

We report here that phosphatidylinositol 3,5-bisphosphate is required for the full activation and localization of mTORC1 by insulin and amino acids, due to the direct interaction of the lipid with the Raptor subunit, which permits efficient activation by GTPases.
Abstract
The kinase complex mechanistic target of rapamycin 1 (mTORC1) plays an important role in controlling growth and metabolism. We report here that the stepwise formation of phosphatidylinositol 3-phosphate (PI(3)P) and phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) regulates the cell type–specific activation and localization of mTORC1. PI(3)P formation depends on the class II phosphatidylinositol 3-kinase (PI3K) PI3K-C2α, as well as the class III PI3K Vps34, while PI(3,5)P2 requires the phosphatidylinositol-3-phosphate-5-kinase PIKFYVE. In this paper, we show that PIKFYVE and PI3K-C2α are necessary for activation of mTORC1 and its translocation to the plasma membrane in 3T3-L1 adipocytes. Furthermore, the mTORC1 component Raptor directly interacts with PI(3,5)P2. Together these results suggest that PI(3,5)P2 is an essential mTORC1 regulator that defines the localization of the complex.
INTRODUCTION
The mechanistic target of rapamycin (mTOR) protein kinase complexes control cell growth and metabolism in response to growth factors, nutrients, and energy levels. mTORC1 is activated by the growth factor–sensitive phosphatidylinositol 3,4,5-triphosphateGTPase-activating protein (GAP) complex that inactivates the GTPase Rheb (Inoki and Guan, 2009). GAP inactivation increases the activity of Rheb, which in turn can activate mTORC1. Amino acids can increase mTORC1 activity through the Rag family of GTPases (Kim et al., 2008; Sancak et al., 2008, 2010).
Insulin regulates the synthesis of polyphosphoinositides, causing an increase in PI(3,4,5)P3, and a plasma membrane–specific pool of PI(3)P (Maffucci et al., 2003; Kong et al., 2006; Falasca et al., 2007; Lodhi et al., 2008). This latter process requires the Rab5 guanine nucleotide exchange factor (GEF) Gapex-5 (also known as GAPVD1; Lodhi et al., 2008) and the class II phosphatidylinositol 3-kinase PI3K-C2α (also known as PIK3CA; Falasca et al., 2007). The polyphosphoinositide, PI(3,5)P2, is formed from the phosphorylation of PI(3)P by the phosphatidylinositol 5-kinase (PI5K) PIKFYVE (also known as FAB1 or PIP5K3; Ho et al., 2011). PIKFYVE exists in a multimeric complex with the adaptor proteins VAC14, VAC7, ATG18, and FIG4. We report here that PI(3,5)P2 is required for the full activation and localization of mTORC1 by insulin and amino acids, due to the direct interaction of the lipid with the Raptor subunit, which permits efficient activation by GTPases.
RESULTS AND DISCUSSION
Amino acids and insulin increase PI(3)P and PI(3,5)P2
We examined the effect of insulin on PI(3,5)P2 by labeling 3T3-L1 adipocytes with tritiated inositol and examining them with high-performance liquid chromatography. Insulin caused a small but significant increase in the abundance of both PI(3)P and PI(3,5)P2 in a wortmannin-sensitive manner (Figure 1A). This was associated with an increase in the appearance of the PI(3)P-sensing reporter green fluorescent protein (GFP)2xFYVE at the periphery of the cells (Figure 1B, quantified in Figure 1C). Small interfering RNA (siRNA)-mediated knockdown of the class II PI kinase PI3K-C2α reduced the insulin-dependent increase in PI(3)P at or near the plasma membrane (Figure 1C). Treatment of cells with amino acids also caused increases in PI(3)P and PI(3,5)P2 (Figure 1, D and E). Likewise, amino acid treatment produced redistribution of the PI(3)P reporter construct to the plasma membrane in 3T3-L1 adipocytes (Figure 1F). In both cases, the distribution of the reporter protein along the surface of the plasma membrane was discontinuous. Likewise, knockdown of PI3K-C2α ablated amino acid–stimulated plasma membrane localization of this reporter construct (Figure 1, C and F). A similar increase in phosphoinositide levels was seen in HEK-293T cells (Figure 1E), although we did not observe the redistribution of GFP-2xFYVE in these cells, suggesting that insulin-stimulated or amino acid–stimulated plasma membrane–specific synthesis of PI(3)P may be restricted to adipocytes and myocytes. We also observed amino acid–induced changes in PI(4)P and PI(4,5)P2 in all cell types tested (Figure 1, D and E). The mechanism underlying these changes, as well as their significance with respect to amino acid signaling, remains unclear.
