

The sulfur assimilation pathway is used to understand how combinatorial transcription coordinates cellular processes. Global gene expression was measured in yeast lacking different combinations of transcription factors in order to determine how these factors coordinate sulfur assimilation with diverse metabolic and physiological processes.
Abstract
Methionine abundance affects diverse cellular functions, including cell division, redox homeostasis, survival under starvation, and oxidative stress response. Regulation of the methionine biosynthetic pathway involves three DNA-binding proteins—Met31p, Met32p, and Cbf1p. We hypothesized that there exists a “division of labor” among these proteins that facilitates coordination of methionine biosynthesis with diverse biological processes. To explore combinatorial control in this regulatory circuit, we deleted CBF1, MET31, and MET32 individually and in combination in a strain lacking methionine synthase. We followed genome-wide gene expression as these strains were starved for methionine. Using a combination of bioinformatic methods, we found that these regulators control genes involved in biological processes downstream of sulfur assimilation; many of these processes had not previously been documented as methionine dependent. We also found that the different factors have overlapping but distinct functions. In particular, Met31p and Met32p are important in regulating methionine metabolism, whereas p functions as a “generalist” transcription factor that is not specific to methionine metabolism. In addition, Met31p and Met32p appear to regulate iron–sulfur cluster biogenesis through direct and indirect mechanisms and have distinguishable target specificities. Finally, CBF1 deletion sometimes has the opposite effect on gene expression from MET31 and MET32 deletion.
INTRODUCTION
Cells must adapt to a great variety of internal and external signals. One way they accomplish this is by using condition-specific, sequence-specific transcription factors (TFs) to make compensatory changes in gene expression. Instead of using a distinct TF for every possible combination of environmental perturbations, the cell uses a parsimonious strategy of transcriptional control in which TFs can be used in combinations that elicit distinct, stimulus-specific, transcriptional responses from their target genes.
The existence of genome-wide expression and TF-binding data in Saccharomyces cerevisiae and other organisms has enabled researchers to systematically catalogue many such combinations, including pairwise combinations (Pilpel et al., 2001), higher-order combinations such as the “multi-input motif,” and more complex TF circuits such as the “regulator chain,” “multicomponent loop,” “feedforward loop,” and “dense-overlapping regulon” (Lee et al., 2002; Milo et al., 2002; Shen-Orr et al., 2002). However, comparatively few examples of combinatorial control have been studied in detail experimentally. In yeast, well-studied examples of combinatorial control include the response to fatty acids by Oaf1p, Oaf3p, Pip2p, and Adr1p (Smith et al., 2007); combinatorial use of Hog1p and Msn2p/Msn4p (Capaldi et al., 2008); and, on a smaller scale, signal integration at the FLO11 promoter through the use of multiple TF-binding sites (TFBS; Rupp et al., 1999).
We developed an integrated experimental and computational framework to systematically investigate combinatorial transcriptional regulation in response to methionine abundance in S. cerevisiae. Many previous studies of combinatorial regulation focused on the use of combinatorial control to integrate multiple environmental signals. Methionine provides an example of special interest because a single signal (methionine abundance) controls a wide variety of intracellular responses in a way that has not been studied. Here we investigate the mechanisms of this control.
Methionine is synthesized by the sulfur assimilation pathway, also known as the methionine (Met) pathway, which occupies a central role in metabolism and growth control in yeast. The pathway synthesizes cysteine, methionine, and S-adenosyl methionine (SAM) from inorganic sulfate and leads to a host of other essential metabolites. Some of these contain sulfur atoms (e.g., glutathione and acetyl-CoA), but others are connected to the pathway because they contain methyl groups derived from SAM (e.g., phosphatidyl choline) or aminopropyl groups from the SAM derivative S-adenosyl methioninamine (e.g., polyamines). Essential macromolecular products of the sulfur assimilation pathway include proteins in general; membranes, which contain phospholipids whose biosynthesis depends on methyl groups derived from SAM; and many specific proteins, notably the diverse iron–sulfur proteins that carry out electron transfer reactions. Methionine and SAM are well situated to be control points for protein synthesis (every polypeptide chain is initiated with methionyl-tRNA), membrane biosynthesis (SAM is required at several steps of the pathways leading not only to phospholipids but also sterols), redox balance (via glutathione), and methylation of histones and DNA itself (although yeast DNA is not known to be methylated).
Much of what is known about the biological connections among methionine biosynthesis, central metabolism, and growth control has been learned through studies of mutants (mostly obtained as methionine auxotrophs) that either abrogate or deregulate the Met pathway. These studies have revealed a complex transcriptional regulatory network (Figure 1A; reviewed in Thomas and Surdin-Kerjan, 1997). At the metabolic level, this circuit is governed by SAM (Kuras and Thomas, 1995a), which acts through an ensemble of TFs. In response to high SAM levels, the SCFMet30 ubiquitin ligase ubiquitinylates the transcriptional activator Met4p, resulting in Met4p inactivation, but not degradation (Cai and Davis, 1990; Blaiseau et al., 1997; Rouillon et al., 2000; Kaiser et al., 2006). Met4p binds DNA with the assistance of three DNA-binding proteins (Met31p, Met32p, and Cbf1p) and one protein (Met28p) that facilitates the interaction between Met4p and the DNA-binding proteins. Met31p and Met32p, which are largely overlapping in function, bind to the DNA sequence motif AAACTGTGG, whereas Cbf1p binds to the motif CACGTG (Cai and Davis, 1990; Blaiseau et al., 1997; Blaiseau and Thomas, 1998). Cbf1p, variously known as CP1, CEP1, and CPF1, also binds centromeres through the same CACGTG sequence and is required for proper chromosome segregation (Bram and Kornberg, 1987; Baker et al., 1989; Baker and Masison, 1990; Thomas and Surdin-Kerjan, 1997). Met4p acts with different DNA-binding proteins to regulate different Met pathway biosynthetic genes (reviewed in Thomas and Surdin-Kerjan, 1997), and Cbf1p was shown early on to be “partly dispensable” for the expression of most of the Met pathway genes (Kuras and Thomas, 1995b). Evidence in the literature, although sparse, suggests that MET31 and MET32 might not be perfectly redundant (Su et al., 2008; Cormier et al., 2010).
FIGURE 1:

Overview of the background and the computational methods. (A) The transcriptional circuit governing sulfur assimilation and methionine biosynthesis. (B) Overview of the preliminary bioinformatic analysis using met6Δ expression data to identify TFBMs for Met31p/Met32p and Cbf1p and to distinguish the functional roles of these TFs using a Web-based tool called Gene Ontology Term Finder (GOTF). (C) Overview of the use of TF-dependency analysis to compare TF-deletion mutants in order to identify and characterize genes that depend specifically on Met31p/Met32p, Cbf1p, or both. (D) Overview of the use of multiple regression and Student's t-test to identify differences between MET31 and MET32.
The more widespread physiological influence of the Met pathway was first observed when Unger and Hartwell (1976) found that methionine starvation causes cell cycle arrest. According to an early microarray-based cell cycle analysis, the Met biosynthetic genes—but not the biosynthetic genes of any other amino acid—are expressed periodically throughout the cell cycle (Spellman et al., 1998). It was subsequently shown that constitutive activation of Met4p (such as by deletion of MET30) causes cell cycle arrest at the G1/S transition and that this arrest depends on the transcriptional activation domain of Met4p (Patton et al., 2000). Methionine starvation, unlike starvation for other amino acids, results in cell cycle arrest and survival; survival during methionine starvation is abolished by double deletion of MET31 and MET32 and is correlated with the ability of cells to mount an effective oxidative stress response (Petti et al., 2011). It is notable in this context that activity of the Met transcription factors is more tightly correlated with the yeast metabolic cycle than any other TF (Tu et al., 2005; Murray et al., 2007). Finally, a detailed study of the slow-growth phenotype of MET4 deletion mutants showed a direct regulatory connection between the sulfur assimilation pathway and the phosphatidylcholine biosynthetic pathway (Hickman et al., 2011), in which the SAM2 gene (encoding one of two SAM synthetases) is regulated by both MET4 and OPI1, which regulates lipid metabolism.
Two recent articles examined in detail the binding of Cbf1p and Met31p/Met32p to promoters in the Met biosynthetic pathway (Lee et al., 2010; Siggers et al., 2011). Nevertheless, neither study looked in depth beyond the sulfur assimilation pathway, and the biological rationale for the complexity of this transcriptional regulatory network remains unclear. We hypothesized that there is some division of labor among these TFs that facilitates the coordination of methionine metabolism with the great variety of cellular processes that depend on sulfur metabolism. We reasoned that by comparing phenotype and gene expression across a panel of isogenic TF-deletion mutants undergoing methionine starvation, we could understand this division of labor and learn how the multiplicity of factors coordinates the many biological functions that depend, directly or indirectly, upon the sulfur assimilation pathway.
We began by surveying the genome-wide effects of methionine starvation in methionine auxotrophs lacking MET6 (which encodes methionine synthase) or MET13 (which encodes methylenetetrahydrofolate reductase). In a preliminary bioinformatic analysis of this data, we found evidence that the Met TFs Met31p/Met32p and Cbf1p are not redundant, lending support to our “division of labor” hypothesis. We also used these data to systematically identify cellular processes that depend on methionine and to generate TF-binding matrices for Met31p/Met32p and Cbf1p, which we use in most subsequent analyses.
To expand upon the “division of labor” result, we designed a set of follow-up experiments and computational analyses to investigate distinctions between these TFs and the potential role of combinatorial regulation in coordinating methionine-dependent processes with methionine abundance. In these experiments, we deleted the genes encoding the three DNA-binding proteins in this system—CBF1, MET31, and MET32—individually and in pairwise combinations. To assure that all the mutants were starving equally for methionine, we made these deletions in the met6Δ background. We measured and compared the gene expression patterns and cell cycle progression of these strains during methionine starvation to those of their met6Δ parent. (We also deleted MET4, but not in the met6Δ background, because met4Δ is growth impaired even when supplemented with excess methionine [ Hickman et al., 2011]; we used results with this strain for qualitative comparisons only.)
We designed these experiments with particular computational methods in mind. Using these methods, we analyzed the expression data to identify differences in specificity among the TFs. The analytical pipeline for several key methods is summarized in Figure 1, B–D; another method, transcription factor activity analysis, is summarized in Lee and Bussemaker (2010). Our analysis focused on two main questions: first, whether the different TFs regulate different genes; second, whether there are functional differences between the genes regulated by each TF, which we would take as evidence that there exists a division of labor that enables different TFs to coordinate Met metabolism with different cellular processes. We focused on differences between the two major TF classes—Cbf1p and Met31p/Met32p—but also examined differences between Met31p and Met32p in two genetic backgrounds (with and without CBF1).
