1158/0008-5472.CAN-08-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo HS, Hsu FN, Chiang MC, You SC, Chen MC, Lo MJ, Lin H. The role of Cdk5 in retinoic acid-induced apoptosis of cervical cancer cell line. Chin J Physiol. 2009;52:23–30. doi: 10.4077/cjp.2009.amg067. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Benavides DR, Kansy JW, Mapelli M, Greengard P, Bibb JA, Eisch AJ. Cdk5 is essential for adult hippocampal neurogenesis. Proc Natl Acad Sci USA. 2008;105:18567–18571. doi: 10.1073/pnas.0810137105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Liebl J, Weitensteiner SB, Vereb G, Takacs L, Fuerst R, Vollmar AM, Zahler S. Cyclin dependent kinase 5 (Cdk5) regulates endothelial cell migration and angiogenesis. J Biol Chem. 2010;285:35932–35943. doi: 10.1074/jbc.M110.126177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja L, Yang SN, Webb DL, Juntti-Berggren L, Berggren PO, Bark C. Cyclin-dependent kinase 5 promotes insulin exocytosis. J Biol Chem. 2001;276:34199–34205. doi: 10.1074/jbc.M103776200. [DOI] [PubMed] [Google Scholar]
- Lin H, Chen MC, Chiu CY, Song YM, Lin SY. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J Biol Chem. 2007;282:2776–2784. doi: 10.1074/jbc.M607234200. [DOI] [PubMed] [Google Scholar]
- Lin H, Juang JL, Wang PS. Involvement of Cdk5/p25 in digoxin-triggered prostate cancer cell apoptosis. J Biol Chem. 2004;279:29302–29307. doi: 10.1074/jbc.M403664200. [DOI] [PubMed] [Google Scholar]
- Linnoila RI. Spectrum of neuroendocrine differentiation in lung cancer cell lines featured by cytomorphology, markers, and their corresponding tumors. J Cell Biochem Suppl. 1996;24:92–106. doi: 10.1002/jcb.240630506. [DOI] [PubMed] [Google Scholar]
- Linnoila RI, Zhao B, DeMayo JL, Nelkin BD, Baylin SB, DeMayo FJ, Ball DW. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res. 2000;60:4005–4009. [PubMed] [Google Scholar]
- Liu JL, Wang XY, Huang BX, Zhu F, Zhang RG, Wu G. Expression of CDK5/p35 in resected patients with non-small cell lung cancer: relation to prognosis. Med Oncol. 2011;28:673–678. doi: 10.1007/s12032-010-9510-7. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. Ablation of p120-catenin enhances invasion and metastasis of human lung cancer cells. Cancer Sci. 2009;100:441–448. doi: 10.1111/j.1349-7006.2008.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Miki M, Ball DW, Linnoila RI. Insights into the achaete-scute homolog-1 gene (hASH1) in normal and neoplastic human lung. Lung Cancer. 2012;75:58–65. doi: 10.1016/j.lungcan.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Moersig W, Horn S, Hilker M, Mayer E, Oelert H. Transfection of E-cadherin cDNA in human lung tumor cells reduces invasive potential of tumors. Thorac Cardiovasc Surg. 2002;50:45–48. doi: 10.1055/s-2002-20161. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YFM, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- Nishikawa E, et al. miR-375 is activated by ASH1 and inhibits YAP1 in a lineage dependent manner in lung cancer. Cancer Res. 2011;71:6165–6173. doi: 10.1158/0008-5472.CAN-11-1020. [DOI] [PubMed] [Google Scholar]
- Osada H, Tomida S, Yatabe Y, Tatematsu Y, Takeuchi T, Murakami H, Kondo Y, Sekido Y, Takahashi T. Roles of achaete-scute homologue 1 in DKK1 and E-cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res. 2008;68:1647–1655. doi: 10.1158/0008-5472.CAN-07-5039. [DOI] [PubMed] [Google Scholar]
- Sriuranpong V, Borges MW, Strock CL, Nakakura EK, Watkins DN, Blaumueller CM, Nelkin BD, Ball DW. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol. 2002;22:3129–3139. doi: 10.1128/MCB.22.9.3129-3139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strock CJ, Park JI, Nakakura EK, Bova GS, Isaacs JT, Ball DW, Nelkin BD. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006;66:7509–7515. doi: 10.1158/0008-5472.CAN-05-3048. [DOI] [PubMed] [Google Scholar]
- Tripathi BK, Zelenka PS. Cdk5-dependent regulation of Rho activity, cytoskeletal contraction, and epithelial cell migration via suppression of Src and p190RhoGAP. Mol Cell Biol. 2009;29:6488–6499. doi: 10.1128/MCB.01098-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi BK, Zelenka PS. Cdk5: A regulator of epithelial cell adhesion and migration. Cell Adh Migr. 2010;4:333–336. doi: 10.4161/cam.4.3.11131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr., Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Kemp DM, Habener JF. Glucose-induced expression of the cyclin-dependent protein kinase 5 activator p35 involved in Alzheimer's disease regulates insulin gene transcription in pancreatic β-cells. Endocrinology. 2004;145:3023–3031. doi: 10.1210/en.2003-1522. [DOI] [PubMed] [Google Scholar]
- Wang XY, Dakir el H, Naizhen X, Jensen-Taubman SM, DeMayo FJ, Linnoila RI. Achaete-scute homolog-1 linked to remodeling and preneoplasia of pulmonary epithelium. Lab Invest. 2007;87:527–539. doi: 10.1038/labinvest.3700552. [DOI] [PubMed] [Google Scholar]
- Wang XY, Demelash A, Kim H, Jensen-Taubman S, Dakir el H, Ozbun L, Birrer MJ, Linnoila RI. Matrilysin-1 mediates bronchiolization of alveoli, a potential premalignant change in lung cancer. Am J Pathol. 2009;175:592–604. doi: 10.2353/ajpath.2009.080461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, Penalva LO, Yuan H, Linnoila RI, Lu J, Okano H, Glazer RI. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol Cancer. 2010;9:221. doi: 10.1186/1476-4598-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Tsai LH. Cdk5 phosphorylation of FAK regulates centrosome-associated miocrotubules and neuronal migration. Cell Cycle. 2004;3:108–110. [PubMed] [Google Scholar]
- Zelenka PS. Regulation of cell adhesion and migration in lens development. Int J Dev Biol. 2004;48:857–865. doi: 10.1387/ijdb.041871pz. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci USA. 2008;105:8772–8777. doi: 10.1073/pnas.0711355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Hu YF, Zhang A, Wang W, Li B, Amin N, Grant P, Pant HC. Overexpression of p35 in Min6 pancreatic beta cells induces a stressed neuron-like apoptosis. J Neurol Sci. 2010;299:101–107. doi: 10.1016/j.jns.2010.08.067. [DOI] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.