FIGURE 1:

Insulin and amino acids induce PI(3)P and PI(3,5)P2 formation. (A) 3T3-L1 adipocytes were labeled with [3H]inositol for 48 h, serum-starved for 3 h prior to stimulation, and then, where indicated, incubated with 100 nM wortmannin for 1 h, which was followed by 100 nM insulin for 15 min. Lipids were extracted and analyzed as described in Materials and Methods. (B) 3T3-L1 adipocytes were electroporated with GFP-2xFYVE. After 16 h, cells were serum-starved (Basal and Insulin) for 3 h or amino acid–deprived (Amino Acids) for 45 min, which was followed by stimulation with 100 nM insulin or amino acids for 15 min. Scale bars: 5 μm. There were no detectable differences between serum- starved and amino acid–deprived cells with respect to GFP-2xFYVE staining. Three representative cells are shown. (C) 3T3-L1 adipocytes were electroporated with siRNA targeting PI3K-C2α or a control siRNA along with GFP-2xFYVE for 72 h. Cells were stimulated with insulin as described in (B), fixed, and then counted. Cells were quantified by counting the percentage of cells with plasma membrane GFP staining. (D) 3T3-L1 cells were [3H]inositol labeled, amino acid–deprived, and then stimulated with amino acids for 5 or 15 min as described in the figure. Asterisks indicate comparisons with untreated cells. (E) HEK-293T cells were labeled at ∼30% confluence and stimulated with amino acids for 15 min. Lipids were extracted as described in Materials and Methods. (F) 3T3-L1 cells were electroporated with GFP-2xFYVE and either control or PI3K-C2α–specific siRNA for 72 h and treated as described in (A). Cells were quantified by counting the percentage of cells with plasma membrane GFP staining. The data in (C) and (D) represent three independent experiments in which >200 cells were quantified. Asterisks indicate a statistically significant difference (p < 0.05).
PI3K-C2α regulates mTOR function and translocation
Because both amino acids and insulin are known activators of mTORC1, we tested the role of PI3K-C2α in mTORC1 function. Knockdown of PI3K-C2α reduced mTORC1 activity, as measured by the phosphorylation of its substrate S6K (Figure 2A). There was no effect on phosphorylation of Akt, confirming that the class II PI3K does not influence the Akt–TSC–mTOR axis.
FIGURE 2:
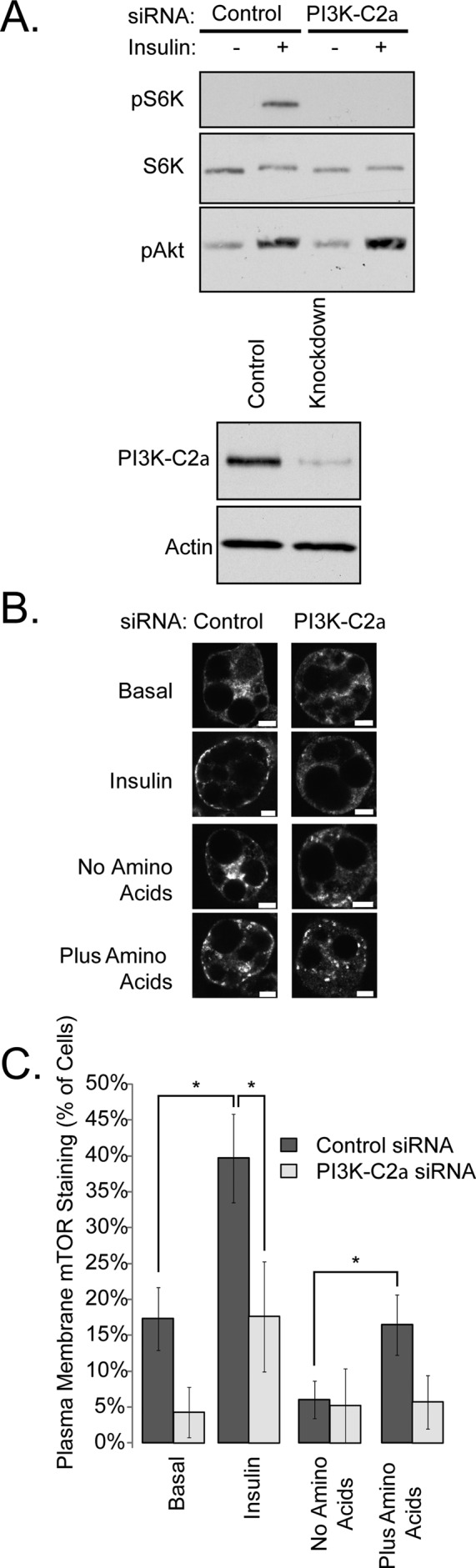
PI3K-C2α is required for mTORC1 activation and translocation. (A) siRNA directed against PI3K-C2α was electroporated into 3T3-L1 adipocytes for 96 h. Cells were serum-deprived for 3 h, stimulated with 100 nM insulin for 10 min, and blotted as indicated. (B) 3T3-L1 adipocytes were electroporated with control or PI3K-C2α siRNA for 96 h. Cells were stimulated as described, fixed, and then stained with anti-mTOR antibodies. Scale bars: 10 μm. (C) Cells were quantified by counting the percentage of cells in which plasma membrane staining of mTOR was visible. These data represent three independent experiments in which >150 cells were counted. Asterisks indicate a statistically significant difference (p < 0.05).