We found abundant evidence that the Met TFs regulate genes involved in biological processes that lie downstream of sulfur assimilation. Some of these were previously known to depend on sulfur or methionine, but many (e.g., copper transport, iron assimilation, iron–sulfur cluster biogenesis, maltose metabolism, and microautophagy) were not. Met4p is involved in virtually all of these; there is a striking similarity between the patterns of gene expression in the met4Δ strain and the double met31Δmet32Δ strains. Consistent with a regulatory circuit that serves to coordinate multiple cellular processes, we found that the different factors regulate overlapping but distinct groups of genes characterized by overlapping but distinct biological functions. We find that Met31p and Met32p have distinguishable target specificities, with significant gene expression differences among the mutant phenotypes under methionine starvation. It appears from our results that, depending on circumstances, all three of the DNA-binding proteins can act as repressors and activators through the same (or very similar) sequence motifs. In many cases, we found that a given target gene is regulated differently by different TFs. Finally, we note that the regulatory consequences of CBF1 deletion sometimes oppose those of MET31 and MET32 deletion, suggesting that Cbf1p and Met31p/Met32p can influence gene expression in opposing ways and that the influence of one is unaffected by deletion of the other.
The experiments in this study are designed to perturb the Met pathway by starving auxotrophic TF-deletion mutants. By comparing phenotype and gene expression across a variety of deletion mutants, one can detect functional distinctions among different TFs (and TF combinations). An alternative approach, based on separately inducing each TF-encoding gene and measuring the effects on gene expression, provides both support and additional information. Those results are presented in an accompanying article (McIsaac et al., 2012).
RESULTS
Methionine starvation influences many diverse cellular processes
To gain a preliminary overview of the cellular response to methionine starvation, we used gene expression data previously collected during starvation of strains that cannot make methionine because they lack a biosynthetic enzyme, as opposed to strains that have a regulatory defect. We had studied two such Met biosynthetic auxotrophs, met6Δ and met13Δ (Petti et al., 2011). Here we used this data in several ways. First, we used it to gain an overview of methionine-dependent genes and processes. Second, we used met6Δ as a control against which to compare the transcription factor–deletion mutants. Third, we used the met6Δ data to identify transcription factor–binding motifs for Met31p/Met32p and Cbf1p.
Before choosing met6Δ as the background and control strain for the regulatory studies described later, we compared the met6Δ and met13Δ data in order to verify that most gene expression is extremely similar in both strains. This allowed us to conclude that the effects we see are indeed the result of methionine depletion and are not specific to the loss of methionine synthase.
Multiple regression was used to identify 466 genes whose expression depends significantly on time, differs significantly between met6Δ and met13Δ, and changes by twofold or more (see Materials and Methods). Minor differences were found in the expression kinetics or amplitude of genes involved in sulfur metabolism (more highly induced in met6Δ), cell cycle and nucleotide metabolism (repressed earlier in met13Δ), and regulation of translation (repressed earlier in met6Δ).
We then characterized the general transcriptional response to Met depletion using the remaining 2669 genes that change by twofold or more, depend significantly on time, and behave similarly in met6Δ and met13Δ (Supplemental Figure S1). We clustered these genes using the k-means algorithm, identified clusters with distinctive expression profiles, and analyzed their Gene Ontology (GO) term enrichment using an online tool called the Generic Gene Ontology Term Finder (GOTF; Supplemental Data Set 1; see Materials and Methods). The most rapidly and highly induced cluster (122 genes, cluster 9 in Supplemental Figure S1) is enriched for processes related to metabolism of sulfur, methionine, and metabolically related amino acids; oxidative stress response; response to heavy metals; and targets of the Met transcription factors Met4p, Cbf1p, Met31p, and Met32p. However, we also observed rapid, highly coordinated induction of genes involved in nonglycolytic energy generation, including storage compound metabolism (cluster 16) and electron transport (cluster 11), implying increased reliance on mitochondrial energy generation during Met starvation. Consistent with the progressive cell cycle arrest observed during methionine starvation (Unger and Hartwell, 1976; Petti et al., 2011), all gene clusters enriched for biological processes related to cell division, the chromosome cycle, and DNA replication are gradually repressed during the starvation (clusters 3, 4, 10, and 19). A small set of markedly coregulated genes with a distinctive expression profile (cluster 4) is enriched for a variety of seemingly disparate functions, including DNA packaging (e.g., the key histone-encoding genes HTA1, HTA2, HTB1, HHF1, HHF2, HHT1 HHT2), purine metabolism, single-carbon metabolism, and amino acid biosynthesis. This is intriguing, given the putative role of methionine in regulating the cell cycle. Both HHF2 and HHT2 contain binding sites for Met4p, suggesting that the transcriptional regulation of these histone-encoding genes may depend directly on Met abundance (MacIsaac et al., 2006). All of the clusters enriched for ribosomal biogenesis genes are repressed, consistent with numerous previous findings that environmental stress represses ribosome biogenesis.
Using gene expression patterns in mutants to dissect the complex combinatorial regulation of the pathway
To understand how the Met pathway TFs might regulate Met-influenced processes that go beyond the sulfur assimilation pathways per se, we first undertook a simple bioinformatic analysis of transcriptional regulation, as outlined in Figure 1B. Here we used the gene expression data we collected for the met6Δ strain to derive key transcription factor–binding motifs (TFBMs), and characterized the biological function of motif-containing genes that change substantially during methionine starvation (Figure 2). We hierarchically clustered genes that change by twofold or more in the met6Δ time course and identified a set of 45 genes that respond early and dramatically to methionine starvation. Most (43 of 45) of these genes are induced within 10 min of methionine removal (Figure 2A), and most change by a factor of 20 or more; a similar result was found by Lee et al. (2010). Primarily located in cluster 9 discussed earlier, these genes function in methionine, cysteine, and S-adenosyl methionine biosynthesis, sulfur metabolism, single-carbon metabolism (CHA1), DNA repair (RAD59), and biosynthesis of the antioxidants glutathione (GTO1) and NADPH (BNA3). Using the motif identification algorithm MEME (see Materials and Methods), we identified two sequence motifs in the promoters of these 45 genes (Figure 2B). The motifs match previously derived motifs for Met31p and Met32p, which are believed to bind the same motif, and Cbf1p (Blaiseau et al., 1997; Lee et al., 2010; Siggers et al., 2011).
FIGURE 2:

Functional specificity of promoter motifs derived from methionine-responsive genes. (A) Expression levels of genes that are induced early during Met starvation of met6Δ. (B) DNA sequence motifs derived from the promoters of the genes in A. (C) GO term enrichment of genes containing the Cbf1p motif only, the Met31p/Met32p motif only, or both motifs.
These sequence motifs have been well studied in the context of methionine metabolism, but the wide variety of methionine-influenced processes suggests that they also influence non-Met pathway genes. We used MAST to search the genome for additional instances of each motif (MAST score <500, as in Lee et al., 2010; see Materials and Methods) and filtered the results for genes that change twofold or more in the met6Δ control data. We classified each MAST hit as a putative direct “target” of Met31p/Met32p, Cbf1p, or both, and calculated the functional enrichment of each class of target genes (Figure 2C). In what follows, we distinguish these probable “direct” targets from probable “indirect” targets, which exhibit expression changes but do not have motifs for Cbf1 or Met31/Met32.
“Joint targets” of both TFs are involved primarily in sulfur/methionine metabolism and closely related processes, such as the metabolism of related amino acids and oxidation–reduction processes. In contrast, targets of only Cbf1p or only Met31p/Met32p are involved in a wider variety of processes including, but not limited to, methionine and sulfur metabolism.
Met31p/Met32p and Cbf1p regulate overlapping but distinct gene sets enriched for different functions
To investigate experimentally the specialization between Met31p/Met32p and Cbf1p, we deleted MET31, MET32, and CBF1 individually and in all pairwise combinations in a met6Δ background. We measured gene expression during the early exponential phase of growth and during methionine depletion in these strains and selected genes that a) depend significantly on time and change by twofold or more in at least one strain, or b) are constitutively expressed ± twofold relative to met6Δ in at least one strain (see Materials and Methods). Based on the met4Δ expression data, this panel of single- and double-TF deletions captures the full range of expression profiles available to Met TF-deletion mutants: compared with the other TF-deletion mutants, we found expression profiles unique to met4Δ in only 14 of 6256 genes assayed (notable examples include HXT1, SNQ2, CAT8, ESF1, SSA2, SPE2, DDI2, and DDI3).
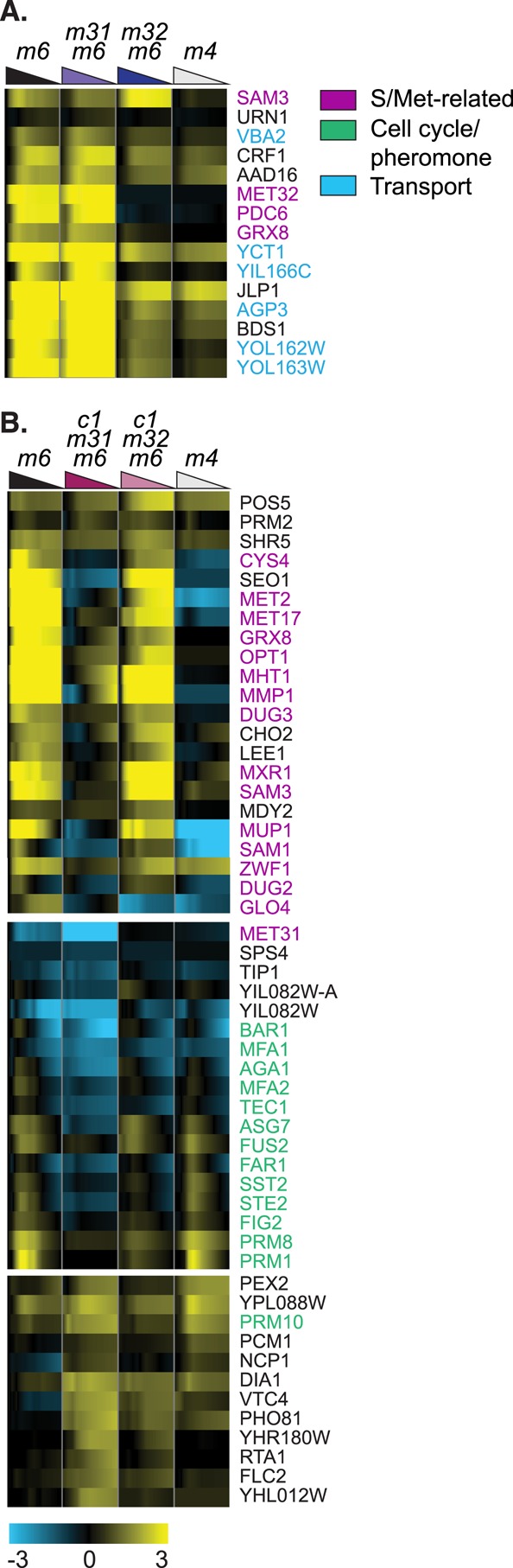
We first examined the specialization between Cbf1p and Met31p/Met32p without distinguishing between Met31p and Met32p. In an initial, coarse-grained analysis, we surveyed all GO categories with at least 10 members to identify those in which met31Δmet32Δmet6Δ and cbf1Δmet6Δ differ most. Specifically, for each GO category i, we computed a distance defined as 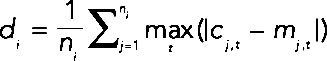 . Here ni is the number of genes in category i, cj,t is the expression level of gene j at time point t in cbf1∆met6∆, and mj,t is the expression level of gene j at time point t in met31∆met32∆met6∆. As shown in Figure 3, the differences between the strains are roughly normally distributed across the GO categories. Gene sets that are differently affected by deletion of MET31/MET32 and CBF1 are located in the tails of the distribution. The right tail of the distribution, where expression is higher in cbf1∆met6∆ than in met31∆met32∆met6∆, is dominated by cysteine and methionine biosynthesis, sulfur metabolism, and growth-related terms such as ribosome biogenesis and assembly, rRNA processing, and translation. In the left tail, we find terms that are mainly related to redox homeostasis, including iron transport and homeostasis, copper transport, metal ion transport, fatty acid metabolism, and glutathione metabolism. This means that Met31p/Met32p and Cbf1p differentially regulate genes related to methionine metabolism, as well as genes related to growth and cellular redox. We infer, as indicated in Figure 3, that the categories on the left are induced by Cbf1p and/or repressed by Met31p/Met32p, whereas the categories on the right are induced by Met31p/Met32p and/or repressed by Cbf1p. Independent evidence for this view can be found in McIsaac et al. (2012).
. Here ni is the number of genes in category i, cj,t is the expression level of gene j at time point t in cbf1∆met6∆, and mj,t is the expression level of gene j at time point t in met31∆met32∆met6∆. As shown in Figure 3, the differences between the strains are roughly normally distributed across the GO categories. Gene sets that are differently affected by deletion of MET31/MET32 and CBF1 are located in the tails of the distribution. The right tail of the distribution, where expression is higher in cbf1∆met6∆ than in met31∆met32∆met6∆, is dominated by cysteine and methionine biosynthesis, sulfur metabolism, and growth-related terms such as ribosome biogenesis and assembly, rRNA processing, and translation. In the left tail, we find terms that are mainly related to redox homeostasis, including iron transport and homeostasis, copper transport, metal ion transport, fatty acid metabolism, and glutathione metabolism. This means that Met31p/Met32p and Cbf1p differentially regulate genes related to methionine metabolism, as well as genes related to growth and cellular redox. We infer, as indicated in Figure 3, that the categories on the left are induced by Cbf1p and/or repressed by Met31p/Met32p, whereas the categories on the right are induced by Met31p/Met32p and/or repressed by Cbf1p. Independent evidence for this view can be found in McIsaac et al. (2012).
FIGURE 3:

Transcription factor specificity among biological processes. Histogram of GO biological processes, showing the average maximum expression difference between cbf1Δmet6Δ and met31Δmet32Δmet6Δ for each GO category with at least 10 members.
Transcription factor-dependency analysis differentiates between the Met31p/Met32p and Cbf1p regulons
Our analysis so far paints a broad picture of functional specialization among the Met pathway TFs. However, we wanted a more detailed understanding of the combinatorial regulation among these TFs and of how each TF affected the response to Met starvation relative to the control strain, met6∆. To answer these questions, we developed a nonparametric statistical method, which we refer to as “TF-dependency analysis,” to identify genes whose expression depends on Cbf1p, Met31p/Met32p, or both. As outlined in Figure 1C, we defined expression signatures corresponding to each scenario and found genes whose expression profiles were significantly correlated with these signatures (see Materials and Methods). At a false discovery rate (FDR) of 5%, we identified 89 genes that depend on both Cbf1p and Met31p/Met32p, 455 that depend primarily on Met31p/Met32p alone, and 88 that depend primarily on Cbf1p alone. Only 15% of the classified genes contain a motif for either TF, but motif prevalence accurately reflects expression dependence within each class (Table 1).
TABLE 1.
Motifs found in each TF-dependency class.
| Percent containing motif(s) | ||||
|---|---|---|---|---|
| Met31p/Met32p motif only | Cbf1p motif only | Both motifs | Any motif | |
| Genes dependent on Met31p/Met32p only | 7.3 | 3.5 | 1.8 | 13 |
| Genes dependent on Cbf1p only | 1.1 | 14 | 2.3 | 14 |
| Genes dependent on both | 6.7 | 9 | 14 | 29 |
Met31p/Met32p induces Met transporters and represses iron homeostasis genes
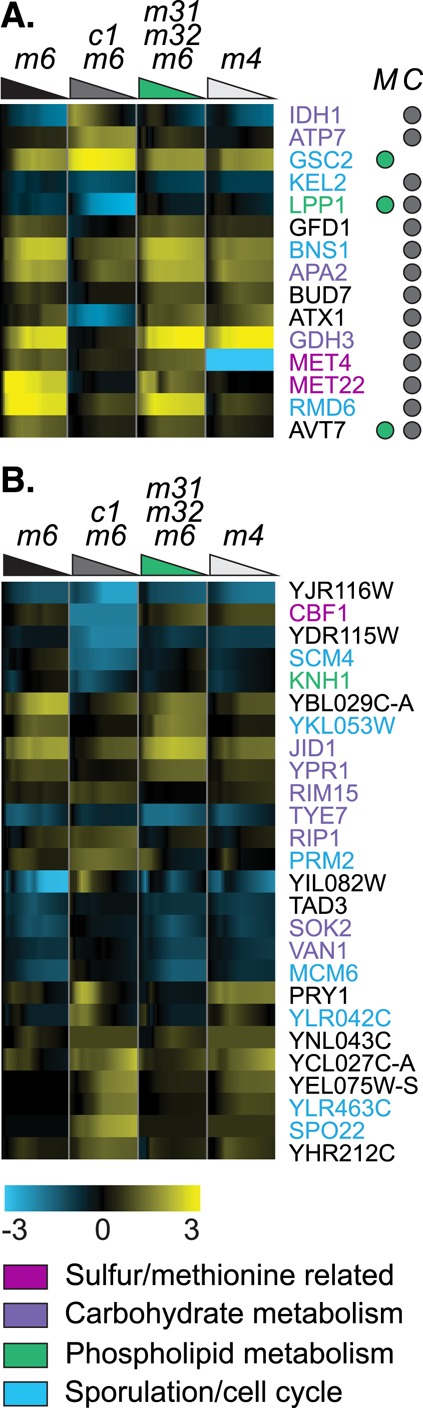
In the Supplement, we provide an overview of the 455 genes that depend more strongly on Met31p/Met32p than on Cbf1p, which are enriched for a variety of processes ranging from sulfur and methionine metabolism to carbohydrate metabolism and iron homeostasis (Supplemental Figure S2 and Supplemental Data Set 1). Here we focus on the 13% that contain a motif for Cbf1p and/or Met31p/Met32p (the “target” genes in Figure 4A), and the “nontarget” genes that lack a motif but show particularly strong, specific dependence on Met31p/Met32p (Figure 4B). There are two striking subsets of target genes. One requires Met31p/Met32p for induction and is enriched for a small number of processes—sulfur assimilation and methionine, aspartate, cysteine, and glutathione metabolism. Of note, only three of the genes involved in the sulfur and methionine pathway—MET3, MET13, and MET17—encode methionine biosynthetic enzymes. The others (e.g., MHT1, MMP1, MET2, MUP3, CYS3, MUP1, SAM1, SAM2, SAM3) are involved in ancillary functions such as methionine transport and the SAM cycle. This supports and refines our previous result: many Met pathway genes, particularly methionine and sulfur transporters, depend on Met31p/Met32p but not on Cbf1p. The second striking subset of genes appears, surprisingly, to be repressed specifically by Met31p/Met32p. These genes are strongly enriched for iron assimilation and iron transport (e.g., SIT1, ENB1, FIT3, ARN2, ISU2) and are dramatically induced in the met31Δmet32Δmet6∆ strain. We also found one Met31p/Met32p target, ISU2, which, by virtue of its role in the synthesis of iron–sulfur (Fe-S) proteins, suggests a compelling connection between sulfur assimilation and iron homeostasis.
FIGURE 4:

Genes whose expression depends on MET31/MET32. (A) Genes that depend statistically significantly on MET31/MET32 (but not CBF1) and contain a TFBM for Cbf1p (gray dot under column C) and/or Met31p/Met32p (green dot under column M). Strain order for both panels is met6Δ, cbf1Δmet6Δ, met31Δmet32Δmet6Δ, met4Δ. The colored triangles are color coded for each strain and represent the decrease in methionine over each time course. (B) Selected genes that depend statistically significantly on MET31/MET32 and lack a TFBM.
These results are further supported by the additional, nondirect target genes involved in the homeostasis of iron (e.g., FET3, TIS11, FTR1, FRE1, FRE2, FRE3, FIT2, AFT1, SCS3, VHT1) and other metals, such as copper (CUP1-1, CUP1-2, ARN1; Figure 4B). We noticed in previous work that deletion of MET31 and MET32 (Petti et al., 2011) or depletion of MET4 (Hickman et al., 2011) induces iron-related genes, but the TF binding site (TFBS) data used in those analyses (MacIsaac et al., 2006) indicated that these genes are not direct targets of any of the Met TFs. In those studies, therefore, we concluded there that these genes are indirectly induced by methionine depletion. In the target genes listed in the preceding paragraph, however, we have evidence that Met31p/Met32p directly regulate iron assimilation.
The influence of MET31 and MET32 is not limited to the overrepresented processes discussed earlier. Key regulatory genes for processes such as carbohydrate metabolism (HXT1, RIM1) and phosphate metabolism (PHO11, PHO12, PHO81, PHO84, PHO86, PHO89) are dramatically affected by MET31/MET32 deletion. The glucose transporter gene HXT1 appears to be strongly repressed in met31∆met32∆met6Δ and more weakly repressed in cbf1∆met6Δ.
Cbf1 regulates glutamate, meiotic, and carbohydrate genes
Fewer genes are affected by CBF1 deletion than by MET31/MET32 deletion, but they span at least as wide a variety of processes, among which methionine metabolism is weakly represented. An overview of Cbf1p-dependent genes (Supplemental Figure S3) shows weak enrichment for deoxyribonucleotide biosynthesis (RNR2, RNR4) and several genes associated with meiosis (RME1, BNS1, SPR6, RMD6, SPO22). In the more focused view of putatively direct Cbf1p targets (Figure 5), it is notable that Cbf1p-dependent genes—even those containing TFBMs— are very weakly enriched for Met metabolism (Figure 5A), although they include MET4, the master transcriptional activator of the Met pathway. Instead, Cbf1-dependent target genes are enriched for glutamate and glutamine metabolism and for nitrogen compound metabolism, and contain diverse metabolic genes such as isocitrate dehydrogenase (IDH1), NADP-dependent glutamate dehydrogenase (GDH3), the ATPase ATP7, and LPP1, which controls levels of the phospholipid-regulator phosphatidic acid.
FIGURE 5:
Genes whose expression depends on CBF1. (A) Genes that depend statistically significantly on CBF1 (but not MET31/MET32) and contain a TFBM for Cbf1p (gray dot under column C) and/or Met31p/Met32p (green dot under column M). Strain order for both panels is met6Δ, cbf1Δmet6Δ, met31Δmet32Δmet6Δ, met4Δ. (B) Selected genes that depend statistically significantly on CBF1 and lack a TFBM.