We examined the cellular localization of mTORC1 in 3T3-L1 adipocytes. Immunostaining of 3T3-L1 adipocytes showed endogenous mTOR in punctate structures in the interior of the cell under basal conditions, whereas insulin shifted mTOR to the plasma membrane (Figure 2B, quantified in Figure 2C), where it formed discontinuous punctate staining similar to that observed with GFP-2xFYVE (Figure 1B). To confirm the specificity of this effect on the GFP-2xFYVE reporter, we pretreated cells with wortmannin for 1 h at the doses indicated, and then stimulated the cells with either insulin or amino acids. As shown in Supplemental Figure S1, A and B, pretreatment of 3T3-L1 adipocytes with either 10 or 100 nM wortmannin blocked GFP-2xFYVE redistribution to the plasma membrane. To test the role of PI3K-C2α, we knocked down this kinase with siRNA, and found that the translocation of mTOR in response to either amino acids or insulin was reduced in these cells, consistent with the effect of this knockdown on the activation of mTORC1 (knockdown efficiency in these experiments is shown in Figure S1C). We note that while less mTOR is detected on the plasma membrane after knockdown relative to the control siRNA, a small insulin effect remains. This may indicate another pathway for mTOR translocation, or a reflection of incomplete inhibition of the kinase by siRNA knockdown. It has been noted that lower doses of wortmannin do not inhibit PI3K-C2α in vitro. Because we observed inhibition of both amino acid–dependent and insulin-dependent plasma membrane PI(3)P formation, even under low (10-nM) doses of wortmannin, it is possible that either 1) the in vivo efficiency of wortmannin is lower than its in vitro efficiency or 2) there is a second PI3K activity that is inhibited by low doses of wortmannin, in addition to the effects which we have attributed to PI3K-C2α.
To test further the translocation of mTOR by PI(3)P-dependent mechanisms, we pretreated 3T3-L1 adipocytes with 100 nM wortmannin and followed this with stimulation with insulin. As described in Figure S1, F and G, wortmannin treatment reduced peripheral mTOR staining produced by insulin. These data support the hypothesis that a PI3K-dependent mechanism regulates mTOR redistribution to the plasma membrane in these cells.
PIKFYVE is required for mTORC1 function
PI(3)P might directly modulate the activity of a protein required for mTORC1 activation, serve as a substrate for the production of another phospholipid involved in this process, or both. We considered a role for the PI5K PIKFYVE, which catalyzes the conversion of PI(3)P to PI(3,5)P2 (Cooke et al., 1998; Gary et al., 1998; Sbrissa et al., 1999; Sbrissa and Shisheva, 2005; Dove et al., 2004; Ikonomov et al., 2011). PIKFYVE resides in a multimeric complex containing the scaffold protein VAC14 and the lipid phosphatase FIG4. Knockdown of any member of the heterotrimeric PIKFYVE complex prevented insulin-stimulated S6K phosphorylation, but did not attenuate insulin-stimulated Akt phosphorylation (Figure 3, A and B, quantified in Figure S1H). PIKFYVE knockdown produced a 36% reduction in PI(3,5)P2 and a 13% increase in PI(3)P (Figure 3C). Similarly, VAC14 knockdown resulted in a 28% decrease in PI(3,5)P2 levels and a 5% increase in PI(3)P levels. Decreases in steady-state PI(3,5)P2 levels are often associated with an increase in PI(3)P, potentially due to decreased flux from the substrate to the product (Duex et al., 2006; Chow et al., 2007; Zhang et al., 2007; Jin et al., 2008).