Consistent with the coarse-grained GO term analysis in Figure 3, we found a number of Cbf1p-dependent carbohydrate metabolism genes, including TYE7, RIM15, SOK2, RIP1, VAN1, IDH1, ATP7, and GDH3, most of which appear to be repressed by Cbf1p (Figure 5). Because there are comparatively few Cbf1p-dependent genes and many of them are poorly annotated, we obtained more information about these genes using bioPIXIE (Myers and Troyanskaya, 2007; http://avis.princeton.edu/pixie/index.php), a program that analyzes the functional enrichment of genes that interact physically or genetically with a query gene. bioPIXIE indicates that additional Cbf1p-dependent genes interact with carbohydrate metabolism genes, including YPR1 (pentose metabolism), JID1 (respiration), and APA2 (regulation of gluconeogenesis). Most of these lack a motif for either Cbf1p or Met31p/Met32p, raising the possibility that Cbf1p affects these genes indirectly or that they represent a general stress response. However, the cbf1∆met6∆ strain grows to a higher cell density during methionine starvation than the met31∆met32∆met6∆ strain, and these carbohydrate genes are not perturbed in the latter strain. This suggests that the effect of CBF1 deletion on these genes is specific, not a general stress or slow-growth response. The influence of Cbf1p on TYE7 could be particularly consequential because TYE7 is a transcriptional activator that binds the E-boxes of glycolytic genes.
The dearth of Cbf1p-dependent methionine biosynthetic genes is striking and supports previous evidence here and elsewhere (Lee et al., 2010) that Cbf1p is not the primary regulator of sulfur assimilation and Met metabolism. MET4 and MET22 are the only specifically Cbf1p-dependent genes with Met-related annotations. However, we found one gene that potentially connects Met abundance with the cell cycle: RMD6, which is required for meiosis, requires CBF1 for induction, contains the Cbf1p TFBM, and interacts physically and/or genetically with 20 genes involved in the metabolism, transport, and utilization of sulfur and Met (bioPIXIE; see Materials and Methods). Given that a number of other genes involved in sporulation or cell division are Cbf1p dependent (RME1, BNS1, SPR6, KEL2, SCM4, GSC2, SPO22, MCM6, YKL053W [bioPIXIE], YLR042C [bioPIXIE], and YLR463C [bioPIXIE]), these data suggest that Cbf1p may help couple methionine abundance with cell division through the regulation of these genes.
Met31p/Met32p and Cbf1p coregulate most methionine biosynthetic genes
Deleting either CBF1 or MET31/MET32 perturbs the expression of 89 genes. These “jointly regulated” genes are enriched for sulfur assimilation and the metabolism of Met and related amino acids, as well as for cell wall organization and lipid metabolism (notable examples include OYE2, IZH4, and YEH1; Supplemental Figure S4). Twenty-nine percent of the genes in this class contain motifs for Cbf1p and/or Met31p/Met32p, and 14% contain motifs for both (Table 1 and Figure 6). This group, which contains most of the enzyme-encoding genes in the Met pathway, is also enriched for siroheme and heme metabolism, oxidation–reduction, cofactor metabolism, and the metabolism of related amino acids (serine, cysteine, aspartate). Other jointly regulated target genes represent a variety of other processes: ADH3 (mitochondrial alcohol dehydrogenase), GIT1 (uptake of glycerol and choline), MNN4 (glycosylation of oligosaccharides), BNA3 (biosynthesis of NADH and cell cycle [bioPIXIE]), ATM1 (Fe-S cluster biosynthesis and respiration [bioPIXIE]), RAD59 (DNA repair, also found in other studies), VHS3 (G1/S cell cycle transition [bioPIXIE] and ion homeostasis), and JIP5 (ribosome biogenesis). These results suggest that Cbf1p and Met31p/Met32p must both be present for full induction of methionine biosynthesis genes and additional genes that may be particularly important in Met metabolism.
FIGURE 6:
Genes whose expression depends on MET31/MET32 and CBF1. (A) Genes that depend statistically significantly on both CBF1 and MET31/MET32 and contain a TFBM for Cbf1p (gray dot under column C) and/or Met31p/Met32p (green dot under column M). Strain order for both panels is met6Δ, cbf1Δmet6Δ, met31Δmet32Δmet6Δ, met4Δ. (B) Selected genes that depend statistically significantly on CBF1 and MET31/MET32 and lack a TFBM.
Synergy between Met31p/Met32p and Cbf1p
Thus far, we have shown that Cbf1p and Met31p/Met32p regulate overlapping but distinct gene sets. In particular, genes that are coregulated by Cbf1p and Met31p/Met32p deviate from wild-type expression roughly equally if either CBF1 or MET31/MET32 is deleted. We next looked for additive effects of CBF1 and MET31/MET32 by asking whether there are genes whose expression is altered more substantially in mutants where both TF classes (CBF1 and either MET31 or MET32) are deleted. To answer this, we searched for genes whose expression is perturbed relative to the control strain (met6Δ) in cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ, but not in cbf1Δmet6Δ and met31Δmet32Δmet6Δ. (As discussed earlier, few genes are uniquely expressed in met4Δ, suggesting that we did not miss many synergistic interactions by ignoring the quadruple mutant met31Δmet32Δcbf1Δmet6Δ.) We used linear regression to compare each TF mutant to met6Δ (see Materials and Methods) and chose genes whose expression in cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ is significantly different from that in met6Δ (F statistic q ≤ 0.05, fold-change difference ≥1.5). We also required that the significance of this difference be at least 10 times greater than that for the difference between met6Δ and either cbf1Δmet6Δ or met31Δmet32Δmet6Δ.
Based on these criteria, 114 genes exhibit subtle synergy between the two TF classes (Supplemental Data Set 1). The vast majority of these genes show diminished response to methionine starvation, although several (PHO5, HHF1, HHT2) show more extreme induction. Most are involved in ribosome biogenesis and translation, with a few notable exceptions such as the carbon metabolism genes TKL1, ALD6, PDC1, ADH1, GDH1, GPM1, and TDH1, the glutathione S-transferase GTT2, the transcription factor YAP5, and the phosphatase PHO5, which is the most differentially regulated gene in met31Δcbf1Δmet6Δ and met32Δcbf1Δmet6Δ. Of note, PHO5, HHT2, and HHF1 interact, according to bioPIXIE.
Very few of these genes have binding sites for either Cbf1p (GAR1, ASP3-1, GTT2, MBF1, UTP21, PIR1, RPP2B, BNS1, GUA1) or Met31p/Met32p (RPL4B, ATG29, RPS7A, NMD3, ECM33), and only one gene, YAP5, has motifs for both TFs. On one hand, the low incidence of TFBMs and the prevalence of ribosomal genes in this list suggest that most of these genes are responding to an indirect, additive effect of methionine starvation in the TF mutants, such as low growth rate due to depletion of sulfur-containing compounds and insufficient methylation. On the other hand, many ribosomal genes display minor expression defects in cbf1Δmet6Δ, suggesting that cbf1Δ may indeed regulate ribosomal genes. This is consistent with our initial, coarse analysis, which showed that numerous ribosome biogenesis GO categories are less repressed in cbf1∆met6∆ than in met31∆met32∆met6∆.
Distinguishing between MET31 and MET32
To pursue possible differences between the functions of MET31 and MET32 (Su et al., 2008; Cormier et al., 2010), we used multiple regression as outlined in Figure 1D to compare met31Δmet6Δ to met32Δmet6Δ (see Materials and Methods). Briefly, we required that the strain pairs differ significantly according to multiple regression (F-test, q ≤ 0.1) or average expression level (Student's t-test, q ≤ 0.1). To remove results that are statistically significant but still modest in magnitude, we also required that the two strains differ by at least 1.5-fold in at least one time point.
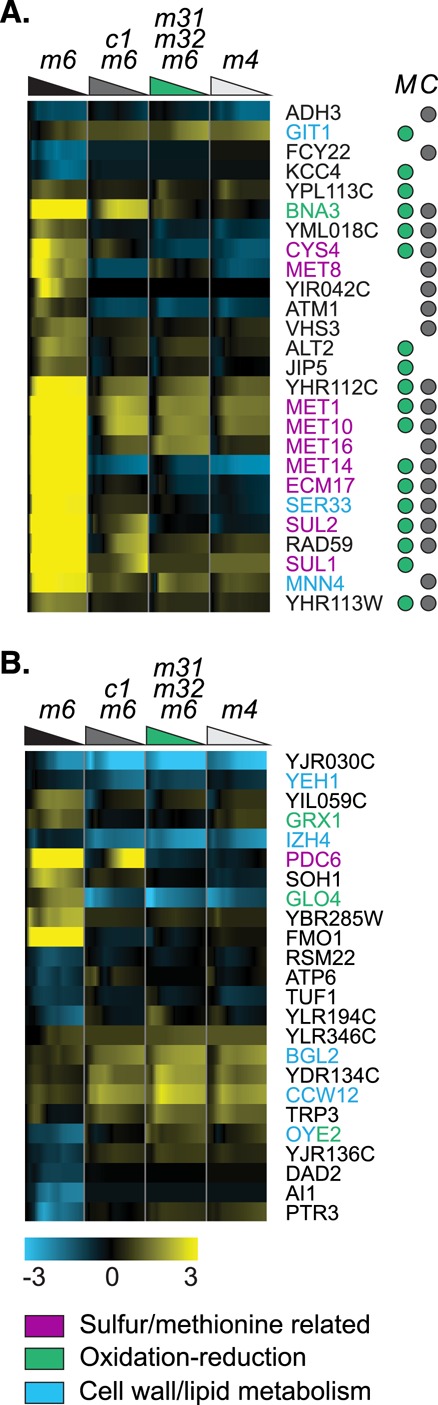
By these criteria, 35 genes differ in expression between met31Δmet6Δ and met32Δmet6Δ. The 15 most striking examples are shown in Figure 7A. (Three additional genes, MXR1, VHT1, and SAM1, also appear to depend differently on Met31p and Met32p but did not meet our strict selection criteria.) For many of these genes, deleting MET31 appears to have the opposite effect of deleting MET32: the genes are constitutively overexpressed in met31Δmet6Δ (note the strong expression at time zero) compared with met6Δ, but underexpressed in met32Δmet6Δ relative to met6Δ. This subset is heavily enriched for genes involved in transport (VBA2, YCT1, YIL166C, AGP3, YOL162W, YOL163W). Although YOL162W and YOL163W are officially unannotated, bioPIXIE indicates that they interact physically and genetically with a group of genes that is heavily enriched for Met biosynthesis (and associated functions) and thiamine metabolism. CRF1 is a TOR-activated transcriptional repressor of the ribosomal genes. Two genes, GRX8 and PDC6, are highly induced (8- and 64-fold, respectively) in met31Δmet6Δ but virtually uninduced in met32Δmet6Δ. GRX8 is one of several glutaredoxins whose expression is regulated by Met abundance. PDC6 is an isoform of pyruvate decarboxylase whose induction during sulfur limitation has also been observed by others (Boer et al., 2003). More recently, PDC6 was shown to be regulated by Met32p through a noncanonical binding motif (Cormier et al., 2010).
FIGURE 7:
Genes that are regulated differently by Met31p and Met32p. (A) Select genes that differ significantly between met31Δmet6Δ and met32Δmet6Δ. Strain order is met6Δ, met31Δmet6Δ, met32Δmet6Δ, met4Δ. (B) Selected genes that differ significantly between cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ. Strain order is met6Δ, cbf1Δmet31Δmet6Δ, cbf1Δmet32Δmet6Δ, met4Δ.
We next examined the Met31p/Met32p-dependent genes described earlier (including genes that also depend on Cbf1p) to determine the extent to which Met31p and Met32p can substitute for each other. With the exception of the genes mentioned previously, Met31p and Met32p appear to be able to substitute for each other in all Met31p/Met32p-dependent genes.
We suspected, on the basis of preliminary data, that the differences between met31Δ and met32Δ might be more pronounced in a cbf1Δ background. To narrow the long list of genes that differ between cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ, we increased the FDR stringency to 1%. Even then, 166 genes pass our criteria, the strongest of which are shown in Figure 7B. The top cluster is strongly enriched for Met and sulfur metabolism, again indicating that Met31 and Met32 play different roles even in the standard pathways with which they are associated. However, only two of the Met/sulfur-associated genes (MET2 and MET17) encode enzymes of the sulfur assimilation pathway. Five regulate glutathione abundance and metabolism (GRX8, DUG2, DUG3, GLO4, OPT1). This cluster also contains genes involved in other metabolic pathways, including NAD (POS5), NADPH (ZWF1), phospholipid biosynthesis (CHO2), and fatty acid metabolism (LEE1, based on bioPIXIE). The second cluster is very strongly enriched for genes involved in pheromone response, including, for example, STE2, FAR1, FUS2, BAR1, MFA1, MFA2, and so on.
Variants of the canonical Met31p/Met32p DNA-binding motif are associated with different functional responses
As described, we found that many genes depend on Met31p/Met32p, including some that require Met31p and Met32p for full induction and some that are only induced in the absence of Met31p and Met32p. Furthermore, we identified some genes that are regulated differently by Met31p and Met32p. Reasoning that there might be variants of the Met31p/Met32p TFBM that account for this wide variety of expression profiles, we looked for motif variants that distinguish between induced or repressed genes in met31Δmet32Δmet6Δ. Using gene sets described earlier, we identified genes that depend on Met31p/Met32p but exhibit no significant differences between met31Δmet6Δ and met32Δmet6Δ (in either the CBF1 or the cbf1Δ background). We divided the most convincing genes in this list (Figure 8A) into two groups, based on whether they are positively regulated (i.e., repressed in the deletion mutant; Figure 8B) or negatively regulated (i.e. induced in the deletion mutant; Figure 8C) by Met31p/Met32p. Using MEME, we derived different versions of the Met31p/Met32p TFBM from the promoters of each group, which we call the “positive variant” (Figure 8B) and the “negative variant” (Figure 8C). The positive variant is almost identical to the canonical motif shown in Figure 8A, with the exception that its upstream adenine residues are much more highly conserved. In contrast, the negative variant is substantially different from the canonical motif and contains an exact match to the Aft1p motif (Zhu et al., 2009).
FIGURE 8:
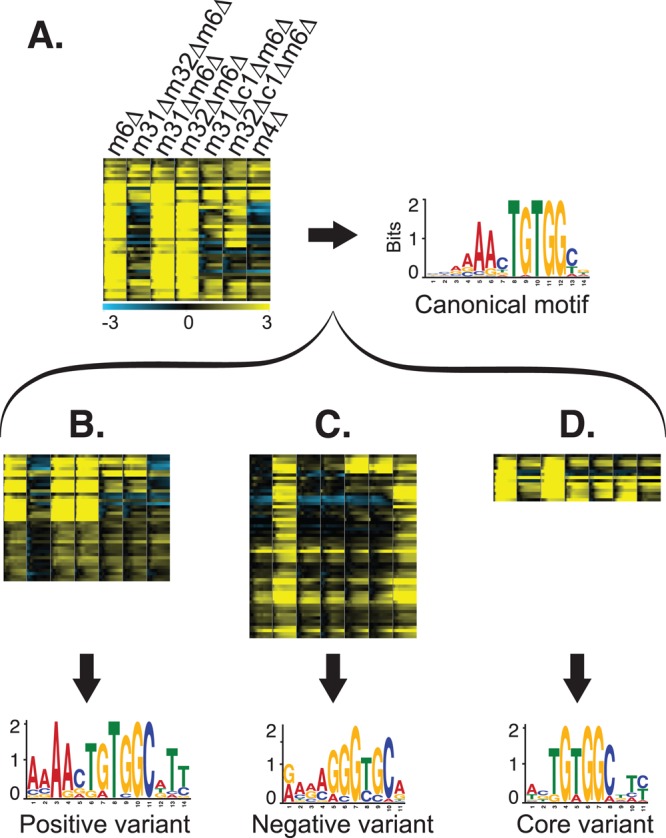
Met31p/Met32p motif variants. (A) Expression levels of genes that are induced early during Met starvation of met6Δ, shown for all strains (left), and the canonical Met31p/Met32p TFBM derived from the promoters of these genes (right). Strain order for all panels is met6Δ, met31Δmet32Δmet6Δ, met31Δmet6Δ, met32Δmet6Δ, cbf1Δmet31Δmet6Δ, cbf1Δmet32Δmet6Δ, met4Δ. (B) Genes induced by Met31p/Met32p (top) and the “positive” motif variant derived from their promoters (bottom). (C) Genes repressed by Met31p/Met32p (top) and the “negative” motif variant derived from their promoters (bottom). (D) Genes that differ between met31Δmet6Δ and met32Δmet6Δ (top) and the “core” motif variant derived from their promoters (bottom).
To search for motifs in genes that respond differently in met31Δmet6Δ and met32Δmet6Δ, we applied MEME to the promoters of the genes in Figure 8D. The result was a third variant, the “core variant,” consisting of the central, conserved TGTGG of the canonical motif. We also searched the promoters of the genes in Figure 7B and derived a motif very similar to the canonical Met31/Met32 motif.
One measure of the validity of a computationally derived TFBM is the functional specificity of the genes in which it occurs. To further characterize the three motif variants, we used MAST to search the genome for additional examples of each variant (using a more stringent MAST score of 50), filtered the MAST results for genes that change at least twofold in our data, and measured the functional enrichment of the resulting genes. Based on this analysis, the positive variant is found preferentially in genes involved in Met and sulfur metabolism, chromatin silencing, mitochondrial degradation glutathione metabolism, and spindle pole separation, whereas the core variant is more specific to Met and sulfur metabolism genes. (One complicating factor is that the core motif is a fragment of the positive variant, so some promoters match both the positive and the core motifs. However, the functional enrichment results hold even when such promoters are eliminated from the functional enrichment analysis.) The negative variant is found almost exclusively in genes involved in iron and ion homeostasis and transport, consistent with its close match to Aft1p, a major transcriptional regulator of iron metabolism (Yamaguchi-Iwai et al., 1995). As discussed later, this suggests that Met31p and Met32p regulate iron metabolism through direct and indirect mechanisms.
Transcription factor activity analysis
The previous analyses were designed to be stringent and restrictive. To obtain a broader overview of the physiological differences between the Met pathway regulatory mutants we studied, we calculated the transcription factor activity of each of 124 non–Met-pathway TFs (see Materials and Methods). Briefly, this metric quantifies the activity of each TF at each time point by performing linear regression of the genome-wide mRNA expression levels on the affinity with which the TF is predicted to bind to each gene's promoter region (Lee and Bussemaker, 2010). It provides information about how each of our deletions affects each regulon in early log phase (the initial time point), as well as during Met depletion. This approach is particularly useful because the biological roles of many yeast regulons have already been fairly well described. Thus we can summarize the functional effects of the TF deletions in terms of previously characterized regulons.
This approach is complementary to the analyses given earlier and differs from them in several key ways. First, a given regulon can span multiple cellular processes. Second, this approach does not tell us whether a Met pathway TF directly or indirectly influences each regulon. Third, this method captures more subtle interstrain differences because we do not impose any thresholds on the extent to which a regulon must differ among strains. Moreover, the analysis was performed on all genes in the genome, including those with subtle changes in gene expression.
The activity of all 124 non-Met TFs is represented as a hierarchically clustered heatmap in Supplemental Figure S5. The figure illustrates the number and diversity of cellular processes influenced by the Met pathway TFs, including Met biosynthesis and sulfur metabolism (MET4, MET31, MET32, MET28, CBF1; cluster 1), carbohydrate metabolism (TYE7 [cluster 1], MIG1), response to metals and metal ion homeostasis (RCS1, AFT2, YAP1, CAD1, YAP7; cluster 2), pH regulation (RIM101), phosphate metabolism (PHO4), ribosome biogenesis (RAP1, FHL1, SFP1; cluster 3), mitochondrial respiration (HAP2, HAP3, HAP5; cluster 4), and the cell cycle (STB1, SWI4, SWI6, MBP1; cluster 5). Importantly, this analysis does not indicate whether the regulation is direct or which genes are perturbed by TF deletion—this information is better supplied by the preceding analyses.
This analysis reveals marked differences among the Met pathway TFs, with striking examples highlighted in Figure 9. We see substantial differences between Cbf1p and Met31p/Met32p, Met31p and Met32p, and Cbf1p/Met31p and Cbf1p/Met32p. Consistent with the TF-dependency classifications given earlier, TF activity reveals that Cbf1p is negligible compared with Met31p/Met32p in methionine and sulfur metabolism: whereas deletion of CBF1 barely affects the methionine regulon, represented by the TFs Met4p, Met31p, Met32p, Cbf1p, and Met28p in Figure 9A, double deletion of MET31 and MET32 nearly obliterates the regulon's response to methionine depletion. met31Δ and met32Δ also differ in their effects on this regulon: met31Δ appears to increase and met32Δ to decrease the steady-state expression level of this regulon, consistent with the differences between Met31p and Met32p identified earlier using regression. In contrast, the activity of the ribosomal regulon TFs appears to depend almost entirely on CBF1 (Figure 9I and Supplemental Figure S5, cluster 3). Although these activities are all negative during the starvation, deleting CBF1 eliminates the time dependence seen in a CBF1 background. Striking differences between Met31p and Met32p can also be seen in the regulons controlled by Cbf1p, Tye7p, Mig1p, and Pho4p. Subtler differences in the respiration regulons (Hap2p, Hap3p, and Hap5p), and the Ino2p and Ino4p regulons are also seen (Figure 9, J and M).
FIGURE 9:

Transcription factor activities. Calculated activity profiles for selected TFs across all strains. Each panel shows TF activity (vertical axis) for the indicated TFs throughout the time courses for all eight strains. The strains are represented in the following order along the horizontal axis: met6Δ, cbf1Δmet6Δ, met31Δmet32Δmet6Δ, cbf1Δmet31Δmet6Δ, cbf1Δmet32Δmet6Δ, met31Δmet6Δ, met32Δmet6Δ, met4Δ. The triangles are color coded for each strain and represent the decrease in methionine over each time course. “TF activity” corresponds to the “t score” in Lee and Bussemaker (2010), which is the regression coefficient obtained from regression of the expression data on the position-specific affinity matrix for the given TF (see Materials and Methods).