Knockdown of PI3K-C2α reduced the phosphorylation of S6K in response to insulin, whereas knockdown of the class III PI3K Vps34 (also known as PI3KC3) was without effect. Interestingly, although knockdown of Vps34 had a stronger effect on total PI(3)P and PI(3,5)P2 abundance compared with PI3K-C2α or FIG4 (Figure 3C), loss of the former protein produced less inhibition of mTORC1 activation (Figure 3A), suggesting that distinct pools of PI(3)P and PI(3,5)P2 in different cellular locations might differentially regulate mTORC1 function. The inhibitory effects of PIKFYVE or PI3K-C2α knockdown on mTORC1 activation were independent of changes in Akt phosphorylation, indicating that the PIKFYVE complex is required for mTORC1, but not mTORC2, activity. We have not detected robust effects of PI3K-C2α knockdown on amino acid–dependent mTORC1 activation.
FIGURE 3:

PIKFYVE is required for mTORC1 activation and translocation. (A) 3T3-L1 adipocytes were electroporated with the indicated siRNAs for 96 h. Cells were serum-starved and stimulated with insulin for 15 min. Lysates were generated and probed with the indicated antibodies. (B) Control (unstimulated) lysates from (A) were blotted with the indicated antibodies. (C) In an experiment parallel to (A) and (B), 3T3-L1 adipocytes were labeled with [3H]inositol and lipids were extracted and analyzed with no serum starvation. Asterisks indicate significant differences from control knockdowns (p < 0.05). (D) HEK-293A cells were transfected with siRNA against PIKFYVE for 96 h, after which lysates were made and blotted as described in the figure. (E) 3T3-L1 adipocytes were electroporated with control of PIKFYVE siRNA for 96 h, and treated as described in Figure 2D. Scale bars: 5 μm. (F) Cells were quantified by counting the percentage of cells in which plasma membrane staining of mTOR was visible. These data represent three independent experiments in which >150 cells were counted. Asterisks indicate a statistically significant difference (p < 0.05).
Similar to what was observed with insulin, knockdown of PIKFYVE also decreased the activation of the mTORC1 complex in response to amino acids (Figure 3D, quantified in Figure S1I). Knockdown of PIKFYVE reduced both the basal and stimulated plasma membrane localization of endogenous mTOR in response to insulin and amino acids (Figure 3, E and F). These data suggest that both PI(3)P (through PI3K-C2α) and PI(3,5)P2 (through PIKFYVE) are required for the localization of mTOR to the plasma membrane in 3T3-L1 adipocytes and that PI(3)P may act through the generation of PI(3,5)P2.
To test directly the role of PI(3,5)P2 in mTORC1 activation, we transfected PI(3)P and PI(3,5)P2 directly into 3T3-L1 adipocytes. Addition of PI(3,5)P2, but not PI(3)P, modestly increased phosphorylation of S6K, although to a lesser extent than did insulin (Figure 4A, quantified in Figure S1J). PI(3,5)P2 transfection also caused a modest redistribution of mTOR to the periphery of 3T3-L1 adipocytes, again with a punctate, discontinuous distribution (Figure 4B, quantified in Figure 4C). Addition of PI(3)P also caused the translocation of the GFP-2xFYVE reporter construct to the plasma membrane, indicating not only transfection efficiency, but that the majority of the added PI(3)P concentrates at the plasma membrane of 3T3-L1 adipocytes (Figure S1, D and E). We were unable to rescue the effects of PIKFYVE or PI3K-C2α knockdown with lipid transfection experiments. Although this may be a technical limitation of these lipid transfection studies, it is also possible that these proteins function in a phospholipid-independent manner to regulate mTORC1 function.
FIGURE 4:

PI(3,5)P2 transfection can activate mTORC1 and cause its translocation to the plasma membrane of 3T3-L1 adipocytes. (A) 3T3-L1 adipocytes were serum-starved and treated with 1 μM carrier alone, insulin, or 1 μM PI(3)P:carrier or PI(3,5)P2:carrier complexes for 30 min. Lysates were generated and blotted as described in the figure. (B) 3T3-L1 adipocytes were transfected with 1 μM carrier alone or carrier:PI(3,5)P2 for 30 min. Three representative cells are shown. Scale bars: 5 μm. Cells were quantified by counting the percentage of cells in which plasma membrane staining of mTOR was visible. (C) Quantification of the experiment described in (B). Carrier 2 was used for all experiments described in Figure 4. These data represent three independent experiments in which >100 cells were counted. Asterisks indicate a statistically significant difference (p < 0.05).
Whether the activity of the PIKFYVE complex is regulated by insulin remains uncertain. The PI5K complex may be constitutively active, such that PI(3,5)P2 production arises from increased flux via PI(3)P. Alternatively, insulin or amino acids might activate this complex, either through receptor-based signaling pathways, or through PI(3)P itself, which can bind directly to PIKFYVE through its FYVE domain.