Consistent with Figure 3, a striking number of regulons depend in opposite ways on Cbf1p and Met31p/Met32p (Figure 9, B, D, E, H, and M). Another unexpected result is the frequency with which Met31p/Met32p behaves as a transcriptional repressor, as indicated by high time-zero expression levels in met31Δmet32Δmet6Δ. Based on exponential-phase TF activities (time 0), Met31p/Met32p represses, and Cbf1p activates, the iron regulons represented by Rcs1p (Aft1p) and Aft2p, the copper regulon represented by Mac1p, the carbohydrate metabolism regulons represented by Tye7p and Mig1p, the Pho4p regulon, and the phospholipid biosynthesis regulons represented by Ino2p and Ino4p. Although our detailed analyses identified individual Cbf1p-dependent and Met31p/Met32p-dependent genes associated with these processes, few genes were regulated by both Cbf1p and Met31p/Met32p. This suggests that Cbf1p and Met31p/Met32p exert opposing effects on different genes that participate in these processes.
Synergy between Cbf1p and either Met31p or Met32p is reflected in regulons that are perturbed (relative to met6Δ) only in cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ. Specifically, Met31p/Met32p and Cbf1p cooperate to regulate respiratory gene expression (Hap2p, Hap3p, and Hap5p), response to oxidative stress (Stb5p), and the cell cycle (Stb1p, Swi4p, Swi6p, and Mbp1p), as well as Dig1p, Ste12p, and Mcm1p (Figure 9, J, K, N, and O).
The TF activity profiles highlight two interesting aspects of TF-binding specificity. First, the Cbf1p motif is almost identical to the Tye7p motif, as indicated by the strong correlation of Cbf1p and Tye7p activity in Figure 9B. This could be a biologically irrelevant coincidence, or, as discussed later, it could support the previously observed connections between sulfur/methionine metabolism and carbohydrate metabolism. Second, the activity profiles in Figure 9A show that the binding profiles of Met31p and Met32p differ slightly. The motif source used in this analysis (MacIsaac et al., 2006) reports the Met31p and Met32p motifs as GTGTGG and AACTGTGGC, respectively. As discussed later, the Met31p motif in (MacIsaac et al., 2006) is similar to the “core variant” that we derived from genes that differ in expression between met31Δmet6Δ and met32Δmet6Δ.
Cbf1 and Met31/Met32 have distinct roles in carbohydrate metabolism
Multiple lines of evidence, as described earlier, suggest that Met abundance regulates carbohydrate metabolism and, moreover, that Cbf1p and Met31p/Met32p might regulate carbohydrate metabolism differently. We used a 96-well plate-reader to measure in triplicate the growth of cbf1∆met6∆ and met31∆met32∆met6∆ in rich media containing 16 different carbon sources (see Materials and Methods). We found small but highly reproducible differences in growth rate and/or steady-state cell density between the two strains. In particular, cbf1∆met6∆ was growth impaired relative to met31∆met32∆met6∆ in glucose, inositol, fructose, and mannose but grew better than met31∆met32∆met6∆ in maltose and galactose (Supplemental Figure S6A). In additional experiments using the same panel of TF mutants in a MET6 background, we found that cbf1Δ, cbf1Δmet31Δ, and cbf1Δmet32Δ cells survive far longer than met6Δ and met31Δmet32Δ during Met starvation when the carbon source is galactose (unpublished data).
Cbf1p may cooperate with Met31p and Met32p to regulate cell cycle progression
The literature is rich in evidence that methionine abundance regulates cell cycle progression. Unlike other auxotrophs, methionine auxotrophs arrest in the G0/G1 phase of the cell cycle during methionine starvation. To understand whether any particular Met TF or TF combination is required for arrest, we measured bud index (the percentage of cells lacking a bud due to arrest in G0/G1) and cell density concurrently with mRNA expression in each of the methionine-depletion experiments described earlier. Bud index differs among strains across the length of the time course, even though each time course was started at the same cell density (an early exponential-phase Klett of ∼22). Consistent with previous findings (Unger and Hartwell, 1976; Petti et al., 2011), met6∆ cells arrest efficiently by 6 h (average 6-h bud index is 96%), whereas met4∆ cells do not (6-h bud index, 82%; Supplemental Figure S6B). Note that the 6-h bud index for met6Δ is comparable to the final, steady-state bud index for met6Δ measured over 27 h in (Petti et al., 2011). The consistently lower bud index of met4Δ was recapitulated in cbf1∆met31∆met6∆ and cbf1∆met32∆met6∆, whose 6-h bud index is significantly lower that of met6∆ (p = 2.5 × 10−5, Student's t-test) but not in cbf1Δmet6Δ, met31Δmet6Δ, met32Δmet6Δ, or met31Δmet32Δmet6Δ. Thus cell cycle arrest is impaired at all methionine concentrations when members of both classes of TF—Cbf1p and Met31p/Met32p—are deleted.
This result is consistent with the TF activity analysis, which shows that Cbf1p cooperates with Met31p and Met32p to regulate cell cycle genes (Figure 9, N and O). To understand in greater detail which cell cycle genes are regulated by Cbf1p/Met31p/Met32p, we examined the expression of genes associated with the “cell cycle” GO term, paying particular attention to those that are repressed during methionine starvation of met6Δ (Supplemental Figure S1, clusters 3, 4, and 19). Consistent with the TF activity analysis, many cell cycle genes repressed in met6∆ are somewhat less repressed in cbf1∆met31∆met6∆ and cbf1∆met32∆met6∆. However, it was difficult to identify particular genes that might contribute to the arrest defect, so we undertook a broader search for genes that are 1) potentially involved in cell cycle regulation and 2) expressed differently in cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ than in the other strains. We found 17 genes that meet these criteria (Supplemental Figure S6C), including nine that are less repressed (or more activated) and 8 that are less activated (or more repressed) in cbf1Δmet31Δmet6Δ and cbf1Δmet32Δmet6Δ. The former includes HHT1, HTA2, HTB1, HTB2, HHF2, YOX1, SCW10, GIC1, and PCL1, and the latter includes YLR112W, FAR1, PRM1, GIP1, BMH1, GLC8, CDC53, and MBF1. Two genes—BMH1 and MBF1—contain Cbf1p-binding motifs and none contains the Met31p/Met32p motif. However, given that cbf1∆met31∆met6∆ and cbf1∆met32∆met6∆ strains differ from met6∆ strains in their failure to arrest the cell cycle, we cannot rule out the possibility that the differences we detect may be the effect, rather than the cause, of the arrest.
DISCUSSION
The motivation for this study was to understand whether and how combinatorial transcriptional regulation enables the coordination of diverse cellular processes with the sulfur assimilation pathway, which synthesizes methionine. Methionine abundance influences many biological processes and is regulated by a relatively complex circuit involving three DNA-binding proteins—Met31p, Met32p, and Cbf1p. We hypothesized that there exists among these TFs a division of labor that facilitates the coordination of diverse processes. We collected a large set of data in which we measured genome-wide gene expression profiles under conditions that perturb the Met pathway: namely, starvation of a methionine auxotroph. We used a MET6 (methionine synthetase) deletion mutant as our control strain and compared this to a series of double- and triple-deletion mutants in which MET31, MET32, and/or CBF1 were also deleted. Data were obtained over a time series beginning in exponential growth phase and continuing beyond the cessation of growth caused by methionine starvation. We used a variety of new and established computational methods to investigate whether and how the TFs differ from each other, including two main complementary approaches. One, TF-dependency analysis, quantifies the dependence of each gene on each TF and characterizes the resulting gene classes according to biological function and DNA-binding motifs; the second, TF activity analysis, measures the aggregate behavior of genes in known regulons. As described, both approaches yield similar general conclusions.
First, we identified biological processes that were not previously known to be regulated by the Met TFs and are not typically associated with sulfur or Met metabolism. Second, we found clear functional distinctions between Met31p/Met32p and Cbf1p that support our hypothesis that there exists a division of labor among these TFs, including a clear biological distinction between the numerous processes activated by Met31p/Met32p and/or repressed by Cbf1p, on one hand, and activated by Cbf1p and/or repressed by Met31p/Met32p, on the other (Figure 2). Third, we found strong evidence for both positive (activator) and negative (repressor) activities for the Met TFs. Finally, we were able to distinguish the activities of MET31 and MET32 from one another and identified a variant of the Met31p/Met32p TFBM that appears to be specific to Met32p. The arrangement we observed—in which different transcription factors regulate overlapping but distinct genes and processes—is analogous to the “dense overlapping regulon” (DOR) described in Shen-Orr et al. (2002). The DOR has been hypothesized to function in the coordination of diverse cellular processes.
How is combinatorial regulation accomplished?
Our results suggest that methionine biosynthesis is coordinated with other biological processes through the use of two comparatively methionine-specific transcription factors—Met31p and Met32p—and a “generalist” transcription factor—Cbf1p. Although all three TFs regulate a wide variety of cellular processes, Met31p and Met32p are more important in methionine metabolism than Cbf1p, whose regulatory roles appear to be more evenly distributed across cellular metabolism and other processes such as sporulation and translation. The metabolism-related regulation we found is summarized in Figure 10, in which direct and indirect TF targets are superimposed on a skeletal backbone of metabolism. Clearly, Met31p and Met32p regulate every gene in the methionine biosynthetic pathway, whereas Cbf1 only regulates—jointly with Met31p/Met32p—the sulfate importers, MET14, and the two steps that depend on NADPH (MET16, MET10, and ECM17).
FIGURE 10:

Overview of metabolic genes regulated by the Met TFs. Key metabolic genes whose expression depends on Met31p/Met32p (green), Cbf1p (blue), or both (red) are highlighted in color. Putatively direct targets (which contain a TBFM and are expressed significantly differently from the met6Δ control) are surrounded by a black border, whereas indirect targets (no TFBM) are surrounded by a yellow border.
This diagram also highlights the broad influence of the Met regulators over central metabolism, including phosphatidylcholine biosynthesis (PSD1, CHO1); cardiolipin biosynthesis (PGS), pyrimidine biosynthesis (URA5); tryptophan (TRP3) and NAD biosynthesis (BNA2,3,4, 5, and 6); central metabolism (PDC6, ADH3, PYK2, IDH1, SDH1, SDH2, DAL7); fatty acid biosynthesis (FAS2, MCT1, HFA1); glutathione metabolism (GDH3); lipid metabolism leading to phosphatidyl choline (SER33); nitrogen metabolism (IDP1, GDH3, GLT1); allantoin degradation (DAL1, DAL2, DAL3); purine biosynthesis (IMD2); and the production of deoxyribonucleotides (RNR2, RNR4).
Different ways of implementing combinatorial regulation are evident from Figure 10. Many metabolic pathways are jointly regulated by Cbf1p and Met31p/Met32p. Sometimes this is accomplished by a joint target (BNA3, SER33, PDC6, and many genes in methionine biosynthesis itself) and sometimes by two different targets in the same pathway (PSD1 and CHO1; BNA2 and BNA4, BNA5, BNA6). Clearly, there must be other ways to accomplish joint regulation that were missed by our experimental design. For instance, SAM2 is jointly regulated by Opi1p and Met4p, and its promoter contains binding sites for Met31p/Met32p, Cbf1p, and Opi1p (Hickman et al., 2011). However, we found no dependence of SAM2 on Cbf1p. An article by McIsaac et al. (2011) explores an independent approach to regulation by inducing the Met pathway TFs one at a time These experiments show that CBF1 induction causes SAM2 repression, suggesting that many (if not all) of the regulatory effects we may have missed in the starvation experiments can be found with an alternative approach.