Raptor directly interacts with PI(3,5)P2
It is tempting to speculate that PI(3,5)P2 may serve as a coregulator of mTORC1 along with the Rheb and Rag GTPases, similar to the interaction of EEA1 with the GTPase Rab5 and PI(3)P (Di Paolo and de Camilli, 2006). We therefore tested whether PI(3,5)P2 directly interacts with mTORC1 complex components. The best-established PI(3,5)P2 binding protein in yeast is the WD40 domain-containing protein Atg18 (Dove et al., 2004; Narayan and Lemmon, 2006). We thus hypothesized that the WD40 domain of Raptor might directly bind to PI(3,5)P2.
We generated and purified glutathione S-transferase (GST) fusion constructs of the Raptor WD40 domain (Figure S2A) to evaluate binding. Dot blot experiments indicated an interaction of GST-WD40 with PI(3,5)P2 and a weaker interaction with PI(3)P and PI(5)P (Figure 5A). Because dot blots can overrepresent interactions with monophosphorylated phosphatidylinositols (Narayan and Lemmon, 2006), we also used liposome precipitation (Figure 5B). While the fusion protein weakly interacted with PI(3)P, there was a strong interaction with PI(3,5)P2 (Figure 5, B and C). Full-length Raptor also interacted with PI(3,5)P2 liposomes compared with control liposomes (Figure 5D). As a positive control for this assay, GST-Atg18 was precipitated with PI(3,5)P2-containing liposomes (Figure S2B).
FIGURE 5:

The WD40 domain of Raptor interacts directly with PI(3,5)P2. (A) Purified GST-Raptor WD40 domain was incubated with nitrocellulose containing the indicated lipids. GST-Raptor WD40 was detected using anti-GST antibodies. (B) GST-Raptor WD40 was incubated with decreasing concentrations of brominated PC or brominated PC with PI(3,5)P2 or PI(3)P, and precipitated by centrifugation. Pellets were resuspended and blotted using anti-GST antibodies. Load represents the amount of protein present if binding were 100%. (C) Liposomes containing 1 μM of the indicated phospholipid were incubated with GST-Raptor WD40 and precipitated by centrifugation. Pellets were resuspended and blotted using anti-GST antibodies. (D) Full-length HA-Raptor was generated as described in Materials and Methods. Eluted protein was incubated with brominated PC or brominated PC with PI(3,5)P2, and precipitated by centrifugation. Pellets were resuspended and blotted using anti-myc antibodies. (E) GST-Raptor WD40 (0.96, 1.45, and 2.6 μM) was passed over surfaces containing PC or PC + 3% PI(3,5)P2. Background signals were subtracted; sensorgrams of increasing concentrations of protein are shown. (F) 293A cells were transfected with a GFP-WD40 fusion and immunostained for mTOR or Lamp2. Scale bars: 10 μm. (G) 3T3-L1 adipocytes were transfected with GFP-WD40 and treated with insulin or amino acids as described in Figure 1B. There were no obvious detectable differences between serum-starved and amino acid–deprived cells with respect to GFP-WD40 staining. Three representative cells are shown. Scale bars: 5 μm. (H) Quantification of the data presented in (G) and also cells that were pretreated with 100 nM Wortmannin for 1 h. Asterisks indicate statistical significance (p < 0.05).
For the third lipid-binding assay, we performed surface plasmon resonance measurements to quantitate the interaction (Figures 5E and S2D), demonstrating that the Raptor WD40 domain interacts with PI(3,5)P2 with an approximate Kd of 600 ± 400 nM, although in some preparations no binding was detected. Positive control interactions were also monitored using GST-Hrs 2xFYVE for PI(3)P, GST-PLCδ1 for PI(4,5)P2, and GST-Atg18 for PI(3,5)P2 interactions (Figure S2C, protein purification shown in Figure S2A). Taken together, these data implicate the WD40 domain of Raptor as a novel PI(3,5)P2 binding module, providing a mechanism whereby PI(3,5)P2 may regulate mTORC1.
To test the cellular localization of the PI(3,5)P2, we generated a GFP-WD40 domain fusion construct. In HEK-293A cells, the GFP-WD40 fusion protein colocalized in large intracellular structures with both Lamp2 and mTOR (Figure 5F). HEK-293A cells were used for this experiment because the Lamp2 staining in these cells is more discrete than in 3T3-L1 adipocytes. These structures were not present in untransfected cells. We also tested the localization of this construct in 3T3-L1 adipocytes stimulated with insulin or amino acids. While normally present in large intracellular structures, insulin or amino acid stimulation caused a shift in GFP-WD40 to regions at or near the plasma membrane (Figure 5G, quantified in Figure 5H). Pretreatment of 3T3-L1 adipocytes for 1 h with 100 nM wortmannin abolished the insulin- or amino acid–stimulated translocation of the GFP-WD40 construct to the plasma membrane (Figure 5H). These data support the hypothesis that this reporter reflects PI3K-dependent changes at the plasma membrane of these cells. While further experiments using cells completely depleted of PI(3,5)P2 will be required to establish the fidelity of this fusion construct as a bona fide PI(3,5)P2 sensor in cells, these data support the idea that the lipid is produced in compartments coincident with mTOR in multiple cell types.
Previous studies indicate that the PI5K PIKFYVE regulates several vacuolar and late endosomal processes (Bonangelino et al., 1997, 2002; Gary et al., 1998; Rudge et al., 2004; Nicot et al., 2006), suggesting that PI(3,5)P2 may localize to vacuolar or late endosomal surfaces. The use of the Raptor WD40 domain as a potential PI(3,5)P2 sensor confirms the predicted localization of the lipid and is consistent with the reported localization of mTORC1 to late endosomal structures in many mammalian cells (Sancak et al., 2008, 2010; Flinn et al., 2010; Korolchuk et al., 2011) and their functional equivalent (vacuoles) in yeast (Cardenas and Heitman, 1995; Zurita-Martinez et al., 2007; Berchtold and Walther, 2009; Binda et al., 2009). Interestingly, unlike what we observed in other cell types, stimulation of 3T3-L1 adipocytes with insulin or amino acids produced plasma membrane localization of mTORC1, coincident with the generation of PI(3)P and PI(3,5)P2 in this region. Taken together, these data suggest a model (Figure S3) in which the PI3K-C2α–dependent and PIKFYVE-dependent sequential generation of PI(3)P and PI(3,5)P2 provides a localization signal for the activation of mTORC1 by both amino acids and insulin. Indeed, while these two stimuli use different activating pathways (Rheb for insulin and Rags for amino acids), both increase PI(3,5)P2 in order to localize Raptor to a discrete cellular location; the pathway may vary depending on cell type. Thus, while the role of PI(3,5)P2 in the regulation of the catalytic activity of mTOR remains uncertain, the phosphoinositide plays a key role in defining subcellular localization of the complex that ultimately may ensure the fidelity and specificity of its activation, perhaps explaining the substrate specificity and different roles of the kinase in different cells.
MATERIALS AND METHODS
All reagents were obtained from Sigma-Aldrich (St. Louis, MO), Invitrogen (Carlsbad, CA), Fisher, or EMD Biomedicals, unless otherwise noted. Antibodies against PI3K-C2α were from BD Biosciences (Franklin Lakes, NJ). Antibodies against phospho-S6K1 (Thr-389), S6K, mTOR (for Western blotting and immunofluorescence), phospho-Akt (Ser-473), phospho-S6 (Ser-235/236), S6, RagC, and Akt were from Cell Signaling (Danvers, MA). Antibodies against Rheb and mTOR (for immunoprecipitation) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against caveolin 1/2 and PI3K-C2α were from BD Biosciences. Antibodies against PIKFYVE and FIG4 were from Pietro De Camilli (Yale University). Antibodies against VAC14 were described in Zhang et al. (2007). GFP-2xFYVE was described in Lodhi et al. (2008).
Cell culture and transfection
3T3-L1 adipocytes were differentiated as described in Chiang et al. (2006). HEK-293A cells were transfected using Lipofectamine 2000 (Invitrogen; for siRNA experiments) according to the manufacturer's instructions. 3T3-L1 adipocytes were transfected via electroporation as described in Chiang et al. (2006). For siRNA knockdown experiments, two oligonucleotides were combined (see Supplemental Table S1 for oligonucleotide sequences). For all knockdown experiments, the control oligonucleotide was the Stealth siRNA Negative Control mid-GC (Invitrogen). For amino acid deprivation, media was replaced with PBS plus magnesium, calcium, and 4.5 g/l glucose. For amino acid stimulation, the media was replaced with DMEM lacking serum but containing penicillin, streptomycin, and glutamine. Lysates were generated in RIPA buffer, with the exception of Figure S2D, in which lysates were generated in a buffer containing 40 mM HEPES, 120 mM NaCl, 2 mM EDTA, 0.3% CHAPS, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, 50 mM sodium fluoride, and protease inhibitors. Lipid transfection experiments were performed using the Intracellular Lipid Delivery System from Echelon Biosciences (Salt Lake City, UT), according to the manufacturer's protocol. The lipids transfected were di-C16 side chain lipids. Carrier 3 was used for transfection of PI(3)P, and carrier 2 was used for the transfection of PI(3,5)P2, as per the manufacturer's instructions.