Methionine-dependent processes reflect the role of sulfur in diverse electron transfer reactions
Redox reactions are central to the mitochondrial electron transfer chain, which contains proteins rich in Fe-S clusters (Voet and Voet, 2010). Our results show that Met31p/Met32p represses genes involved in iron homeostasis (particularly iron transport) and Fe-S cluster biogenesis through both direct and indirect mechanisms. In support of direct repression, several iron-homeostasis genes and one gene involved in Fe-S biogenesis depend on MET/MET32 and contain the Met31p/Met32p TFBM. In support of indirect repression, we derived the Aft1p TFBM from genes that are significantly induced in met31Δmet32Δmet6Δ. Aft1p induces iron-related gene expression specifically in response to defects in Fe-S cluster biogenesis (Chen et al., 2004). Thus, deletion of MET31 and MET32 appears to cause defects in Fe-S biogenesis that ultimately activate Aft1p and the iron-homeostasis genes under its control. Taken together, these results suggest that methionine (or sulfur) deprivation, which activates Met31p and Met32p (via Met4p), may concomitantly repress iron import in order to reflect the lack of sulfur atoms available for Fe-S biogenesis. The electron transport chain also contains two copper atoms in complex IV. Consistent with this, Met31p/Met32p-dependent genes are enriched for copper ion homeostasis, suggesting that Met31p/Met32p more generally regulates the abundance of electron transport chain metals in response to sulfur availability.
Sulfur is also critical to the redox reactions central to the activity of antioxidants such as glutathione and glutaredoxin (Supplemental Figure S7). We found that many antioxidant biosynthetic genes are induced jointly by Met31p/Met32p and Cbf1p (GRX1, GLO4, FMO1, OYE2) or by Met31p/Met32p alone (GRX8, OPT1, GTO3, GLO2, MXR1, OYE3, DUG2, DUG3, CCP1). In turn, glutathione synthesis and function requires glutamate (found here to be regulated by Cbf1p), cysteine (regulated by Met31p/Met32p and Cbf1p), and NADPH generated by the pentose phosphate pathway (regulated by ZWF1 [also known as MET19]), which is in turn regulated by Cbf1p and Met31p/Met32p). We previously showed that met31Δmet32Δ is highly sensitive to oxidative stress resulting from hydrogen peroxide (Petti et al., 2011).
The connection between methionine and growth
The connection between methionine and translation is well known: the “start” codon AUG codes for methionyl-tRNA, ensuring that methionine is the first amino acid in the vast majority of proteins. Our data suggest that Cbf1p, with some help from Met31p and Met32p, contributes to the transcription of ribosomal genes. Cbf1p regulates ribosomal genes in Candida albicans (Lavoie et al., 2010). Thus S. cerevisiae may have retained some of this ancestral transcriptional coordination between Met metabolism and translational control.
MATERIALS AND METHODS
PCR-mediated gene replacement was used to delete MET6, CBF1, MET31, MET32, and MET4 in the S288C derivatives FY4 (MATa) and FY5 (MATα). Single-deletion mutants were crossed using standard methods to create the double and triple mutants listed in Table 2. Methionine-starvation filter-switching time courses were performed as described in detail in Petti et al. (2011). Briefly, for each strain, a single colony was grown to stationary phase in minimal medium containing limiting methionine (7.5 mg/l), set back in fresh methionine-limited medium, grown to early exponential phase, filtered in 5-ml aliquots onto nylon filters that were placed on methionine-limited Petri dishes, and grown to mid-exponential phase. Filters were transferred to Petri dishes lacking methionine and collected at 0, 10, 30, 60, 90, 120, 150, 180, 210, 240, and 360 min posttransfer for the MET6-background strains, and at 0, 30, 60, 90, 120, 180, and 360 min posttransfer for the met6Δ-background strains.
TABLE 2:
Strains used.
| Strain | Description |
|---|---|
| DBY11152 | met6::KanMX MATa |
| DBY11173 | met13::KanMX MATa |
| Spore from freshly dissected DBY11388 | met4::NatMX, freshly dissected from met4::NatMX/MET4 |
| 11197 | cbf1::KanMX MATa |
| 11242 | met31::KanMX;met32::KanMX MATα |
| 11243 | cbf1::KanMX;met31::KanMX MATa |
| 11244 | cbf1::KanMX;met32::KanMX MATa |
| 11396 | cbf1::KanMX;met6::KanMX MATa |
| 11398 | met31::NatMX;met6::KanMX MATa |
| 11385 | met32::NatMX;met6::KanMX MATa |
| 11409 | met31::NatMX;met32::KanMX;met6::KanMX MATα |
| 11412 | cbf1::KanMX;met31::NatMX;met6::KanMX MATa |
| 11413 | cbf1::KanMX;met32::NatMX;met6::KanMX MATa |
At each time point, one filter was used to measure cell density (by Klett and Coulter count) and bud index (by manual scoring of sonicated culture). A second filter was flash-frozen in liquid nitrogen and later used for genome-wide mRNA abundance measurements. As described further in Petti et al. (2011), RNA was isolated from the filters using phenol-chloroform extraction, labeled cRNA was synthesized using the Agilent Low-Input Linear Amplification Kit (Agilent Technologies, Santa Clara, CA), and the labeled cRNA was hybridized to 4 × 44k or 8 × 15k Agilent Yeast Oligo V2 microarrays together with reference cRNA from FY4 grown in a phosphate-limited chemostat.
Raw mRNA abundance measurements were processed using Perl, as described further in Petti et al. (2011). Briefly, for each gene in each time course, the final processed signal intensity of nonoutlier probes was floored to 350. Then the log base 2 of the ratio of red intensity to green intensity was computed and averaged across replicate probes, and every measurement in every strain was normalized (by subtraction) to the zero time point of met6Δ. As described further in Petti et al. (2011) and Hickman et al. (2011), a regression model with quadratic terms was used to identify differentially expressed genes and to estimate the maximum fold change for each gene. The statistical significance of differential expression was determined using an F-test to assess the fit of the expression profile to the regression model. An F-test p-value < 0.05 was taken to indicate time-dependence.
Comparison of met6Δ and met13Δ was accomplished using the regression model specified in the following equations. In the full regression model (Eq. 1), D classifies the genotype, such that D = 1 for met13Δ and D = 0 for met6Δ. Regression significance was calculated from the F statistic comparing the fit of the full model (Eq. 1) with the fit of the reduced model (Eq. 2). Q-VALUE software was used to calculate q-values from the p-values (Storey and Tibshirani, 2003). A gene was considered to differ between met6Δ and met13Δ if the q-value for the F statistic was <0.01.
Full regression model:
|
|
Reduced regression model:
|
|
For analysis of the met6Δ data in isolation, we used the 2669 genes that change by twofold or more in the met6Δ time course, depend significantly on time (q < 0.01, F-test), and behave similarly in met6Δ and met13Δ. For analysis of the total data set, we used the 3671 genes that change by twofold or more in at least one strain and either 1) depend significantly on time (p ≤ 0.05, F-test) or 2) are constitutively expressed ± twofold relative to met6Δ (p ≤ 0.05, t-test). All regression, correlation, and randomization analyses used in this work were coded in Matlab, version R2011a (The MathWorks, Natick, MA).
We developed a correlation-based method that we refer to as TF-dependency analysis to identify genes whose “wild-type” expression, as measured in met6Δ, depends on Cbf1 only (case 1), Met31/Met32 only (case 2), or both (case 3). First, an artificial gene expression template was constructed to match each case. For instance, the template for case 1 reflects that, for a gene that depends on Cbf1 but not Met31/Met32, expression levels in cbf1Δ differ from those in met6Δ and met31Δmet32Δmet6Δ. The three templates are as follows:
Case 1: met6Δ = 0 (at all time points), cbf1Δmet6Δ = 1, met31Δmet32Δmet6Δ = 0.
Case 2: met6Δ = 0 (at all time points), cbf1Δmet6Δ = 0, met31Δmet32Δmet6Δ = 1.
Case 3: met6Δ = 0 (at all time points), cbf1Δmet6Δ = 1, met31Δmet32Δmet6Δ = 1.
Second, the Pearson correlation between each gene and each template was calculated. A bootstrapped p-value for each correlation coefficient was calculated by repeating the correlation calculation on 1 × 104 randomly permuted sets of expression data and determining the fraction of “random” correlation coefficients that exceeded the “real” r in absolute value. Q-VALUE software was used to calculate q-values from the p-values (Storey and Tibshirani, 2003). Finally, a gene was assigned to a particular TF-dependency class if the q-value for the correlation between the gene and the template was at most 0.05 (corresponding to p ≤ 0.007).
Pairwise comparison of strains (comparison of met31Δmet6Δ to met32Δmet6Δ and comparison of cbf1Δmet31Δmet6Δ to cbf1Δmet32Δmet6Δ) was performed using the regression model specified in Eqs. 1 and 2, as well as a two-tailed Student's t-test. In the full regression model (Eq. 1, D classifies the genotype with respect to met31Δ and met32Δ (or cbf1Δmet31Δ and cbf1Δmet32Δ), such that D = 1 for met32Δ (or cbf1Δmet32Δ) and D = 0 for met31Δ (or cbf1Δmet31Δ). Regression significance was calculated from the F statistic comparing the fit of the full model (Eq. 1) with the fit of the reduced model (Eq. 2). Pairwise comparison of strains for the synergy analysis was performed analogously, using a regression model that allowed for simultaneous pairwise comparisons between more than two strains. Nonparametric p-values were calculated for the F statistic and the t statistic by randomly permuting the expression data 1 × 105 times. Q-VALUE software was used to calculate q-values from the p-values (Storey and Tibshirani, 2003). A gene was considered to differ between two strains if the q-value for the F statistic or the t statistic was <0.1 and the maximum expression difference between the strains was at least 1.5-fold. In some cases, as noted in the text, the stringency of the q-value cutoff was increased in order to limit the results to the strongest, most plausible, candidate genes. Regardless of the q-value threshold, the maximum p-value used in any filtering step was 0.002.
Hierarchical and k-means clustering were performed using the Multiple Experiment Viewer (Saeed et al., 2006). For both types of clustering, the Pearson correlation coefficient was used as the distance metric. The k-means clustering was performed using a range of values for k. The value giving the largest fraction of functionally enriched clusters and best discrimination between distinct expression profiles was used in subsequent analyses.
Functional enrichment of gene clusters was measured with respect to the “Biological Process” classifications specified in the GO (Ashburner et al., 2000). Enrichment for GO terms was measured using the GOTF (Boyle et al., 2004; available at http://go.princeton.edu/cgi-bin/GOTermFinder), using the FDR option for multiple hypothesis correction. Here we report enrichments with FDR at most 0.1.