Immunofluorescence microscopy
Mammalian cells were fixed in 4% Formalin and visualized directly or stained with antibodies. After incubation with primary antibodies, coverslips were incubated with Alexa Fluor 488 or Alexa Fluor 594 goat anti–mouse or anti–rabbit immunoglobulin G (Invitrogen) at 2 μg/ml. Coverslips were mounted with Vectashield mounting media (Vector Laboratories, Burlingame, CA). Images were captured at room temperature by using an Olympus FV300 laser-scanning confocal microscope and acquired using Fluoview software. Cells were denoted as plasma membrane–positive by a nearly complete ring of staining around the periphery after examination of several confocal planes.
Cloning and recombinant protein production
GST-Raptor WD40 and GFP-Raptor WD40 were generated by amplifying amino acids 1013–1335 of human Raptor (NM_020761.2) from a flag-Raptor template, adding BamHI and XhoI restriction sites, and subcloning into the same sites of pGEX-4T1 (GE Healthcare, Waukesha, WI) or the BglI and SalI sites of pEGFP-C1. GST-Raptor WD40 and other positive controls were transformed and induced in Rosetta-gami2 DE3 (pLysS) bacterial cells (Novagen) and purified on glutathione-Sepharose (GE Healthcare) according to the manufacturer's instructions.
Analysis of phosphoinositides
Phosphatidylinositol phosphates were determined as described in Lodhi et al. (2008) with two minor changes. Cells were labeled in inositol-free media with [3H]inositol for 48 h, and phospholipids were extracted and deacylated with a solution of 26% methylamine, 45% methanol, and 11% butanol in water for 45 min. All phosphatidylinositol measurements are presented as a percentage of total phosphatidylinositols (phosphatidylinositol + phosphatidylinositol phosphates). The total phosphatidylinositol levels within experiments ranged from 94 to 96% of total PI, depending on the cell type and treatment. Neither this percentage nor the total amount of labeled phosphatidylinositol changed.
Lipid-binding assays
Liposome binding assays and surface plasmon resonance analysis of protein interactions were performed and analyzed as described in Narayan and Lemmon (2006). Dot blots were obtained from Echelon Biosciences. Interaction surfaces were verified by interactions of GST-Hrs, GST-PLCδ1, and GST-Atg18 with PI(3)P, PI(4,5)P2, and PI(3,5)P2, respectively. Between 5,000 and 10,000 RU of lipid was loaded onto each of four chip surfaces. Binding constants were determined by fitting equilibrium binding values to a single-site, ligand-binding curve. Dissociation constants from four experiments were combined to determine the average dissociation constant.
Statistical analysis
All error bars are depicted as SE of the mean. Statistical significance was determined via Student's t test calculated using the R statistical package. Significance (p value of < 0.05) is denoted on figures with an asterisk. All data presented are representative of three or more independent experiments.
Supplementary Material
Acknowledgments
We thank Jason Gestwicki and Srikanth Patury for assistance with Surface Plasmon Resonance. We also thank Irfan Lodhi, Greg Moorhead, Diane Fingar, Alan Cheng, and the members of the Saltiel, Inoki, and Weisman laboratories for helpful discussions. We also thank Allison Darland, Emily Kaufman, and Jordan Pecherer for technical assistance. This work was supported by an American Diabetes Association Mentor-Based Postdoctoral Fellowship (to D.B.) and National Institutes of Health grants 5R01DK061618 and DK060597 (to A.R.S.), 1R01DK083491 (to K.I.), and 1R01GM5040316 (to L.S.W.).
Abbreviations used:
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- mTORC1
mechanistic target of rapamycin 1
- PI(3)P
phosphatidylinositol 3-phosphate
- PI(3,5)P2
phosphatidylinositol 3,5-bisphosphate
- PI3K
phosphatidylinositol 3-kinase
- PI5K
phosphatidylinositol 5-kinase
- PIKFYVE
phosphatidylinositol-3-phosphate-5-kinase
- siRNA
small interfering RNA
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-1034) on June 13, 2012.
D.B. designed and performed all the experiments in Figures 1–4 and Supplemental Figure S1. D.B. and J.-T.M. designed and performed the experiments in Figures 5 and S2. D.B., J.-T.M., S.P., K.I., L.S.W., and A.R.S. participated in the design and analysis of the study.