DNA sequence motifs were identified using the Web-based motif-detection algorithm MEME (http://meme.sdsc.edu/meme/intro.html; Bailey et al., 2009). Before using MEME, Regulatory Sequence Analysis Tools (http://rsat.ulb.ac.be) were used to obtain the promoter sequence, ranging from positions −800 to −1 and excluding overlapping open reading frames, upstream of every gene (Thomas-Chollier et al., 2011). This genome-wide set of promoter sequences was used to generate the nucleotide-frequency background model used by MEME. In MEME, motif width was allowed to range from 6 to 12 nucleotides. All other MEME parameters were set to default values. To identify additional instances of each motif throughout the genome, the motifs identified by MEME were used as input for the companion program MAST (http://meme.sdsc.edu/meme/cgi-bin/mast.cgi). MAST takes as input a list of motifs represented as position-specific scoring matrices (such as those produced by MEME) and searches a user-specified sequence database (here the S. cerevisiae upstream sequence database supplied with the MEME suite) for instances of those motifs. MAST assigns each input sequence an “E value,” which reflects the expected number of sequences in a random sequence database that match the input motifs at least as well as the actual sequence. Although the E value is not a p-value, it is calculated from the p-values of the motif matches in the input sequence. We applied two different E value cutoffs, depending on the nature of the analysis: for MAST hits that were subsequently filtered using additional criteria (e.g., those in Figure 1), we used hits with an E value of <500, as in the analysis of Met TFs by Lee et al. (2010). For more stringent motif identification, we used MAST hits with an E value of <50. An E value of 500 corresponds roughly to a p-value of 0.08 in our analysis, and an E value of 50 roughly corresponds to a p-value of 0.008.
Transcription factor activities were calculated using R and the REDUCE software suite (http://bussemakerlab.org/REDUCE), following the approach presented in Lee and Bussemaker (2010). These calculations are based on the assumption that the activity of a transcription factor in a given condition can be inferred from the transcriptional response of its target genes. The method takes as input 1) a genome-wide promoter sequence, 2) a position-specific affinity matrix for each TF derived from the data in MacIsaac et al. (2006), and 3) a matrix of expression levels whose rows correspond to genes and whose columns, in our case, represent the different time points in our time course. The first two inputs were combined to construct a promoter affinity matrix that records the affinity of each TF (column) for each promoter (row). Next, for each time point, multivariate linear regression of the expression level on the promoter affinity was performed, and the regression coefficient corresponding to the slope was taken to represent the TF activity. “TF activity” is the same as the “t score” in Lee and Bussemaker (2010).
Growth curves in assorted carbon sources were performed using a BioTek plate reader (BioTek, Winooski, VT). For a given strain, a single colony was grown to saturation (15–19 h with shaking at 30°C) in 5 ml of rich medium made with 2% sucrose (instead of glucose). The cells were then washed in phosphate-buffered saline and diluted to 5 × 105 cells/ml in 200 μl of rich medium containing the desired carbon source in a flat-bottomed, polystyrene 96-well plate covered with a Breathe-Easy membrane (Sigma-Aldrich, St. Louis, MO). Carbon sources were supplied at a final concentration of 2%. Each strain was grown in triplicate in each carbon source with continuous shaking at 30°C, and the OD600 was recorded every 30 min for 70 h.
Supplementary Material
Acknowledgments
This research was supported by National Institute of General Medical Sciences Center for Quantitative Biology Grant GM071508 and National Institutes of Health Grant GM046406 (to D.B.). A.A.P. acknowledges support from a Ruth Kirschstein Cancer Training Grant (T32 CA-009528). R.S.M. acknowledges support from a National Science Foundation Graduate Research Fellowship. H.J.B. acknowledges support from National Institutes of Health Grant HG003008 and a John Simon Guggenheim Foundation Fellowship. We thank Jonathan L. Weinstein for advice on statistical analysis and Sanford Silverman and Viktor Boer for discussions of experimental design.
Abbreviations used:
- Fe-S
iron–sulfur
- Met
methionine
- TF
transcription factor
- TFBM
transcription factor–binding motif
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-03-0233) on June 13, 2012.
REFERENCES
- Ashburner M, et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RE, Masison DC. Isolation of the gene encoding the Saccharomyces cerevisiae centromere-binding protein CP1. Mol Cell Biol. 1990;10:2458–2467. doi: 10.1128/mcb.10.6.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RE, Fitzgerald-Hayes M, O'Brien TC. Purification of the yeast centromere binding protein CP1 and a mutational analysis of its binding site. J Biol Chem. 1989;264:10843–10850. [PubMed] [Google Scholar]
- Blaiseau PL, Thomas D. Multiple transcriptional activation complexes tether the yeast activator Met4 to DNA. EMBO J. 1998;17:6327–6336. doi: 10.1093/emboj/17.21.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaiseau PL, Isnard AD, Surdin-Kerjan Y, Thomas D. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol Cell Biol. 1997;17:3640–3648. doi: 10.1128/mcb.17.7.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer VM, De Winde JH, Pronk JT, Piper MDW. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem. 2003;278:3265–3274. doi: 10.1074/jbc.M209759200. [DOI] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bram R, Kornberg R. Isolation of a Saccharomyces cerevisiae centromere DNA-binding protein, its human homolog, and its possible role as a transcription factor. Mol Cell Biol. 1987;7:403–409. doi: 10.1128/mcb.7.1.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Davis RW. Yeast centromere binding protein CBF1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell. 1990;61:437–446. doi: 10.1016/0092-8674(90)90525-j. [DOI] [PubMed] [Google Scholar]
- Capaldi AP, Kaplan T, Liu Y, Habib N, Regev A, Friedman N, O'shea EK. Structure and function of a transcriptional network activated by the MAPK Hog1. Nat Genet. 2008;40:1300–1306. doi: 10.1038/ng.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen OS, Crisp RJ, Valachovic M, Bard M, Winge DR, Kaplan J. Transcription of the yeast iron regulon does not respond directly to iron but rather to iron-sulfur cluster biosynthesis. J Biol Chem. 2004;279:29513–29518. doi: 10.1074/jbc.M403209200. [DOI] [PubMed] [Google Scholar]
- Cormier L, Barbey R, Kuras L. Transcriptional plasticity through differential assembly of a multiprotein activation complex. Nucleic Acids Res. 2010;38:4998–5014. doi: 10.1093/nar/gkq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MJ, Petti AA, Ho-Shing O, Silverman SJ, McIsaac RS, Lee TA, Botstein D. Coordinated regulation of sulfur and phospholipid metabolism reflects the importance of methylation in the growth of yeast. Mol Biol Cell. 2011;22:4192–4204. doi: 10.1091/mbc.E11-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser P, Su N-Y, Yen JL, Ouni I, Flick K. The yeast ubiquitin ligase SCFMet30: connecting environmental and intracellular conditions to cell division. Cell Div. 2006;1:16. doi: 10.1186/1747-1028-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras L, Thomas D. Functional analysis of Met4, a yeast transcriptional activator responsive to S-adenosylmethionine. Mol Cell Biol. 1995a;15:208–216. doi: 10.1128/mcb.15.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras L, Thomas D. Identification of the yeast methionine biosynthetic genes that require the centromere binding factor 1 for their transcriptional activation. FEBS Lett. 1995b;367:15–18. doi: 10.1016/0014-5793(95)00528-h. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol. 2010;8:e1000329. doi: 10.1371/journal.pbio.1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Bussemaker HJ. Identifying the genetic determinants of transcription factor activity. Mol Syst Biol. 2010;6:412. doi: 10.1038/msb.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TA, Jorgensen P, Bognar AL, Peyraud C, Thomas D, Tyers M. Dissection of combinatorial control by the Met4 transcriptional complex. Mol Biol Cell. 2010;21:456–469. doi: 10.1091/mbc.E09-05-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, Petti AA, Bussemaker HJ, Botstein D. Perturbation-based analysis and modeling of combinatorial regulation in the yeast sulfur assimilation pathway. Mol Biol Cell. 2012;23:2993–3007. doi: 10.1091/mbc.E12-03-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, Silverman SJ, Mcclean MN, Gibney PA, Macinskas J, Hickman MJ, Petti AA, Botstein D. Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae. Mol Biol Cell. 2011;22:4447–4459. doi: 10.1091/mbc.E11-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Murray DB, Beckmann M, Kitano H. Regulation of yeast oscillatory dynamics. Proc Natl Acad Sci USA. 2007;104:2241–2246. doi: 10.1073/pnas.0606677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CL, Troyanskaya OG. Context-sensitive data integration and prediction of biological networks. Bioinformatics. 2007;23:2322–2330. doi: 10.1093/bioinformatics/btm332. [DOI] [PubMed] [Google Scholar]
- Patton EE, Peyraud C, Rouillon A, Surdin-Kerjan Y, Tyers M, Thomas D. SCF(Met30)-mediated control of the transcriptional activator Met4 is required for the G(1)-S transition. EMBO J. 2000;19:1613–1624. doi: 10.1093/emboj/19.7.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti AA, Crutchfield CA, Rabinowitz JD, Botstein D. Survival of starving yeast is correlated with oxidative stress response and nonrespiratory mitochondrial function. Proc Natl Acad Sci USA. 2011;108:E1089–E1098. doi: 10.1073/pnas.1101494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilpel Y, Sudarsanam P, Church GM Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- Rouillon A, Barbey R, Patton EE, Tyers M, Thomas D. Feedback-regulated degradation of the transcriptional activator Met4 is triggered by the SCFMet30 complex. EMBO J. 2000;19:282–294. doi: 10.1093/emboj/19.2.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp S, Summers E, Lo H-J, Madhani H, Fink G. MAP kinase and cAMP filamentation signaling pathways converge on the unusually large promoter of the yeast FLO11 gene. EMBO J. 1999;18:1257–1269. doi: 10.1093/emboj/18.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- Siggers T, Duyzend MH, Reddy J, Khan S, Bulyk ML. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol Syst Biol. 2011;7:1–14. doi: 10.1038/msb.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Ramsey SA, Marelli M, Marzolf B, Hwang D, Saleem RA, Rachubinski RA, Aitchison JD. Transcriptional responses to fatty acid are coordinated by combinatorial control. Mol Syst Biol. 2007:3, 115. doi: 10.1038/msb4100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N-Y, Ouni I, Papagiannis CV, Kaiser P. A dominant suppressor mutation of the met30 cell cycle defect suggests regulation of the Saccharomyces cerevisiae Met4-Cbf1 transcription complex by Met32. J Biol Chem. 2008;283:11615–11624. doi: 10.1074/jbc.M708230200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Surdin-Kerjan Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1997;61:503–532. doi: 10.1128/mmbr.61.4.503-532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, Defrance M, Medina-Rivera A, Sand O, Herrmann C, Thieffry D, van Helden J. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39:W86–W91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Kudlicki A, Rowicka M, McKnight SL. Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science. 2005;310:1152–1158. doi: 10.1126/science.1120499. [DOI] [PubMed] [Google Scholar]
- Unger MW, Hartwell LH. Control of cell division in Saccharomyces cerevisiae by methionyl-tRNA. Proc Natl Acad Sci USA. 1976;73:1664–1668. doi: 10.1073/pnas.73.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet D, Voet JG. Biochemistry. New York: John Wiley and Sons; 2010. [Google Scholar]
- Yamaguchi-Iwai Y, Dancis A, Klausner RD. AFT1: a mediator of iron regulated transcriptional control in Saccharomyces cerevisiae. EMBO J. 1995;14:1231–1239. doi: 10.1002/j.1460-2075.1995.tb07106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, et al. High-resolution DNA binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.