REFERENCES
- Berchtold D, Walther TC. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol Biol Cell. 2009;20:1565–1575. doi: 10.1091/mbc.E08-10-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda M, Péli-Gulli M-P, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell. 2009;35:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Bonangelino C, Catlett NL, Weisman LS. Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol Cell Biol. 1997;17:6847–6858. doi: 10.1128/mcb.17.12.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonangelino C, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol. 2002;156:1015–1028. doi: 10.1083/jcb.200201002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas ME, Heitman J. FKBP12-rapamycin target TOR2 is a vacuolar protein with an associated phosphatidylinositol-4 kinase activity. EMBO J. 1995;14:5892–5907. doi: 10.1002/j.1460-2075.1995.tb00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S-HH, Chang L, Saltiel AR. TC10 and insulin-stimulated glucose transport. Methods Enzymol. 2006;406:701–714. doi: 10.1016/S0076-6879(06)06055-1. [DOI] [PubMed] [Google Scholar]
- Chow C, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke FT, Dove SK, McEwen RK, Painter G, Holmes AB, Hall MN, Michell R, Parker PJ. The stress-activated phosphatidylinositol 3-phosphate 5-kinase Fab1p is essential for vacuole function in S. cerevisiae. Curr Biol. 1998;8:1219–1222. doi: 10.1016/s0960-9822(07)00513-1. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, de Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dove SK, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–1933. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falasca M, Hughes WE, Dominguez V, Sala G, Fostira F, Fang MQ, Cazzolli R, Shepherd PR, James DE, Maffucci T. The role of phosphoinositide 3-kinase C2α in insulin signaling. J Biol Chem. 2007;282:28226–28236. doi: 10.1074/jbc.M704357200. [DOI] [PubMed] [Google Scholar]
- Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM. The late endosome is essential for mTORC1 signaling. Mol Biol Cell. 2010;21:833–841. doi: 10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary JD, Wurmser AE, Bonangelino C, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998;143:65–79. doi: 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3, 5-bisphosphate: no longer the poor PIP2. Traffic. 2011;13:1–8. doi: 10.1111/j.1600-0854.2011.01246.x. [DOI] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Delvecchio K, Xie Y, Jin J-P, Rappolee D, Shisheva A. The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve−/− embryos but normality of PIKfyve+/− mice. J Biol Chem. 2011;286:13404–13413. doi: 10.1074/jbc.M111.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Guan K-L. Tuberous sclerosis complex, implication from a rare genetic disease to common cancer treatment. Hum Mol Genet. 2009;18:R94–R100. doi: 10.1093/hmg/ddp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AM, et al. Phosphatidylinositol 3-phosphate [PtdIns3P] is generated at the plasma membrane by an inositol polyphosphate 5-phosphatase: endogenous PtdIns3P can promote GLUT4 translocation to the plasma membrane. Mol Cell Biol. 2006;26:6065–6081. doi: 10.1128/MCB.00203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:1–10. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi I, Bridges D, Chiang S-HH, Zhang Y, Cheng A, Geletka L, Weisman LS, Saltiel AR. Insulin stimulates phosphatidylinositol 3-phosphate production via the activation of Rab5. Mol Biol Cell. 2008;19:2718–2728. doi: 10.1091/mbc.E08-01-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 2003;22:4178–4189. doi: 10.1093/emboj/cdg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K, Lemmon MA. Determining selectivity of phosphoinositide-binding domains. Methods. 2006;39:122–133. doi: 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot AS, Fares H, Payrastre B, Chisholm AD, Labouesse M, Laporte J. The phosphoinositide kinase PIKfyve/Fab1p regulates terminal lysosome maturation in Caenorhabditis elegans. Mol Biol Cell. 2006;17:3062–3074. doi: 10.1091/mbc.E05-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudge S, Anderson DH, Emr SD. Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15:24–36. doi: 10.1091/mbc.E03-05-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;14:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbrissa D, Ikonomov OC, Shisheva A. PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. Effect of insulin. J Biol Chem. 1999;274:21589–21597. doi: 10.1074/jbc.274.31.21589. [DOI] [PubMed] [Google Scholar]
- Sbrissa D, Shisheva A. Acquisition of unprecedented phosphatidylinositol 3,5-bisphosphate rise in hyperosmotically stressed 3T3-L1 adipocytes, mediated by ArPIKfyve-PIKfyve pathway. J Biol Chem. 2005;280:7883–7889. doi: 10.1074/jbc.M412729200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci USA. 2007;104:17518–17523. doi: 10.1073/pnas.0702275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita-Martinez SA, Puria R, Pan X, Boeke JD, Cardenas ME. Efficient Tor signaling requires a functional class C Vps protein complex in Saccharomyces cerevisiae. Genetics. 2007;176:2139–2150. doi: 10.1534/genetics.107.072835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.